

Abstract
The tubby mouse is a phenotypic model for sensorineural deafness and retinal dystrophy including Usher Syndrome Type 1. Thioredoxin is a small 13 kDa protein which, when ubiquitously expressed as a transgene in the mouse, provides protection against multiple disease states including light-induced and oxidative stress-induced neurodegeneration and is down-regulated in the tubby retina. We tested if overexpression of human Thioredoxin in the tubby mouse inhibits retinal degeneration and loss of visual function. Electroretinography, immunocytochemistry, quantitative histology, RTPCR and Western blots were used to obtain data which showed that Thioredoxin overexpression prevented loss of photoreceptors and retinal function. Analysis of signal pathways showed that Thioredoxin upregulated neurotrophic factors BDNF and GDNF and activated survival signaling pathways Akt, Ras/Raf1/ and the ERKs while inhibiting the ASK1/JNK apoptosis pathway. Relationships between the tubby gene, its pathological phenotype and regulation of the Thioredoxin system remain to be established.
Keywords: Trx, tubby, retina, degeneration, oxidative stress, neurotrophins, neuroprotection, survival, apoptosis, transgene
INTRODUCTION
Thioredoxin (Trx) is a small (13-kDa), ubiquitous protein with two redox-active cysteine residues, -Cys-Gly-Pro-Cys-, in its active center (Holmgren, 1985). Trx plays an important role in various signaling events involved in the control of numerous physiological processes including cell growth, survival/apoptosis, development, differentiation and proliferation(Hirota et al., 1999; Saitoh et al., 1998). In response to various stress conditions, Trx increases the activity of several transcription factors, such as nuclear factor kappa B (NF-kB), activator protein (AP-1), p53 and related co-activators such as Ref-1, which depend on reduced sulfhydryl groups for their function(Ando et al., 2008; Henderson, Jr. et al., 2002; Ueno et al., 2007). Trx can also be secreted to the extracellular matrix where it functions as an autocrine growth factor and co-cytokine(Rosen et al., 1995; Silberstein et al., 1993). The Trx transgenic (Tg-Trx) mouse, which ubiquitously overexpresses human Trx under the control of beta-actin promoter, has been generated and shown to display various phenotypes including an elongated lifespan (Cho et al., 2003) and protection against multiple disease states such as ischemic injury(Turoczi et al., 2003); cigarette smoke-induced lung inflammation and emphysema (Sato et al., 2008; Turoczi et al., 2003); congestive heart failure(Satoh et al., 2007); diabetes mellitus(Yamamoto et al., 2008), chronic pancreatitis(Ohashi et al., 2006) and gastric mucosal injury(Tan et al., 2007). In recent years, data show that Trx is not only a trophic factor for neuronal cells homeostasis(Akterin et al., 2006; Masutani et al., 2004; Matsuura et al., 2007), but is also a neuroprotective factor. Intravenous administration of recombinant hTrx in mice was reported to decrease brain damage subsequent to transient focal cerebral ischemia(Hattori et al., 2004). Moreover, overexpression of Trx and intravitreal injection of recombinant human Trx protein prevent light-induced photoreceptor degeneration(Tanito et al., 2005; Tanito et al., 2007), and protect retinal ganglion cells against oxidative stress-induced neurodegeneration in vitro and in vivo(Munemasa et al., 2008).
The homozygous tubby (tub/tub) mouse has been described as a phenotypic model of Usher syndrome type I found in humans and characterized by a progressive loss of photoreceptor cells beginning 2-3 weeks after birth. The detailed mechanism underlying photoreceptor degeneration in this mouse remains unknown. Our data show that Trx in the retinas of tubby mice was significantly lower than that of age-matched C57BL/6J mice. It has been suggested that the lower levels of Trx in the retinas of tubby mice may be related to the degeneration of the photoreceptors(Kong et al., 2007). We further demonstrated that systemic administration of sulforaphane is able to effectively up-regulate the Trx system and delay inherited photoreceptor degeneration(Kong et al., 2007) in the tubby retina. However, the mechanisms underlying the protective effect of Trx in the tubby mouse remain unknown.
In our present study, we make novel observations showing that overexpression of Trx delays the inherited photoreceptor degeneration and preserves functional vision via activating survival signaling pathways including Akt (protein kinase B), Ras/Raf/, extracellular signal-regulated kinase (ERKs) signal cascade, inhibition of apoptosis signal-regulating kinase 1 (ASK1) and c-Jun N-terminal kinase (JNK) signal pathway. In addition, overexpression of Trx increases the expression of brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF).
MATERIALS AND METHODS
Animals
The tubby and C57BL/6J mice used in this study were from the Jackson Laboratory. Transgenic Trx (Tg-Trx) mice were generated by Dr. Junji Yodoi, Kyoto University, Kyoto, Japan. The tubby Tg-Trx mouse was generated by cross-breeding these two lines (tub/Trx). All animals were born and raised in a 12-h on versus 12-h off cyclic light environment at an illumination of 50-60 lux. Animals were cared for and handled according to the Association for Research in Vision and Ophthalmology statement for the use of animals in vision and ophthalmic research and with IACUC approved animal use protocols, which comply with the University of Oklahoma Faculty of Medicine guidelines for use of animals in research. Prior to conducting experiments routine polymerase chain reaction (PCR) was used to identify the genotypes of various mice as described previously (Kong et al., 2006). Genomic DNA from mouse tail was amplified by PCR using the following two sets of primers: tubby (forward primer, 5′-ccgtgtcacacaggcttct-3′ and reverse primer, 5′-ctgggcaccatgcgtaca-3′) and Tg-Trx (Forward, 5’-AAGCAGATCGAGAGCAAGACT-3’, Rev, 5’-TCCTTATTGGCTCCAGAAAAT-3’. After 94°C for15 min, 35 cycles were run (94°C 30 s, 55°C 30 s, 72°C 30 sec terminating with 72°C 2 min, 10°C 10 min) and the PCR products of tubby were digested with the SmlI restriction enzyme (New England Biolabs, Ipswich, MA) and analyzed by 5% MetaPhor agarose gel (Lonza, Rockland, ME) electrophoresis. The 101 bp wild-type (+/+) product is undigested, the tubby mutant product is digested to 52 bp and 49 bp products, and tub/+ shows all three products of 101 bp, 52 bp, and 49 bp. The PCR product of Tg-Trx were analyzed by 1.5% agarose gel and is 277 bp.
Morphological evaluation of photoreceptor rescue by quantitative histology
Morphological analysis was performed as described(Cao et al., 2001; Yu et al., 2004). After electroretinography, mice were killed by an overdose of carbon dioxide. The eyes were enucleated, fixed, embedded in paraffin and 5 μm thick sections were cut along the vertical meridian to allow comparison of all regions of the eye. In each of the superior and inferior hemispheres, outer nuclear layer (ONL) thickness was measured at nine defined points. Each point was centered on adjacent 220 μm lengths of retina. The first point of measurement was taken at approximately 220 μm from the optic nerve head, and subsequent points were located more peripherally. In addition to mean ONL thickness for the entire retinal section, ONL thickness of the region of retina most sensitive to the damaging effects of light was compared among different groups of mice. In each of the experiments where ONL thickness was quantified, a single section from each of 10 eyes was measured. The regression analysis with power calculation was performed using Microsoft Excel program.
Evaluation of photoreceptor cell function by electroretinography
Electroretinograms (ERG) was performed as described previously(Kong et al., 2006; Yu et al., 2004). Briefly, animals were kept in total darkness overnight before ERG recording. Pupils were dilated with 1% atropine and 2.5% phenylephrine HCl. Animals were anesthetized intramuscularly with a ketamine–xylazine mixture. ERG responses were recorded with a silver chloride needle electrode placed on the cornea with 1% tetracaine topical anesthesia. A reference electrode was positioned at the nasal fornix, and a ground electrode on the foot. The duration of light stimulation was 10 ms. The band pass was set at 0.3–500 Hz. The light intensity used in this study was 3.49 log cd-s/m2 preset by an LKC Electroretinogram system (EPIC-2000, LKC Technologies, Inc., Gaithersburg, MD 20879). Five responses were averaged with flash intervals of 20 s. For quantitative analysis, the B-wave amplitude was measured between a- and b-wave peaks. The ERG waveforms for both eyes in the same animal were simultaneously recorded and compared as right/left eye ratio of the b-wave amplitude.
Semiquantitative Reverse Transcription (RT)-PCR
Reverse transcription (RT)-PCR was performed as described previously(Zhou et al., 2005). For RT-PCR, 4μg RNA was mixed in water with random hexamers (50 ng/μl) and 10 mM dNTPs heated to 65 °C for 5 min. The 20-μl RT reaction contained RNA, primers, 5 mM MgCl2, 10 mM dithiothreitol, 0.5 mM dNTPs, 40 units of RNase inhibitor (RNase OUT), and 50 units of Superscript II reverse transcriptase (Invitrogen; Carlsbad, CA). The mixture was heated at 42 °C for 1 h and then at 70 °C for 15 min. After chilling on ice, 2 units of RNase H were added to each reaction and incubated at 37 °C for 20 min. PCR was carried out in 50-μl reaction volumes, containing 2 μl of cDNA, 2 units of TaqDNA polymerase (Promega, Madison, WI), 0.2 mM dNTPs, 10 μM each of forward and reverse primers, and 1.5 mM MgCl2. Amplification cycles were: one cycle at 94 °C for 3 min, followed by 35 cycles of 94 °C for 20 s, 58 °C for 45 s, and 72 °C for 1 min, terminating with 72 °C for 10 min. The products were run on 1% agarose gel containing 10ng/ml ethidium bromides and visualized under UV light. Gene expression was examined by semi-quantitative RT-PCR using the following primers:
| Trx (mouse) | forward, 5’-CAAATGCATGCCGACCTTCCAGTT-3’ |
| reverse, 5’-TGGCAGTTGGGTATAGACTCTCCA-3’ | |
| Trx (human) | forward, 5’-TGGTGAAGCAGATCGAGAGCAAGA-3’ |
| reverse, 5’-CACGCAGATGGCAACTGGTTATGT-3’ | |
| Nrf2 | forward, 5’-AGTTCTCGCTGCTCGGACTA-3’ |
| reverse, 5’-AGGCATCTTGTTTGGGAATG-3’; | |
| BDNF | forward, 5’-GATGCTCAGCAGTCAAGTGCCTTT-3’ |
| reverse, 5’-GACATGTTTGCGGCATCCAGGTAA-3’; | |
| GDNF | forward, 5’-AGTCATTCCCTGTTGCCTTCTCCA-3’ |
| reverse, 5’-GCACCAGCCTTCCACATAAAGCAA-3’; | |
| Raf | forward, 5’-TGCAATTGGGAACTTGGCTCCTTG-3’ |
| reverse, 5’-AGGCCATCCACACAGGACACCTTA-3’; | |
| GAPDH | forward, 5’- TGTGATGGGTGTGAACCACGAGAA-3’ |
| reverse, 5’- GAGCCCTTCCACAATGCCAAAGTT-3’; |
Western Blot Analysis
Western blot analysis was performed as described previously(Kong et al., 2006; Yu et al., 2004). The retina tissues were sonicated in 0.0625 M Tris-HCl, pH 6.8, then centrifuged for 15 min and the supernatant assayed for protein using a Bradford assay. Aliquots (30 μg) of the sonicated supernatant were loaded onto SDS-PAGE mini-gels, electrophoresed, and transferred to nitrocellulose paper. After transferring, blots were washed for 2 × 10 min in TTBS (0.1% Tween 20 in 20mM Tris-HCl, pH 7.4, and 410mM NaCl) and blocked with 10% BSA in TTBS or with 1% milk and 1% BSA in TTBS for 2 h at room temperature or overnight at 4 °C. Blots were incubated for 2 h at room temperature with primary antibody. Sources of antibodies are: rabbit anti-BDNF polyclonal antibody (Alomone Labs, Ltd, Israel), rabbit anti-GDNF polyclonal antibody (Biovision, Mountain View, CA), rabbit anti-Nrf2 polyclonal antibody (Santa Cruz; Delaware, CA), rabbit anti-JNK polyclonal antibody (Abcam; Cambridge, MA), rabbit anti-ERK1/2 antibody and rabbit anti-phospho-Akt(ser 473) antibody (Cell Signaling Technology, Inc. Danvers, MA). Mouse anti-Ras monoclonal antibody (Millipore, Billerica, MA), rabbit anti-Trx polyclonal antibody (Abcam; Cambridge, MA), mouse anti-human Trx monoclonal antibody, rabbit anti-mouse Trx polyclonal antibody (kindly provided by Dr. Masaki Tanito, Kyoto University, Kyoto, Japan). Following primary antibody incubations, blots were washed three times for 5 min each with TTBS, then incubated for 1 h with HRP-linked secondary antibodies, washed four times for 10 min each with TTBS, and developed by ECL. In some instances, membranes were stripped by incubation in stripping buffer (62.5mM Tris-HCl (pH 6.8), 2% SDS, and 100mM 2-mercaptoethanol) for 30 min at 50 °C and reused.
Histopathology
Mouse eyes were enucleated, rinsed in PBS, and fixed with 10% neutral buffered formalin for 24 hours at room temperature. After paraffin embedding, whole eyes were cut into 5μm thick sections, mounted on positively charged slides and air dried overnight. After deparaffinization and rehydration, slides were stained with hematoxylin and eosin.
Immunohistochemistry
Immunohistochemistry staining was performed as described previously(Cao et al., 2000; Kong et al., 2007). Briefly, the eye was enucleated then fixed with 4% paraformaldehyde in phosphate-buffered saline for 4 h. The whole eye was cut along the vertical meridian. The sections were incubated in primary antibodies. The secondary antibody (Vector Laboratories, Burlingame, CA) was labeled with fluorescent isothiocyanate (green). Control sections were treated in the same way with omission of primary antibody or with normal rabbit or mouse serum. Sections were viewed and photo-graphed with a confocal laser scanning microscope (IX81-FV500; Olympus, Melville, NY). The microscope system software (FluoView; Olympus) was used for analysis.
TdT-Mediated Digoxigenin-dUTP Nick-End Labeling Assay
Detection of apoptosis using the TUNEL (TdT-mediated digoxigenin-dUTP nick-end labeling) method was completed using a commercially available in situ apoptosis detection kit (Biovision, Mountain View, CA) as described previously(Zhou et al., 2007). Staining for the TUNEL assay was performed according to the manufacturer’s protocol. TUNEL-positive cells were identified with a Nikon Eclipse 800 microscope, and images were captured by a digital camera and stored on a hard drive. The percentage of apoptotic cells was calculated by dividing TUNEL-positive cells by the total number of cells visualized by Nomarski optics in the same field. Three digitized images of similar total cell numbers were selected from each section for counting and averaging and were considered as one independent experiment. Three independent experiments were then averaged.
Statistical Analysis
The experiments were each performed three times. Mean values from three experiments were calculated and are expressed as means ± SD. Statistical analyses were performed using one way ANOVA and a statistical difference was considered significant at a P value of <0.05.
RESULTS
Animal genotype
To verify the genotype of the Tubby, Tub/Trx, Tg-Trx, Tub/WT, and WT/WT mice, DNA from tail clips were subjected to PCR using primers that produced amplimers distinct from the normal, as well as from the human Trx gene and the mouse tubby gene. The results of such monitoring are seen in Figure 1. The Trx genotype has the 277 bp band in Tub/Trx and Tg/Trx mice, no band in Tubby, Tub/WT and WT/WT mice (Fig.1A). For the Tubby genotype, the normal gene fragment was101 bp and was not sensitive to cutting by SmlI, whereas the tubby gene amplimer had an internal SmlI site and yielded two fragments of 49 bp and 52 bp when subjected to restriction digest. The 101 bp band in WT/WT and Tg-Trx mice is undigested; the Tubby and Tub/Trx mice have both the 52 bp and 49 bp products, whereas the Tub/WT genotype shows all three products of 101 bp, 52 bp and 49 bp(see Fig.1B).
Figure 1. Genotype analysis.
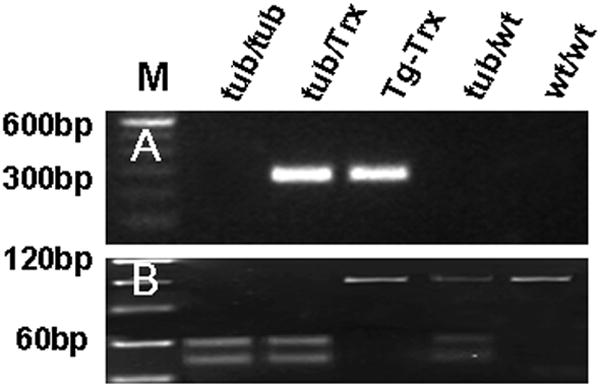
(A) Trx genotyping. The 277 bp band is Tub/Trx and Tg/Trx mice. (B) Tubby genotyping. The 101 bp WT/WT and Tg-Trx mice product is undigested, the Tubby and Tub/Trx mice mutant product is digested to 52 bp and 49 bp products, and Tub/WT shows all three products of 101 bp, 52 bp and 49 bp.
Overexpression and localization of Trx in the retina of Tubby and Tub/Trx mice
Representative data for expression of the Trx gene and protein are presented in Figure 2. Semi-quantitative RT-PCR was used to amplify the Trx gene of retinas from P14 mice. The mouse Trx gene was expressed in the Tubby, Tub/Trx, Tg-Trx, WT/WT and Tub/WT, but the human Trx gene was only expressed in the Tub/Trx and Tg-Trx mice (Fig. 2A). Western immunoblots of Trx retinas from P14 mice indicated that mouse Trx was down regulated in the retina of Tubby and Tub/Trx mice whereas human Trx was over expressed in the retina of Tub/Trx and Tg-Trx mice(Fig. 2B). We also detected the Trx protein by immunofluorescence microscopy of retinal sections from Tubby (Fig. 2C-a), Tub/Trx(Fig. 2C-b), Tg-Trx(Fig. 2C-c),Tub/WT(Fig. 2C-d) and WT/WT(image not shown) at P34. Trx staining was observed in inner segment (I), outer plexiform layer (OPL), inner plexiform layer (IPL), and ganglion cell layer (GCL). Fluorescence Intensity of Trx labeling was remarkably lower in the retinal sections from tubby (Fig. 2C-a) compared to heterozygous Tub/WT (Fig. 2C-d) mice. The Trx transgene clearly increased the labeling intensity of Trx in the Tub/Trx mice (Fig. 2C-b) almost to the levels seen in the mouse with the Trx transgene expressed on the wildtype background (Fig. 2C-c) and in the heterozygous tubby retina (Fig. 2C-d).
Figure 2. Retinal expression of mouse Trx and human Trx in the Tub/Trx mice.
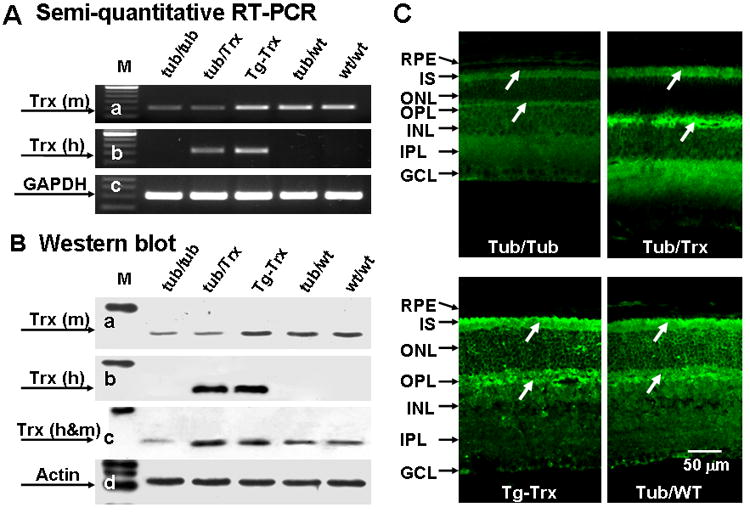
Representative results of (A) Semi-quantitative RT-PCR of retinas from P14 mice. Intensity of each band was standardized by the band intensity of GAPDH. (B) Western blots of retinas from P14 mice. Intensity of each band was standardized by the band intensity of actin. (C) Immunofluorescent localization of retinas from (a) Tubby, (b) Tub/Trx, (c) Tg-Trx and (d) Tub/WT mice at P14. GCL, ganglion cell layer; IPL, inner plexiform layer; INL, inner nuclear layer; OPL, outer plexiform layer; ONL, outer nuclear layer; I, inner segment; OS, outer segment; and RPE, retinal pigment epithelium. The scale bar represents 50 μm.
Trx expression provides protection at the morphological and functional level to Tub/Trx photoreceptor cells
As shown in figure 3, the extent of photoreceptor degeneration in Tubby, Tub/Trx, Tg-Trx, Tub/WT and WT/WT mice at P34 was evaluated histologically by measuring the thickness of outer nuclear layers (ONL) (Fig. 3A[a-e] and Fig. 3B. By P34, about 60% (17.63 ± 5.26 μm) of the photoreceptor cells had degenerated in the Tubby mouse retina (Fig. 3A-a) compared to WT/WT mice(44.66 ± 12.57 μm) (Fig. 3A-e) whereas, only about 25%% (33.65 ± 9.47 μm) of the photoreceptor cells were missing in the Tub/Trx mice Fig. 3A-b at P34. The protection provided by Trx is readily apparent when the Tubby (Fig. 3A-a) retina is compared to Tub/Trx (Fig. 3A-b). The quantitation of the ONL thickness (Fig. 3B) provides a visual representation of the pan-retinal protection provided by Trx in the Tubby retina. Averaging the thickness of the ONL across the whole retina shows (Fig. 3C) that the thickness of the Tub/Trx retinas is significantly higher than that of the Tubby mice at P34 (p<0.001).
Figure 3. Morphological change of retinas from Tubby mice and Tub/Trx mice.
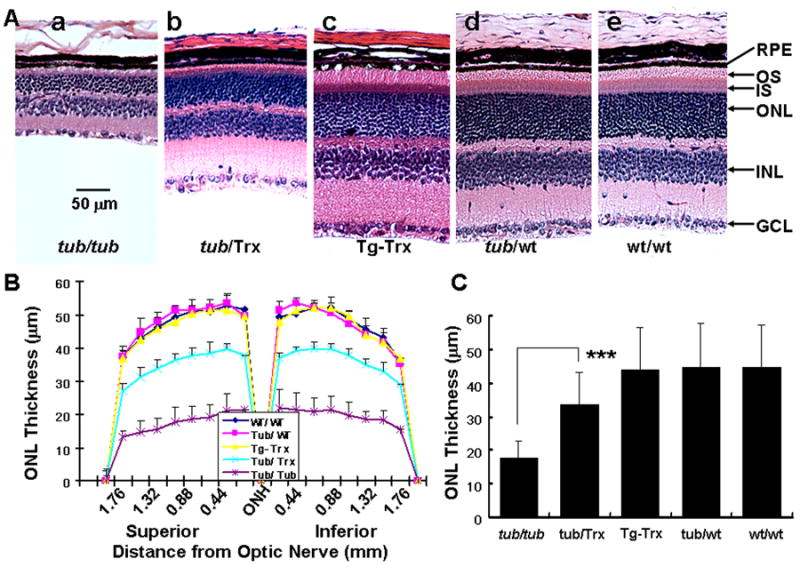
(A) Morphological change (H.E) of retina from Tubby mice (a), Tub/Trx mice (b), Tg-Trx mice (c), Tub/WT mice (d) and WT/WT mice (e) mice at age of P34. GCL, ganglion cell layer; INL, inner nuclear layer; ONL, outer nuclear layer; I, inner segment; and RPE, retinal pigment epithelium. (B) ONL thickness measurement in retinal sections from Tubby and Tub/Trx mice at various points at postnatal 34 days. (C) Analysis of ONL thickness in Tub/Trx mice compared to Tubby mice. Data are expressed as mean ± SD (n=6 in each group). The asterisks *** indicate p<0.001. The scale bar represents 50 μm.
Overexpression of the Trx transgene preserves functional vision of tubby mice
Electroretinography (ERG) waveform data show that B-wave and A-wave amplitudes of rod photoreceptor cells are significantly higher in Tub/Trx compared to those in Tubby (p<0.001) (Fig. 4A, B and D) at P34. Also, B-wave and A-wave amplitudes of cone photoreceptor cells were significantly higher in Tub/Trx compared to Tubby mice (p<0.001, Fig. 4C) and (p<0.05, Fig. 4E) at P34. These results clearly indicate that upregulation of Trx preserves visual function in Tubby mice.
Figure 4. Evaluation of retinal function at postnatal day 34 by electroretinography (ERG).
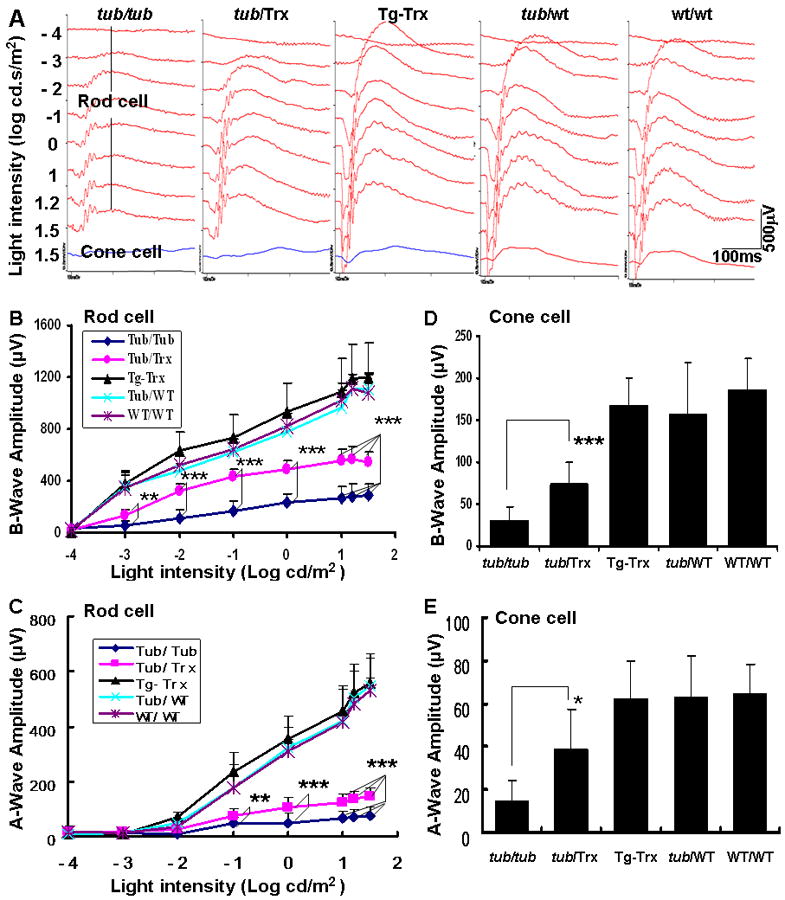
(A) Each panel shows a representative ERG recording from individual strains of mice: Tubby, Tub/Trx, Tg-Trx, Tub/WT and WT/WT mice. The scale bar represents x: 100 ms and y: 500μV. ERGs were recorded from 6 mice per group and the average (B) b-wave amplitudes and (C) a-wave amplitudes were plotted for rods. For cone activity, the ERGs were recorded from 6 mice per group and the average (D) b-wave amplitudes and (E) a-wave amplitudes were plotted for cones. Data were analyzed by one way ANOVA and are expressed as mean ± SD (n=6 in each group). * p<0.05, ** p<0.01, *** p<0.001
Effects of Trx on the Ras/Raf1/ERK signal pathway and expression of Nrf2 in Tub/Trx mice
In an attempt to define the mechanism(s) by which the tubby defect is causing degeneration of the retina and to correlate it with Trx induced changes, the expression of genes involved in cell survival and proliferation was examined. Figure 5 presents expression levels of the mRNAs (Fig. 5A, B) in the retinas of the five strains of mice studied. Semi-quantitative RT-PCR analysis of Raf1 shows that its expression was decreased to about 50% (0.53-fold) in the Tubby mice but increased to about a 75% (0.72-fold) in the Tub/Trx mice compared to WT/WT mice at P34. Similarly, the expression of Nrf2, a major regulator of antioxidant pathways, shows it to be down to 40% in the tubby retina whereas the presence of the Trx gene increases this to about 75% of normal. The western blot analysis of protein expression of Ras (Fig. 5 C,D) in the Tubby mouse shows about 50% of normal whereas the Tub/Trx mouse has about 120% of normal at P34. Similarly, the Nrf2 protein (Fig. 5 C,D) was reduced to about 50% of the Tubby mouse but was fully restored in the Tub/Trx mouse.
Figure 5. Retinal expression of Raf1, Ras, ERK1/2, p-ERK1/2 and Nrf2 in Tubby and Tub/Trx mice.
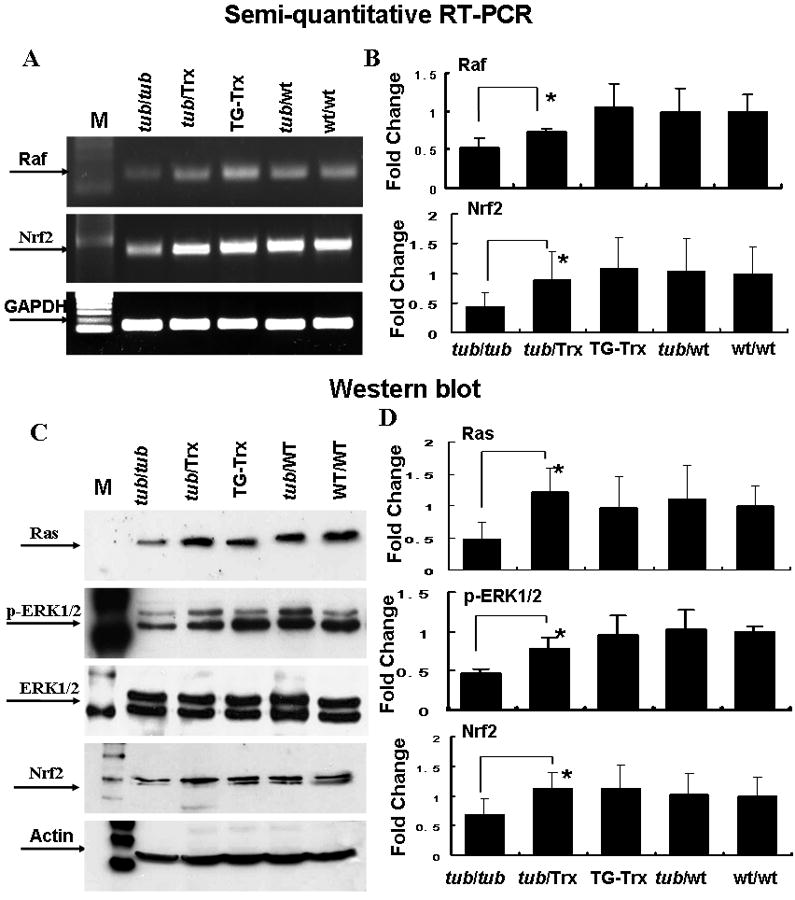
Retinas were removed at P34. Representative results (A, B) of semi-quantitative RT-PCR for Raf1 and Nrf2 (A) and densitometric analysis of the bands (B). The intensity of each band was standardized to the band intensity of GAPDH. Data was expressed as mean ± SD. Representative Western blots of Ras, ERK, p-ERK and Nrf2 (C) and densitometric analysis of protein bands (D) are shown. The intensity of each band was standardized to the band intensity of actin. Data are expressed as mean ± SD (n=4 for each genotype). The * indicate p<0.05.
To examine the role of ERK signal pathway in up-regulation of expression of Nrf2 and the photoreceptor protection in Tub/Trx mice, the total amount of ERK protein and its phosphorylation was determined by western blot (Fig. 5 C,D). The homozygous presence of the tubby gene resulted in about a 50% reduction in the expression and phosphorylation of ERK1/2 which were both restored to normal levels by the Trx transgene.
Inhibition of photoreceptor apoptosis and ASK1/JNK activity
Our data show that there is a significantly higher number of TUNEL-positive cells within the ONL of the Tubby retina (Fig. 6A-a). Expression of the Trx gene on the Tubby background (Fig. 6A-b) significantly decreased the number of TUNEL-positive cells to a level indistinguishable from that found in the Trx/Wt retina (Fig. 6A-c) and the WT/WT retina (image not shown). We also used immunofluorescent labeling to detect the expression and localization of phosphorylated apoptosis stimulating kinase 1 (pASK1). Our study shows that p-Ask1 is significantly higher in the Tubby mice (Fig. 6B-a) compared to the Trx/Tub mice(Fig. 6B-b) and the Tubby heterozygous Tub/WT (Fig. 6B-c) mouse. The p-Ask1 is present at high concentrations in the ganglion cell layer (GCL) and in regions of the ONL of the Tubby retina and significantly less is detected in the Tub/Trx mice (Fig. 6B-b) and the heterozygous Tubby retinas (Fig. 6B-c). The labeling of p-Ask1 in the Tub/Wt mice was essentially identical to that in the Tg-Trx, and WT/WT mice (data not show). Western blot data(Fig. 6C, D) also demonstrated that the phosphorylated form of Akt (p-Akt) in the Tubby mice was significantly down-regulated (0.58-fold) compared to Tub/Trx mice (1.07-fold) at P34. However, phosphorylated JNK (p-JNK) (Fig. 6C, D) in the Tubby mice was almost six fold higher than in the control mice (Tg/Trx, Tub/Wt, Wt/Wt) and this was reduced to fourfold by the presence of the Trx gene.
Figure 6. Trx decreases apoptosis and modifies phosphorylation of Ask-1, Akt and JNK1 in the tubby retina.
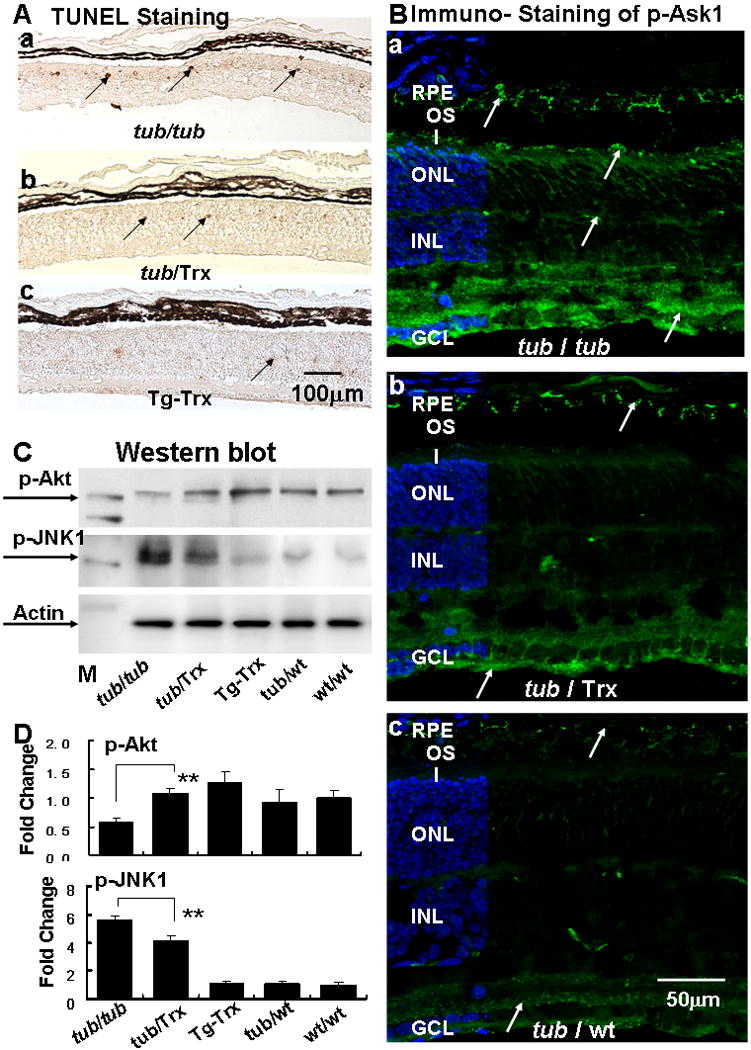
(A) TUNEL staining of retinas from the (a) Tubby, (b) Tub/Trx and (c) Tg-Trx mice. TUNEL-positive cells are seen in the ONL (arrows). The scale bar represents 100 μm. (B) Immunofluorescent labeling (arrows) of phosphorylated-Ask1 in the retinas of (a) Tubby and (b) Tub/Trx and (c) Tub/WT mice (arrow). The scale bar represents 50 μm. (C) Phosphorylation of Akt is increased whereas phosphorylation of JNK1 is decreased by the presence of Trx. (D) Densitometric analysis of p-Akt and p-JNK1 protein bands are shown. Intensity of each band was standardized to the band intensity of actin. Data are expressed as mean ± SD (n=4 in each genotype). ** indicates p<0.01.
Trx increases the production of neurotrophins in the tubby mice
BDNF and GDNF are both proteins which exert a protective effect on photoreceptor cells and whose expression is known to be regulated by Trx. Figure 7 presents the expression of BDNF and GDNF at both the mRNA (Fig. 7A,C,E) and protein level (Fig. 7B, D,F) in the Tubby and Tub/Trx mice and controls at P14. The gene expression and protein levels were 0.42-fold and 0.35-fold respectively (Fig. 7A-a, 7B-a) for BDNF, and 0.50-fold and 0.40-fold respectively (Fig. 7A-b, 7B-b) for GDNF in the Tubby mice compared to wild type retinas. Densitometry (Fig. 7C,D,E,F) shows that the mRNA and the protein for both BDNF and GDNF are down-regulated by the tubby/tubby gene and up-regulated when Trx is expressed on the Tubby background, although the expression levels do not reach those of the controls.
Figure 7. BDNF and GDNF expression in the retina are down-regulated by the tubby gene and up-regulated by the presence of the Trx transgene.
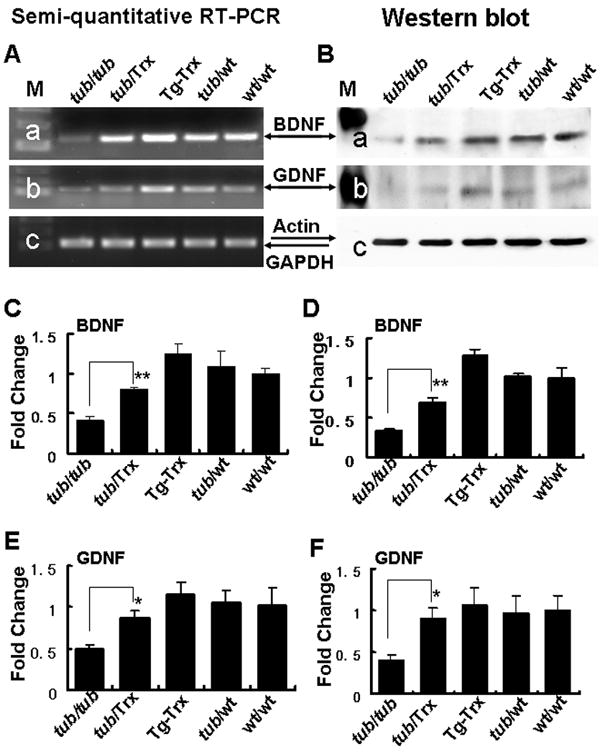
Representative results of (A) Semi-quantitative RT-PCR and (B) Western blot analysis shows significant decreases in BDNF (a) and GDNF (b) mRNAs and proteins in the retinas of Tubby mice at 14 days. Densitometric analysis of BDNF (C) semi-quantitative RT-PCR and (D) Western blot bands confirm the band displays in A and B. Similar analysis of GDNF bands (E) and (F) confirm the observed changes. Intensity of each band was standardized by the band intensity of GAPDH or actin. Data are expressed as mean ± SD (n=3 in each group). The * and ** indicate p<0.05 and p<0.01.
DISCUSSION
We have made three novel observations. The first is that overexpression of Trx delays the inherited photoreceptor degeneration and preserves the visual function in the Tubby mouse. The second is that the mechanism by which Trx acts to protect photoreceptors involves the activation of the Akt, Ras/Raf1/ERKs survival signal transduction cascade and the inhibition of the ASK1/ JNK apoptosis signal pathway. The third observation is that two major contributors to neuroprotection, BDNF and GDNF, are both up-regulated by the expression of Trx in the Tubby mouse.
At P34 in Tubby mice, there is a 61% loss of photoreceptor cells whereas only 24% of the photoreceptors have degenerated in the tub/Trx mice compared to no loss of photoreceptors in tub/wt, wt/wt, and Tg-Trx mice. We previously reported(Kong et al., 2007) that during the early post-natal period, down-regulation of Trx preceded significant photoreceptor cell loss in the tubby mouse, suggesting a causal relationship between attenuation of Trx and Trx reductase expression and the death of photoreceptor cells in the tubby mouse. Human Trx mRNA and Trx protein were demonstrated to be upregulated in the retinas from tub/Trx and Tg-Trx mice compared with tubby or tub/wt or wt/wt animals as determined by RT-PCR, western blots and immunofluorescent staining. In addition, our data support the conclusion that Trx is over expressed not only in photoreceptors, but also in other cell types. The outer plexiform layer (OPL), a region where retinal synapses of photoreceptor, horizontal and bipolar cells are located, showed a significant presence of Trx in tub/Trx mice compared to the Tubby or tub/wt or wt/wt mice. Furthermore, at P34, the ONL thickness and the amplitudes of ERGs were significantly higher in tub/Trx mice compared with Tubby mice. Collectively, the morphological and functional data clearly demonstrate that overexpression of Trx delays hereditary retinal degeneration in Tubby mice and thereby preserves their visual function.
We previously reported that tubby mice born and raised in a bright cyclic light environment had an accelerated loss of photoreceptors compared with mice born and raised in a constant dark environment(Kong et al., 2006). It is known that light exposure enhances lipid peroxidation of the photoreceptor outer segments (Organisciak et al., 1992) and because ‘free-radical’ trapping agents inhibit the damage (Ranchon et al., 2003), free radicals are thought to be involved in light-induced photoreceptor cell death. Trx directly scavenges singlet oxygen and hydroxyl radicals (Das and Das, 2000) and the Trx system is a specific hydrogen donor for peroxiredoxin, which eliminates H2O2 (Chae et al., 1994). Overexpression of Trx (Tanito et al., 2002b) and intravitreal injection of recombinant Trx protein (Tanito et al., 2002a) prevent intense light-induced photoreceptor cell damage in mice. Therefore, the Trx system appears to be involved in the neuroprotective mechanism by attenuation of environmental light-induced retinal damage.
The thioredoxin system helps maintain a reducing environment in cells, but thioredoxin functions as more than simply an antioxidant. Recent data (Tanito et al., 2005) suggested that Trx has neurotrophic factor-like activity and plays a crucial role in maintaining neuronal cell integrity including that of photoreceptor cells. Apoptosis is the common fate of photoreceptors in retinitis pigmentosa and age-related macular degeneration (Wenzel et al., 2005) and it is also a major mechanism for the cell death in the Tubby mouse (Bode and Wolfrum, 2003; Kong et al., 2006). It has been shown that the reduced form of thioredoxin (Trx) interacts with the N-terminal portion of apoptosis stimulating kinase 1 (ASK1) in vitro and in vivo thereby inhibiting the activity of this serine-threonine kinase, a member of the MKKK family(Saitoh et al., 1998). Furthermore, because formation of the Trx-ASK1 complex occurs only with reduced Trx oxidants, our data suggest that reduced Trx is a physiological inhibitor of ASK1 and that its oxidation and release of ASK1 is a common node by which cytotoxic stresses such as TNFalpha, Fas and H2O2 activate the p38 MAPK and SAPK/JNK stress response pathways(Saitoh et al., 1998; Tobiume et al., 2001). Our data demonstrates the inhibition of the inherited programmed cell death of photoreceptors in the tubby retina by overexpression of Trx; the reduction in the activation of ASK1 and JNK in the photoreceptors of tub/Trx mice compared with Tubby. Collectively, these data support the conclusion that the expression and activation of ASK1/JNK are involved in photoreceptor cell degeneration in the tubby retina and that overexpression of Trx inhibits these events. Degeneration of photoreceptors in the Tubby mice has been shown to proceed through the activation of the death signaling pathway and/or by inhibiting the survival signaling pathway(Bode and Wolfrum, 2003; Ikeda et al., 2002). Intracellular signaling cascades that lead to the survival of neurons include the PI3K (phosphoinositide 3-kinase)/Akt (protein kinase B) pathway and the Ras/Raf1 (Ras-activated factor)/MAPK (mitogen-activated protein kinase) pathway (Frebel and Wiese, 2006). Akt is a critical regulator of PI-3-kinase-mediated cell survival and constitutive activation of Akt is sufficient to block cell death by a variety of apoptotic stimuli (Gurusamy et al., 2007). Interestingly, we have shown that overexpression of Trx up-regulates activation of Akt in the Tubby retina suggesting that the up-regulation of Trx in the photoreceptors of the Tubby mice occurs via the PI-3-kinase-Akt survival pathway.
Phosphorylation of members of the mitogen-activated protein kinase (MAP) kinase Ras-Raf1-ERK pathway is critical for neuroprotective signaling and suppression of apoptosis (Gray et al., 2005). In this study, both Ras and Raf1 were up regulated by overexpression of Trx. We have also examined ERKs because they act as crucial intracellular molecular signals. ERKs are involved in highly conserved signaling pathways that regulate diverse cellular functions including proliferation, differentiation, migration, and apoptosis. They are activated through phosphorylation by distinct pathways depending on stimulus and cell type. When activated, they can phosphorylate a wide range of substrates, including transcription factors and cytoskeletal proteins, resulting in specific cellular responses. Our study shows that ERKs are down-regulated in Tubby retinas and are significantly activated by Trx. We have also demonstrated that the level of Nrf2 in whole retinal lysates is significantly down-regulated in Tubby compared with tub/+ retinas and that the activation of Nrf2 is also impaired in Tubby retinas. Furthermore, our data show that both the amount of Nrf2 and its activation in the Tubby retina are increased by the overexpression of Trx. The exact mechanisms by which these changes occur will require further studies.
Thioredoxin is essential for the NGF-mediated neurite outgrowth in neuronal PC12 cells(Bai et al., 2003). Overexpression of Trx in mice provides neural tissue protection from light-induced photoreceptor cell damage(Tanito et al., 2002a) and ischemia-reperfusion injury in the brain(Takagi et al., 1999). Because BDNF and GDNF are neuroprotective proteins, we sought to determine if overexpression of Trx also caused their upregulation. Our data demonstrate that the expression of both BDNF and GDNF is decreased in the retinas of Tubby mice but increased by overexpression of Trx suggesting that Trx has a role in regulating the concentration of BDNF and GDNF and photoreceptor cell survival.
In conclusion, we have demonstrated that overexpression of Trx strongly delays inherited photoreceptor cell degeneration and preserves the visual function via activating survival signaling pathways such as Akt, Ras/Raf1/ extracellular signal-regulated kinase (ERKs) signal cascade and by inhibition of the ASK1/JNK apoptosis signal pathway. In addition, overexpression of Trx increases the expression of brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF). However, the underlying molecular mechanism by which the tubby gene mutation results in its pathological phenotype and the relationship between the tubby gene and the regulation of Trx system remain to be established.
Acknowledgments
Supported in part by grants from: Foundation Fighting Blindness (FFB C-NP-0707-0404-UOK08, FFB TA-NP-1107-0434-UOK;), NIH: (R21EY018306, R01EY018724 COBRE/Core P20 RR17703, NEI/Core P30- EY12190), EY014427, and a an unrestricted grant from Research to Prevent Blindness, New York, NY to the Department of Ophthalmology, University of Oklahoma Health Sciences Center, Oklahoma City, OK. JF McGinnis is a recipient of a Senior Scientific Investigator award from RPB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCE LIST
- Akterin S, Cowburn RF, Miranda-Vizuete A, Jimenez A, Bogdanovic N, Winblad B, Cedazo-Minguez A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer’s disease. Cell Death Differ. 2006;13:1454–1465. doi: 10.1038/sj.cdd.4401818. [DOI] [PubMed] [Google Scholar]
- Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, Wada T, Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res. 2008;36:4327–4336. doi: 10.1093/nar/gkn416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Nakamura H, Kwon YW, Hattori I, Yamaguchi Y, Kim YC, Kondo N, Oka S, Ueda S, Masutani H, Yodoi J. Critical roles of thioredoxin in nerve growth factor-mediated signal transduction and neurite outgrowth in PC12 cells. J Neurosci. 2003;23:503–509. doi: 10.1523/JNEUROSCI.23-02-00503.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode C, Wolfrum U. Caspase-3 inhibitor reduces apototic photoreceptor cell death during inherited retinal degeneration in tubby mice. Mol Vis. 2003;9:144–150. [PubMed] [Google Scholar]
- Cao W, Chen W, Elias R, McGinnis JF. Recoverin negative photoreceptor cells. J Neurosci Res. 2000;60:195–201. doi: 10.1002/(SICI)1097-4547(20000415)60:2<195::AID-JNR8>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Cao W, Tombran-Tink J, Elias R, Sezate S, Mrazek D, McGinnis JF. In vivo protection of photoreceptors from light damage by pigment epithelium-derived factor. Invest Ophthalmol Vis Sci. 2001;42:1646–1652. [PubMed] [Google Scholar]
- Chae HZ, Robison K, Poole LB, Church G, Storz G, Rhee SG. Cloning and sequencing of thiol-specific antioxidant from mammalian brain: alkyl hydroperoxide reductase and thiol-specific antioxidant define a large family of antioxidant enzymes. Proc Natl Acad Sci U S A. 1994;91:7017–7021. doi: 10.1073/pnas.91.15.7017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho CG, Kim HJ, Chung SW, Jung KJ, Shim KH, Yu BP, Yodoi J, Chung HY. Modulation of glutathione and thioredoxin systems by calorie restriction during the aging process. Exp Gerontol. 2003;38:539–548. doi: 10.1016/s0531-5565(03)00005-6. [DOI] [PubMed] [Google Scholar]
- Das KC, Das CK. Thioredoxin, a singlet oxygen quencher and hydroxyl radical scavenger: redox independent functions. Biochem Biophys Res Commun. 2000;277:443–447. doi: 10.1006/bbrc.2000.3689. [DOI] [PubMed] [Google Scholar]
- Frebel K, Wiese S. Signalling molecules essential for neuronal survival and differentiation. Biochem Soc Trans. 2006;34:1287–1290. doi: 10.1042/BST0341287. [DOI] [PubMed] [Google Scholar]
- Gray JJ, Bickler PE, Fahlman CS, Zhan X, Schuyler JA. Isoflurane neuroprotection in hypoxic hippocampal slice cultures involves increases in intracellular Ca2+ and mitogen-activated protein kinases. Anesthesiology. 2005;102:606–615. doi: 10.1097/00000542-200503000-00020. [DOI] [PubMed] [Google Scholar]
- Gurusamy N, Malik G, Gorbunov NV, Das DK. Redox activation of Ref-1 potentiates cell survival following myocardial ischemia reperfusion injury. Free Radic Biol Med. 2007;43:397– 407. doi: 10.1016/j.freeradbiomed.2007.04.025. [DOI] [PubMed] [Google Scholar]
- Hattori I, Takagi Y, Nakamura H, Nozaki K, Bai J, Kondo N, Sugino T, Nishimura M, Hashimoto N, Yodoi J. Intravenous administration of thioredoxin decreases brain damage following transient focal cerebral ischemia in mice. Antioxid Redox Signal. 2004;6:81–87. doi: 10.1089/152308604771978372. [DOI] [PubMed] [Google Scholar]
- Henderson WR, Jr, Chi EY, Teo JL, Nguyen C, Kahn M. A small molecule inhibitor of redox-regulated NF-kappa B and activator protein-1 transcription blocks allergic airway inflammation in a mouse asthma model. J Immunol. 2002;169:5294–5299. doi: 10.4049/jimmunol.169.9.5294. [DOI] [PubMed] [Google Scholar]
- Hirota K, Murata M, Sachi Y, Nakamura H, Takeuchi J, Mori K, Yodoi J. Distinct roles of thioredoxin in the cytoplasm and in the nucleus. A two-step mechanism of redox regulation of transcription factor NF-kappaB. J Biol Chem. 1999;274:27891–27897. doi: 10.1074/jbc.274.39.27891. [DOI] [PubMed] [Google Scholar]
- Holmgren A. Thioredoxin. Annu Rev Biochem. 1985;54:237–271. doi: 10.1146/annurev.bi.54.070185.001321. [DOI] [PubMed] [Google Scholar]
- Ikeda A, Naggert JK, Nishina PM. Genetic modification of retinal degeneration in tubby mice. Exp Eye Res. 2002;74:455–461. doi: 10.1006/exer.2001.1139. [DOI] [PubMed] [Google Scholar]
- Kong L, Li F, Soleman CE, Li S, Elias RV, Zhou X, Lewis DA, McGinnis JF, Cao W. Bright cyclic light accelerates photoreceptor cell degeneration in tubby mice. Neurobiol Dis. 2006;21:468–477. doi: 10.1016/j.nbd.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Kong L, Tanito M, Huang Z, Li F, Zhou X, Zaharia A, Yodoi J, McGinnis JF, Cao W. Delay of photoreceptor degeneration in tubby mouse by sulforaphane. J Neurochem. 2007;101:1041–1052. doi: 10.1111/j.1471-4159.2007.04481.x. [DOI] [PubMed] [Google Scholar]
- Masutani H, Bai J, Kim YC, Yodoi J. Thioredoxin as a neurotrophic cofactor and an important regulator of neuroprotection. Mol Neurobiol. 2004;29:229–242. doi: 10.1385/MN:29:3:229. [DOI] [PubMed] [Google Scholar]
- Matsuura T, Harrison RA, Westwell AD, Nakamura H, Martynyuk AE, Sumners C. Basal and angiotensin II-inhibited neuronal delayed-rectifier K+ current are regulated by thioredoxin. Am J Physiol Cell Physiol. 2007;293:C211–C217. doi: 10.1152/ajpcell.00615.2006. [DOI] [PubMed] [Google Scholar]
- Munemasa Y, Kim SH, Ahn JH, Kwong JM, Caprioli J, Piri N. Protective effect of thioredoxins 1 and 2 in retinal ganglion cells after optic nerve transection and oxidative stress. Invest Ophthalmol Vis Sci. 2008;49:3535–3543. doi: 10.1167/iovs.08-1716. [DOI] [PubMed] [Google Scholar]
- Ohashi S, Nishio A, Nakamura H, Kido M, Ueno S, Uza N, Inoue S, Kitamura H, Kiriya K, Asada M, Tamaki H, Matsuura M, Kawasaki K, Fukui T, Watanabe N, Nakase H, Yodoi J, Okazaki K, Chiba T. Protective roles of redox-active protein thioredoxin-1 for severe acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2006;290:G772–G781. doi: 10.1152/ajpgi.00425.2005. [DOI] [PubMed] [Google Scholar]
- Rosen A, Lundman P, Carlsson M, Bhavani K, Srinivasa BR, Kjellstrom G, Nilsson K, Holmgren A. A CD4+ T cell line-secreted factor, growth promoting for normal and leukemic B cells, identified as thioredoxin. Int Immunol. 1995;7:625–633. doi: 10.1093/intimm/7.4.625. [DOI] [PubMed] [Google Scholar]
- Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. Mammalian thioredoxin is a direct inhibitor of apoptosis signal-regulating kinase (ASK) 1. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Hoshino Y, Hara T, Muro S, Nakamura H, Mishima M, Yodoi J. Thioredoxin-1 ameliorates cigarette smoke-induced lung inflammation and emphysema in mice. J Pharmacol Exp Ther. 2008;325:380–388. doi: 10.1124/jpet.107.134007. [DOI] [PubMed] [Google Scholar]
- Satoh M, Matter CM, Ogita H, Takeshita K, Wang CY, Dorn GW, Liao JK. Inhibition of apoptosis-regulated signaling kinase-1 and prevention of congestive heart failure by estrogen. Circulation. 2007;115:3197–3204. doi: 10.1161/CIRCULATIONAHA.106.657981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberstein DS, McDonough S, Minkoff MS, Balcewicz-Sablinska MK. Human eosinophil cytotoxicity-enhancing factor. Eosinophil-stimulating and dithiol reductase activities of biosynthetic (recombinant) species with COOH-terminal deletions. J Biol Chem. 1993;268:9138–9142. [PubMed] [Google Scholar]
- Takagi Y, Mitsui A, Nishiyama A, Nozaki K, Sono H, Gon Y, Hashimoto N, Yodoi J. Overexpression of thioredoxin in transgenic mice attenuates focal ischemic brain damage. Proc Natl Acad Sci U S A. 1999;96:4131–4136. doi: 10.1073/pnas.96.7.4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A, Nakamura H, Kondo N, Tanito M, Kwon YW, Ahsan MK, Matsui H, Narita M, Yodoi J. Thioredoxin-1 attenuates indomethacin-induced gastric mucosal injury in mice. Free Radic Res. 2007;41:861–869. doi: 10.1080/10715760701199618. [DOI] [PubMed] [Google Scholar]
- Tanito M, Agbaga MP, Anderson RE. Upregulation of thioredoxin system via Nrf2-antioxidant responsive element pathway in adaptive-retinal neuroprotection in vivo and in vitro. Free Radic Biol Med. 2007;42:1838–1850. doi: 10.1016/j.freeradbiomed.2007.03.018. [DOI] [PubMed] [Google Scholar]
- Tanito M, Masutani H, Kim YC, Nishikawa M, Ohira A, Yodoi J. Sulforaphane induces thioredoxin through the antioxidant-responsive element and attenuates retinal light damage in mice. Invest Ophthalmol Vis Sci. 2005;46:979–987. doi: 10.1167/iovs.04-1120. [DOI] [PubMed] [Google Scholar]
- Tanito M, Masutani H, Nakamura H, Ohira A, Yodoi J. Cytoprotective effect of thioredoxin against retinal photic injury in mice. Invest Ophthalmol Vis Sci. 2002a;43:1162–1167. [PubMed] [Google Scholar]
- Tanito M, Masutani H, Nakamura H, Oka S, Ohira A, Yodoi J. Attenuation of retinal photooxidative damage in thioredoxin transgenic mice. Neurosci Lett. 2002b;326:142–146. doi: 10.1016/s0304-3940(02)00314-2. [DOI] [PubMed] [Google Scholar]
- Tobiume K, Matsuzawa A, Takahashi T, Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T, Ichijo H. ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2001;2:222–228. doi: 10.1093/embo-reports/kve046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turoczi T, Chang VW, Engelman RM, Maulik N, Ho YS, Das DK. Thioredoxin redox signaling in the ischemic heart: an insight with transgenic mice overexpressing Trx1. J Mol Cell Cardiol. 2003;35:695–704. doi: 10.1016/s0022-2828(03)00117-2. [DOI] [PubMed] [Google Scholar]
- Ueno H, Kajihara H, Nakamura H, Yodoi J, Nakamuro K. Contribution of thioredoxin reductase to T-cell mitogenesis and NF-kappaB DNA-binding promoted by selenite. Antioxid Redox Signal. 2007;9:115–121. doi: 10.1089/ars.2007.9.115. [DOI] [PubMed] [Google Scholar]
- Wenzel A, Grimm C, Samardzija M, Reme CE. Molecular mechanisms of light-induced photoreceptor apoptosis and neuroprotection for retinal degeneration. Prog Retin Eye Res. 2005;24:275–306. doi: 10.1016/j.preteyeres.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Yamato E, Toyoda S, Tashiro F, Ikegami H, Yodoi J, Miyazaki J. Transgenic expression of antioxidant protein thioredoxin in pancreatic beta cells prevents progression of type 2 diabetes mellitus. Antioxid Redox Signal. 2008;10:43–49. doi: 10.1089/ars.2007.1586. [DOI] [PubMed] [Google Scholar]
- Yu X, Rajala RV, McGinnis JF, Li F, Anderson RE, Yan X, Li S, Elias RV, Knapp RR, Zhou X, Cao W. Involvement of insulin/phosphoinositide 3-kinase/Akt signal pathway in 17 beta-estradiol-mediated neuroprotection. J Biol Chem. 2004;279:13086–13094. doi: 10.1074/jbc.M313283200. [DOI] [PubMed] [Google Scholar]
- Zhou X, Li F, Ge J, Sarkisian SR, Jr, Tomita H, Zaharia A, Chodosh J, Cao W. Retinal ganglion cell protection by 17-beta-estradiol in a mouse model of inherited glaucoma. Dev Neurobiol. 2007;67:603–616. doi: 10.1002/dneu.20373. [DOI] [PubMed] [Google Scholar]
- Zhou X, Li F, Kong L, Tomita H, Li C, Cao W. Involvement of inflammation, degradation, and apoptosis in a mouse model of glaucoma. J Biol Chem. 2005;280:31240–31248. doi: 10.1074/jbc.M502641200. [DOI] [PubMed] [Google Scholar]