

SYNOPSIS
Injectable and implantable porosified silicon (pSi) carriers and devices for prolonged and controlled delivery of biotherapeutics offer great promise for treatment of various chronic ailments and acute conditions. Polyethylene glycols (PEGs) are important surface modifiers currently used in clinic mostly to avoid uptake of particlulates by reticulo-endothelial system (RES). In this work we show for the first time that covalent attachment of PEGs to the pSi surface can be used as a means to finely tune degradation kinetics of silicon structures. Seven PEGs with varying molecular weights (245, 333, 509, 686, 1214, 3400 and 5000Da) were employed and the degradation of PEGylated pSi hemispherical microparticles in simulated physiological conditions was monitored by means of ICP-AES, SEM and fluorimetry. Biocompatibility of the systems with human macrophages in vitro was also evaluated. The results clearly indicate that controlled PEGylation of silicon microparticles can offer a sensitive tool to finely tune their degradation kinetics and that the systems do not induce release of proinflammatory cytokines IL-6 and IL-8 in THP1 human macrophages.
Keywords: mesoporous silicon, polyethylene glycol, biodegradation, biocompatibility
INTRODUCTION
Porous silicon (pSi) was discovered by Uhlir at Bell Laboratories in the mid 1950s, 1 and is currently being employed in various fields of biomedical research with diverse applications including biomolecular screening, 2–3 optical biosensing, 4–5 drug delivery through injectable carriers 6–7 and implantable devices 8 as well as orally administered medications with improved bioavailability 9. There are already several FDA approved and marketed products based on pSi technology which found their niche in ophthalmology 10 and other, based on radioactive 32P doped pSi is currently in clinical trials, as a potential new brachytherapy treatment for inoperable liver cancer 11. This paper presents PEGylation as a method to control degradation kinetics of porous silicon structures (pSi) with important biomedical applications, e.g. in controlled drug delivery and osteoregeneration.
Important requirements for injectable drug delivery carriers are (i) biocompatibility, (ii) efficient clearance or biodegradability, and (iii) favorable biodistribution, for example avoidance or control of uptake by the reticulo- endothelial system (RES). pSi for drug delivery applications mainly features drug molecules that are directly loaded within the matrix of pSi membranes or microcarriers and are trapped within the pores via specific or non-specific interactions. Drug release is then achieved mainly through pSi degradation over time. Canham and colleagues 12–13 have reported that the biodegradation rates of pSi structures can be controlled by the pore size: the larger the pore the faster the degradation and thus the release of the drug molecule.
More recently, our group 7 proposed to use pSi microparticles (first stage particles), with pore sizes ranging from 10 to 50 nm, as a multistage and multifunctional delivery systems where various nano-sized particulates (second stage particles) with different payloads or functions (therapeutic, imaging, thermal ablation and magnetic guidance agents) can be loaded simultaneously. In such applications, the pore size is not only playing an important role in the degradation and release kinetics but it would also significantly affect the loading efficiency of the second stage nanoparticles. It is thus fundamental to decouple the dependence of loading, degradation and release, on particles pore size, in order to optimize the whole system.
Polyethylene glycols (PEGs) represent the major category of surface modifying agents used in classical drug delivery systems and in pharmaceutical dosage forms, mainly due to avoidance of RES uptake and thus a possibility to control biodistribution and circulation time 14. These hydrophilic polymers are approved by FDA for use in food, cosmetics and pharmaceuticals, including injectable, topical, rectal and nasal formulations. PEG molecules demonstrate little toxicity, and are cleared from the body, without being metabolized, by either the kidneys for PEGs < 30 kDa or in the feces for longer PEGs. PEG molecules generally lack immunogenicity and no production of the antibodies against PEGs was reported in the literature under regular clinical administration of pegylated proteins 15. We hypothesized that the degradation of mesoporous silicon based carriers can be regulated not only by controlling the pores size, but also through surface modifications. In this study we evaluated the effect of PEGylation of the mesoporous silicon particles on their degradation. We propose to fine tune the biodegradation kinetics of pSi particles using surface functionalization with PEGs of various molecular weights. In the present work, the degradation of pSi hemispherical microparticles was monitored by means of ICP-AES, SEM and fluorimetry. The biocompatibility of the systems with human macrophages was evaluated. The results clearly indicate that controlled PEGylation of silicon microparticles can offer a sensitive tool to tune their degradation kinetics.
MATERIALS AND METHODS
Fabrication, surface modification and characterization of porous silicon particles
Mesoporous silicon microparticles were fabricated by photolithography and electrochemical etching in the Microelectronics Research Center at The University of Texas at Austin as previously described [6]. The large pore (LP, 30–40nm pores) silicon particles were formed in a mixture of hydrofluoric acid (49% HF) and ethanol (3:7 v/v) by applying a current density of 80 mA cm−2 for 25 s. A high porosity layer was formed by applying a current density of 320 mA cm−2 for 6 s. For fabrication of small pores (SP, 10nm) silicon particles, a solution of HF and ethanol was used with a ratio of 1:1 (v/v), with a current density applied of 6 mA cm−2 for 1.75 min. After removing the nitride layer by HF, particles were released by ultrasound in isopropyl alcohol (IPA) for 1 min.
Silicon microparticles in IPA were dried in a glass beaker by heating (80–90 °C) and then oxidized in a piranha solution (1:2 H2O2:concentrated H2SO4 (v/v), Sigma) at 100–110 °C for 2 h, with intermittent sonication to disperse the aggregates, washed in DI water and stored at 4 °C in DI water until further use. Prior to modification with 3-Aminopropyltriethoxysilane (APTES, Sigma). The particles were then washed with DI water followed by IPA, suspended in IPA containing APTES (0.5% v/v) for 45 min at room temperature, washed 5 times with IPA and stored in IPA at 4°C.
APTES modified large pore particles were reacted with 10 mM mPEG-SCM or NHS-m-dPEG (QuantaBiodesign, USA) in 400–500μL acetonitrile for 1.5 hours. The succinimidyl ester on the PEGs reacts with an amino group that is exposed on the surface of the APTES particles giving a stable chemical linkage of PEGs to the particles. The particles were then washed (by centrifugation) in deionized water 4–6 times to remove any unreacted PEGs. The particles were stored in deionized water or IPA at 4 °C till further use.
Volumetric particle size, size distribution and count was obtained using a Z2 Coulter® Particle Counter and Size Analyzer (Beckman Coulter, Fullerton, CA, USA). Prior to the analysis, the samples were dispersed in the balanced electrolyte solution (ISOTON® II Diluent, Beckman Coulter Fullerton, CA, USA) and sonicated for 5 seconds to ensure a homogenous dispersion.
The zeta potential of the silicon particles was analyzed using a Zetasizer nano ZS (Malvern Instruments Ltd., Southborough, MA, USA). For the analysis, 2 μL particle suspension containing at least 2×105 particles to give a stable zeta value evaluation were injected into a sample cell countering filed with phosphate buffer (PB, 1.4 mL, pH 7.3). The cell was sonicated for 2min, and then an electrode-probe was put into the cell. Measurements were conducted at room temperature (23°C) in triplicates.
Degradation study in simulated physiological conditions
To evaluate degradation kinetics, 107 of the particles were added to PBS (1.5mL, pH 7.2) or 100% fetal bovine serum (FBS, Hyclone, USA). The samples (n=3) were incubated at 37°C and constantly mixed using a rotary shaker until the appropriate time points had elapsed. Aliquots (85μL) were taken from the tubes: 75 μL were filter-spun (0.45 μm filter) to separate the undegraded particles from the degradation medium and the resulting liquid was stored at 4°C for later analysis of total silicon by Inductively Coupled Plasma Atomic Emission Spectrometer (ICP-AES). The remaining 10 μL were extensively washed with DI water to remove the salts, placed on the grid, dried in a dessicator and further analyzed for particles morphology by Scanning Electron Microscopy (SEM). In the case of fluorescent PEG conjugated to the surface of the particles, the samples (150 μL) were spun down at 4500rpm × 20min, the supernatant was collected into 96 wells plates and analyzed for the quantity of fluorophore released from the particles by fluorimetry and for Si contents by ICP-AES.
Silicon contents released to from the particles during the degradation process was measured using a Varian Vista-Pro ICP–AES. Si was detected at 250.69, 251. 43, 251.61 and 288.158nm. A calibration run including the internal control (Yttrium, 1 ppm) was made before each group of 1wsample (100%), the particles were dissolved in 1N NaOH for 4 hours in 37C. Further, all results were expressed as % of the silicic acid released to the medium.
SEM was applied to examine the structure and morphology of the particles. Samples were sputter-coated with gold for 2 min at 10nm using a CrC-150 Sputtering System (Torr International, New Windsor, NY) and observed under a FEI Quanta 400 field emission scanning electron microscope (FEI Company, Hillsboro, OR) at an accelerating voltage of 20 kV, chamber pressure of 0.45 Torr and spot size 5.0.
Fluorescence of the particles conjugated to FITC-PEG (MW 3400) was assessed using a FACScalibur (Becton Dickinson). Bivariate dot-plots defining logarithmic side scatter (SSC) versus logarithmic forward scatter (FSC) were used to evaluate the size and shape of the unlabeled silicon particles (3 μm in diameter, 1.5 μm in height) and to exclude non-specific events from the analysis. A polygonal region (R1) was defined as an electronic gate around the centre of the major population of interest for undegraded particles, which excluded events that were too close to the signal-to-noise ratio limits of the cytometer. The peaks identified in each of the samples were analyzed in the corresponding fluorescent histogram and the geometric mean values recorded. For particle detection, the detectors used were FSC E- 1 and SSC with a voltage setting of 474 volts (V). The fluorescent detector FL1 was set at 800 V and green fluorescence was detected with FL1 using a 530/30 nm band-pass filter. For each analysis, 50,000–200,000 gated events were collected. Instrument calibration was carried out before, in between, and after each series of experiments for data acquisition using BD Calibrite™ beads (3.5 μm in size).
For the analysis of fluorescence intensity analysis the samples were placed on a 96-well plate (Nunclon, Denmark) and quantities of FITC-PEG released from the particles surface were determined in triplicates using BMG FluoSTAR microplate variable wavelength fluorescence spectrophotometer (Galaxy, excitation 488nm, emission 523 nm).
Based on the observed experimental results, a mathematical model was identified and used to get further insight into the underlying physical and chemical processes which are involved in the effect of PEGylation on particles degradation.
Evaluation of biocompatibility of PEGylated particles with human macrophages in vitro
THP-1 monocyte cell line was obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured at 0.4–2×106 cells/mL in RPMI 1640 containing heat-inactivated FCS (10 % w/v), glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL), and maintained at 37 °C under 5 % CO2. All reagents and medium were purchased from ATCC and Gibco BRL (Gaithersburg, MD). THP-1 cells (0.2×106 cells/well) were differentiated into macrophages in 24 well plates containing 1 mL medium/well with phorbol ester (80 ng, PMA, Sigma USA) over 72 h. A stock solution of PMA was prepared by dissolving PMA in sterile dimethylsulfoxide (Sigma). The stock solution was stored frozen at −20°C. Immediately prior to use, the PMA stock solution was diluted in RPMI medium. The differentiation-inducing dose of PMA for THP-1 cells was determined in preliminary dose-response experiments (data not shown). The criteria for differentiation of THP-1 cells were cell adherence, changes in cell morphology, and changes in the cell surface marker expression profile. Following 72 hours incubation, the cells were washed two times with the medium and incubated with particles (5 particles/cell). The supernatants were collected and stored at −70 °C until the cytokine analysis. Proinflammatory cytokines, interleukin-6 (IL-6) and interleukin-8 (IL-8) were analyzed using commercial ELISA kits (BD Biosciences, USA).
RESULTS AND DISCUSSION
Mesoporous hemispherical silicon microparticles were fabricated by photolithography and electrochemical etching as previously described 7. In our study, we used standard surface modification procedures developed for silicon-based materials (schematically presented in Figure 1). During the oxidation process, partial erosion of the particle surface led to an introduction of free hydroxyl groups, imparting to the particles a negative zeta potential (−31.5mV). Through silane chemistry the hydroxyl surface groups were covalently coupled to positively charged 3-Aminopropyltriethoxysilane (APTES), reversing the net surface charge of the particles to +14.73mV. APTES amine groups further served as a background for linking molecules to the particles surface. First, to estimate the range of molar ratios suitable for further conjugation of surface modifiers, we evaluated the effect of fluorescent probe concentration in the reaction medium on the fluorescence of the silicon particles. In the concentration range of 3.75–15mM of the 488-Dylight in the reaction medium, the net fluorescence intensity of the particles reached a plateau, which can be attributed to saturation of the bindings sites on the particles surface (Supplementary data, Figure 1S). A slight reduction in the fluorescence intensity of the particles was observed at higher concentrations of the probe, which could be related to the quenching effect of the probe on the surface. This general behavior was consistent and repetitive among different experiments, though numerical values of fluorescent intensity slightly vary, due to the slightly different surface area and properties of pSi microparticles. Based on these results, a concentration of 10mM of PEG was chosen in order to obtain a saturation of the modifier on the particle surface. As in the case of the fluorescent probe, PEG molecules (MWs from 245 to 5000) were bound to the particles through APTES amine groups. No direct correlation was observed between the length of the PEG molecule and the zeta potential values (Table 1), though all PEGs and fluorescent probes bound to APTES amine groups resulted in a neutralization of the positive charges introduced by APTES thus causing a slightly negative zeta potential, which could be partially explained by the charge-shielding effect of PEG backbones.
Figure 1.
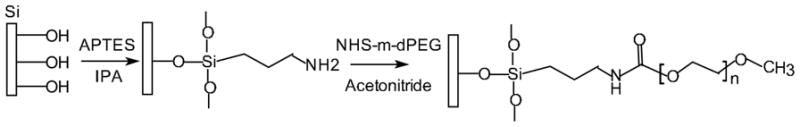
Schematic presentation of chemical modification of pSi microparticles with APTES and PEG molecules.
Table 1.
Description and zeta potential values of the investigated microparticles
| Sample name | Particles zeta potential, mV Mean ± SD (n=4) |
|---|---|
| Oxidized | −31.05±2.73 |
| APTES | +14.73±1.62 |
| PEG 245 | −15.58±3.58 |
| PEG 333 | −3.69±2.23 |
| PEG 509 | −9.68±2.49 |
| PEG 686 | −13.44±3.40 |
| PEG 862 | −3.94±1.91 |
| PEG 1214 | −11.29±1.93 |
| PEG 3400-Dylight 488 | −8.34±2.51 |
| PEG 5000 | −6.48±2.84 |
| Dylight 488 | −15.44±3.40 |
To evaluate the degradation rate of the particles under the simulated physiological conditions, we initially tested the degradation of small pores (10nm) and large pores (30–50nm) non-PEGylated APTES particles in phosphate buffered saline (PBS, pH 7.2) and fetal bovine serum serum (FBS) (Figure 2). In agreement with the published literature, degradation kinetics of the mesoporous Si particles was strongly dependant on the pore size 13. Particles having small pores degraded much slower than the particles with large pores. As the following step, we evaluated the influence of a modification with various PEGs on the degradation kinetics of the particles. Seven PEGs with varying molecular weights were employed: 245, 333, 509, 686, 1214, 3400 and 5000Da. Figure 3 shows degradation profiles of PEGylated particles with 30–50nm pores in PBS and 100% serum in vitro at 37°C. Generally, particles degraded faster in serum, and the higher was PEG’s molecular weight, the slower the degradation profile of the particles was obtained in both physiological media. The conjugation of the PEG with lowest molecular weight to the particles surface did not induce any change in the degradation kinetics in serum, but inhibited degradation and consequently the release of orthosilicic acid into buffer. When PEGs with the longer chains were evaluated, Si mass loss from the particles was slowed down, and the almost fully degraded within 18 to 24 hours in serum and within 48 hours in PBS. The most dramatic effect was observed for PEGs 3400 and 5000 which inhibited the degradation of the systems very prominently, with complete degradation achieved after four days. For these particles during the early stages of the degradation, there was a “lag” period of little or no mass loss.
Figure 2.

Degradation kinetics of large pores (pores size 30–50nm) and small pores (10nm) Si microparticles as evaluated by ICP-AES. The degradation kinetic profile is expressed as a percentage of the total Si contents released to the degradation medium.
Figure 3.


Degradation kinetics of large pores PEGylated pSi microparticles as evaluated by ICP-AES. The degradation kinetic profile is expressed as a percentage of the total Si contents released to the degradation medium: (A) PBS pH 7.2; (B) Fetal Bovine Serum (FBS).
The degradation process as a function of time as shown in Figure 3, can be separated into two phases: phase I, up to about 24 hours; and phase II, from 24 hours onward. The percentage of Si released (Mt) in solution over time can be described quite accurately in both phases employing a general power law αtβ with different scaling coefficients. Regarding the phase I, the APTES modified surface and short PEG chains (PEG245) behave similarly with Mt growing with time following a square root relationship ( ) with α=23.10 and 23.48 (R2=0.965 and 0.984 as from Table 2), respectively. For coating made with longer PEG chains, the exponent β grows with the length of the polymer as listed in Tab. 2S, with β ranging from 0.7 to 1.5; whereas α decreases leading to longer degradation times. Higher-order degradation laws with Mt = αt3 have been observed for PEG3400 and PEG5000 with α =0.0047 and α=0.0020, respectively with R2=0.999 in both cases as from Table 2 (Figure 4, Table 2). For phase II, only particles coated with PEG3400 and PEG5000 exhibit a significant degradation, whereas APTES modified and particles with short PEG chains (up to PEG1214) have almost fully degraded after 18 hours. For PEG3400 and PEG5000, the degradation law can be again described through a general power law of the type αtβ with β=0.6 and α=6.87 (R2=0.971) and α=5.50 (R2=0.992), respectively.
Table 2.
Power law coefficients and R2 values for the first degradation phase.
| Scaling coefficients | R2 | ||
|---|---|---|---|
| APTES | α = 23.10 | β = 0.5 | 0.965 |
| PEG 245 | α = 23.48 | β = 0.5 | 0.984 |
| PEG 333 | α = 11.81 | β = 0.7 | 0.999 |
| PEG 509 | α = 7.03 | β = 0.9 | 0.994 |
| PEG 686 | α = 2.02 | β = 1.3 | 0.986 |
| PEG 862 | α =1.46 | β = 1.4 | 0.986 |
| PEG 1214 | α = 0.94 | β = 1.5 | 0.998 |
| PEG 3400 | α = 0.0047 | β = 3.5 | 0.999 |
| PEG 5000 | α = 0.0020 | β = 3.0 | 0.999 |
Figure 4.

Comparison between the experimental data (dots) and the best fitting power laws αtβ for the first phase of the degradation process according to scaling low Mt =α·tβ
Interestingly, for APTES modified and PEG245 coated particles, the degradation laws exhibit a square root behavior which is possibly associated with a diffusive release of silicic acid from the porous silicon matrix into the surrounding solution. As the length of the PEG chains attached on the particle surface increases, the diffusion of the silicic acid from the pores, where most of the degradation occurs, to the surrounding media is more and more hindered possibly by surface steric interactions with the polymer chains. Notably, a similar behavior is observed for PEG3400 and PEG5000 during phase II, with degradation laws exhibiting an exponent β=0.6, which is very close to that associated with pure diffusion (β=0.5). This would suggest that, during phase II, most of the PEG chains decorating the particle surface have been removed and released in the surrounding medium because of the degradation of the first porous layers.
The deterioration of the pSi microparticle surface morphology over time was evaluated by Scanning Electron Microscopy (SEM). Figure 5 presents SEM micrographs of the particles during the degradation process. The rate of deterioration of the microparticles was associated with the rate of Si chemical degradation, and microparticles conjugated to higher molecular weight PEGs exhibited surface deterioration at a much slower rate. It can be seen that the degradation of the APTES modified (non-PEGylated) particles over time occurred by means of erosion of the particles surface as well as of the pores. As the study progressed, the pore sizes became wider and the surface of the particle less smooth and more irregular. This degradation profile is in general agreement with the published reports on degradation of porous silicon structures [10]. With intermediate PEG (MW 861), the appearance of the particles during the in vitro degradation process changed. The most prominent erosion is seen in the pores in comparison to the particle’s outer surface. This different degradation pattern could be attributed to the steric hindrance of the hydrophilic polymer molecules which probably cover particle surface more efficiently outside the pores, thus preventing penetration of water and other components, which play an important role in the degradation process. In the case of high MW PEG (5000) almost no degradation is seen within the first 48 hours, which confirms the data obtained by ICP-AES analysis.
Figure 5.
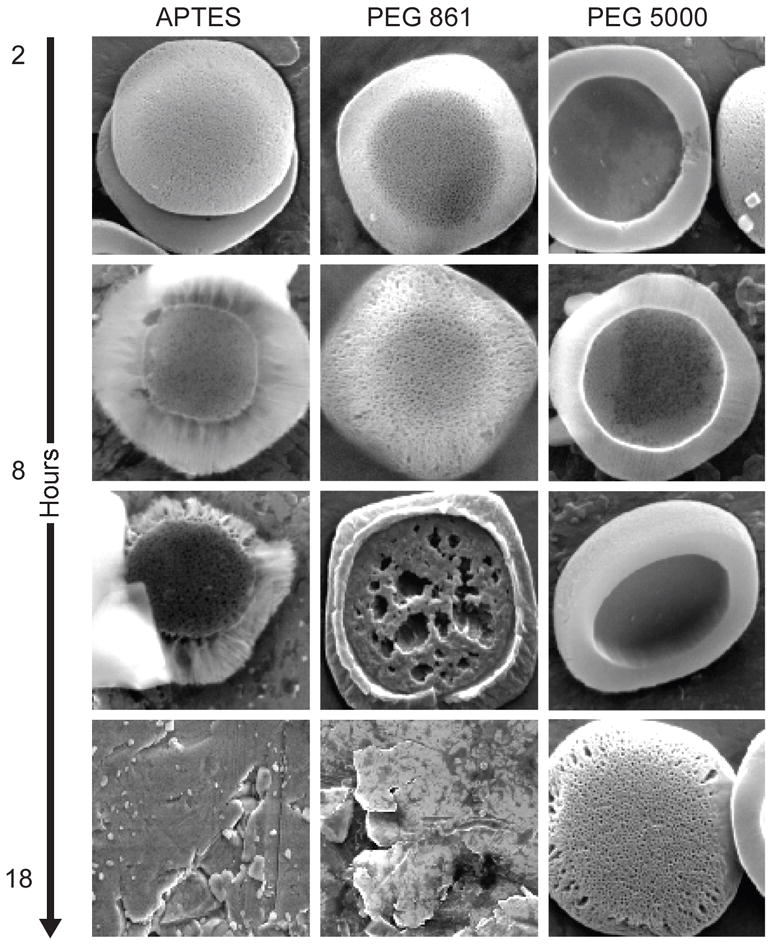
SEM images of the pSi particles during the degradation process in PBS pH 7.2. Systems shown: a) APTES particles; b) Particles modified with PEG 861; c) Particles modified with PEG 5000. Timepoints: 2, 8, 18 and 48 hours.
To evaluate the kinetics of surface degradation of the particles, APTES and PEG3400 particles were labeled with the Dylight 488 fluorescent probe. The release kinetics of the probe from particles surface into the degradation media was followed by fluorescence intensity and FACS. Based on the fluoremetric analysis, for non-PEGylated particles, the fluorescent probe conjugated to the surface was released into the degradation medium within 8–16 hours depending on the degradation medium. For PEGylated particles the surface erosion rate was significantly extended and the fluorescent probe was released from the particle surface only after 24–48 hours (Figure 6) as was also confirmed by FACS analysis (data not shown). The obtained profiles were in agreement with the data on degradation kinetics of the particles surface as evaluated by ICP-AES and SEM.
Figure 6.

Erosion of fluorescent PEG vs low MW probe from the pSi particle surface as followed up by fluorimetry in the degradation medium in PBS and FBS. The degradation kinetic profile is expressed as a percentage of the total fluorescence released to the degradation medium at 72 hours.
The ability to control the release of drug and imaging agents from pharmaceutical systems is critical for many clinical applications. In the case of the multistage delivery carrier 7,16–17 comprised of 1st stage microparticles bearing 2nd stage nanoparticles within the pores of pSi, the release of the second stage nanoparticles from the 1st stage pSi microparticles will depend on several mechanisms, including their diffusion outside the pores, as well as on the simultaneous Si erosion and degradation of the matrix. The mechanism of degradation and drug release from biodegradable controlled release systems can generally be described in terms of three basic parameters. First, the type of the hydrolytically unstable linkage in the system and its position. Second, the way the system biodegrades, either at the surface or uniformly throughout the matrix, affects device performance substantially. The third significant factor is the design of the drug delivery system encountering for system geometry and morphology as well as for the mechanism of loading of therapeutic agents. For example, the active agent may be covalently attached to the particle matrix and released as the bond between drug and polymer cleaves.
The size and number of pores in pSi affects its physiochemical properties, and as a consequence different types of mesoporous Si particles degrade in aqueous solutions and biological fluids at different rates, which could be directly translated to release of free drugs or second stage microparticles from the matrix. The pores of the particles could be considered as a void fraction, being in constant contact with the degradation fluids and presumably originating the orthosilicic acid - the degradation product of porous silicon. Orthosilicic acid, Si(OH)4, is the biologically relevant water soluble form of silicon (Si), recently proven to be play a significant role in bone and collagen growth. Porous Si films release Si(OH)4 (silicic acid) in aqueous solutions in the physiological pH range through hydrolysis of the Si-O bonds, 18 which is harmlessly excreted in the urine through the kidneys 19. In this study we addressed the question how the surface modification of pSi surface with PEG affect the degradation kinetics. APTES particles are a subject of homogenous surface degradation, where the erosion occurs homogeneously throughout the whole surface of the particle as well as the pores. In the case of PEGylated Si particles, the obtained degradation profile could be defined as heterogeneous erosion which besides the surface area, geometry and morphology of the particles is also defined by the length of the polymer chains covering the particle surface. PEGylation in this case appears to be the factor which controls penetration of solutes into the Si matrix of the particles. Derivatization can dramatically affect light-emitting properties of pSi and is degradation. For example, Buriak and co-workers have demonstrated that hydrosilyation with 1-dodecyne may sufficiently stabilize the material to render it corrosion-resistant 20. It is also noteworthy that the higher degradation rates are observed for the particles incubated with serum as compared to PBS, indicating the stronger ionic strength or involvement of some biological processes such as enzymatic degradation.
Events that follow the administration of foreign material into the body could provoke acute or chronic inflammation, while the last one is characterized by the presence of macrophages and release of inflammatory cytokines. Injectable biomaterials are expected to be biocompatible in terms of lack of immunogenic and inflammatory responses. Though silicon has been recognized as an essential trace element in the body which participates in connective tissue, especially cartilage and bone formation 19, some forms of crystalline silicon dioxide are known as a cytotoxic agent in macrophages 21–23. Thus, it was important to assess the effect of pSi microparticles with various surface modifications on human immune cells. Keeping this in mind, we evaluated the biocompatibility of the systems with human monocyte derived differentiated cultured macrophages. Data clearly demonstrate that the tested systems did not induce release of proinflammatory cytokines IL-6 and IL-8 over 48 hours period time in THP-1 macrophages (Figure 7). On contrary, when the cells were incubated with zymosan particles, a positive control, a very prominent increase in the cytokines release was observed. Phagocytic receptors on macrophages bind zymosan, stimulate particles engulfment and cytokines release. This agent is well known to induce inflammatory signals in macrophages through toll-like receptors TLR2 and TLR6. These results are in agreement with previously published studies on zymosan and other Si based systems. As an example, silica based Sol-gel glass was reported not to induce a significant inflammatory response by polymorphonuclear leukocytes 24 and oxidized nanoporous silicon was found to be compatible with primary hepatocytes 25–26. In our recent study, we have also shown that vascular endothelial cells, following internalization of silicon microparticles, maintain cellular integrity, viability and normal mitotic division 17. More explicit biocompatibility studies of the pSi microparticles for biomedical applications are currently underway in various in vitro and in vivo models in our laboratory.
Figure 7.

Release of proinflammatry cytokines IL-6 and IL-8 by human cultured THP-1 macrophages following incubation with pSi particles with various surface modifications.
CONCLUSIONS
A fine control of the degradation and release kinetics of mesoporous silicon structures is of fundamental importance in the development of multistage and multifunctional delivery systems. pSi microcarriers can be administered systemically and used to deliver the payload of different nature (therapeutic, imaging and so on). The size of the pores and the surface chemistry of the pSi structure can be controlled during the fabrication process and thereafter. We found that by conjugation of PEGs with various backbone length to porosified silicon microparticles, it is possible to finely tune the degradation kinetics of the material possessing large pores size. The most dramatic effect was observed for PEGs 3400 and 5000 which inhibited the degradation of the systems over more than 3 days. These data point toward the possibility to control degradation of mesoporous silicon microparticles and devices by means of PEGylation and may have important clinical implications.
Supplementary Material
Acknowledgments
Matt Landry is gratefully recognized for his assistance in preparing the artwork for this manuscipt. The authors acknowledges a financial support from the following sources: NIH U54CA143837, DoDW81XWH-04-2-0035 Project 16, NASA SA23-06-017, State of Texas, Emerging Technology Fund, DoD W31P4Q-07-1-0008 and NIH 1R01CA128797-01.
References
- 1.Uhlir A. Electrolytic shaping of Germanium and Silicon. Bell System Technical Journal. 1956;35:333–347. [Google Scholar]
- 2.Nijdam AJ, Ming-Cheng Cheng M, Geho DH, Fedele R, Herrmann P, Killian K, Espina V, Petricoin EF, 3rd, Liotta LA, Ferrari M. Physicochemically modified silicon as a substrate for protein microarrays. Biomaterials. 2007;28(3):550–8. doi: 10.1016/j.biomaterials.2006.08.051. [DOI] [PubMed] [Google Scholar]
- 3.Nijdam AJ, Zianni MR, Herderick EE, Cheng MM, Prosperi JR, Robertson FA, Petricoin EF, Liotta LA, Ferrari M. Application of physicochemically modified silicon substrates as reverse-phase protein microarrays. J Proteome Res. 2009;8(3):1247–54. doi: 10.1021/pr800455y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lehmann V, Gosele U. Porous silicon formation: A quantum wire effect. Applied Physical Letters. 1991;58:656–658. [Google Scholar]
- 5.Lin VS, Motesharei K, Dancil KP, Sailor MJ, Ghadiri MR. A porous silicon-based optical interferometric biosensor. Science. 1997;278(5339):840–3. doi: 10.1126/science.278.5339.840. [DOI] [PubMed] [Google Scholar]
- 6.Li YY, Cunin F, Link JR, Gao T, Betts RE, Reiver SH, Chin V, Bhatia SN, Sailor MJ. Polymer replicas of photonic porous silicon for sensing and drug delivery applications. Science. 2003;299(5615):2045–7. doi: 10.1126/science.1081298. [DOI] [PubMed] [Google Scholar]
- 7.Tasciotti E, Liu X, Bhavane R, Plant K, Leonard AD, Price BK, Cheng MM, Decuzzi P, Tour JM, Robertson F, et al. Mesoporous silicon particles as a multistage delivery system for imaging and therapeutic applications. Nat Nanotechnol. 2008;3(3):151–7. doi: 10.1038/nnano.2008.34. [DOI] [PubMed] [Google Scholar]
- 8.Sharma S, Nijdam AJ, Sinha PM, Walczak RJ, Liu X, Cheng MM, Ferrari M. Controlled-release microchips. Expert Opin Drug Deliv. 2006;3(3):379–94. doi: 10.1517/17425247.3.3.379. [DOI] [PubMed] [Google Scholar]
- 9.Salonen J, Laitinen L, Kaukonen AM, Tuura J, Bjorkqvist M, Heikkila T, Vaha-Heikkila K, Hirvonen J, Lehto VP. Mesoporous silicon microparticles for oral drug delivery: loading and release of five model drugs. J Control Release. 2005;108(2–3):362–74. doi: 10.1016/j.jconrel.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 10.Brumm MV, Nguyen QD. Fluocinolone acetonide intravitreal sustained release device--a new addition to the armamentarium of uveitic management. Int J Nanomedicine. 2007;2(1):55–64. doi: 10.2147/nano.2007.2.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goh AS, Chung AY, Lo RH, Lau TN, Yu SW, Chng M, Satchithanantham S, Loong SL, Ng DC, Lim BC, et al. A novel approach to brachytherapy in hepatocellular carcinoma using a phosphorous32 (32P) brachytherapy delivery device--a first-in-man study. Int J Radiat Oncol Biol Phys. 2007;67(3):786–92. doi: 10.1016/j.ijrobp.2006.09.011. [DOI] [PubMed] [Google Scholar]
- 12.Canham LT, Reeves CL, Newey JP, Houlton MR, Cox TI, Buriak JM, Stewart MP. Derivatized Mesoporous Silicon with Dramatically Improved Stability in Simulated Human Blood Plasma. Advanced Materials. 1999;11(18):1505–1507. [Google Scholar]
- 13.Canham LT. Bioactive silicon structure fabrication through nanoetching techniques. Advanced Materials. 1995;7(12):1033–1037. [Google Scholar]
- 14.Immordino ML, Dosio F, Cattel L. Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 2006;1(3):297–315. [PMC free article] [PubMed] [Google Scholar]
- 15.Wattendorf U, Merkle HP. PEGylation as a tool for the biomedical engineering of surface modified microparticles. Journal of Pharmaceutical Sciences. 2008;97(11):4655–4669. doi: 10.1002/jps.21350. [DOI] [PubMed] [Google Scholar]
- 16.Riehemann K, Schneider SW, Luger TA, Godin B, Ferrari M, Fuchs H. Nanomedicine--challenge and perspectives. Angew Chem Int Ed Engl. 2009;48(5):872–97. doi: 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serda RE, Gu J, Bhavane RC, Liu X, Chiappini C, Decuzzi P, Ferrari M. The association of silicon microparticles with endothelial cells in drug delivery to the vasculature. Biomaterials. 2009;30(13):2440–8. doi: 10.1016/j.biomaterials.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 18.Jugdaohsingh R, Anderson SH, Tucker KL, Elliott H, Kiel DP, Thompson RP, Powell JJ. Dietary silicon intake and absorption. Am J Clin Nutr. 2002;75(5):887–93. doi: 10.1093/ajcn/75.5.887. [DOI] [PubMed] [Google Scholar]
- 19.Carlisle EM. Silicon: a possible factor in bone calcification. Science. 1970;167(916):279–80. doi: 10.1126/science.167.3916.279. [DOI] [PubMed] [Google Scholar]
- 20.Buriak JM, Allen MJ. Lewis Acid Mediated Functionalization of Porous Silicon with Substituted Alkenes and Alkynes. Journal of the American Chemical Society. 1998;120(6):1339–1340. [Google Scholar]
- 21.Absher MP, Trombley L, Hemenway DR, Mickey RM, Leslie KO. Biphasic cellular and tissue response of rat lungs after eight-day aerosol exposure to the silicon dioxide cristobalite. Am J Pathol. 1989;134(6):1243–51. [PMC free article] [PubMed] [Google Scholar]
- 22.Kolb-Bachofen V. Uptake of toxic silica particles by isolated rat liver macrophages (Kupffer cells) is receptor mediated and can be blocked by competition. J Clin Invest. 1992;90(5):1819–24. doi: 10.1172/JCI116057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wilson J, Pigott GH, Schoen FJ, Hench LL. Toxicology and biocompatibility of bioglasses. J Biomed Mater Res. 1981;15(6):805–17. doi: 10.1002/jbm.820150605. [DOI] [PubMed] [Google Scholar]
- 24.Palumbo G, Avigliano L, Strukul G, Pinna F, Del Principe D, D’Angelo I, Annicchiarico-Petruzzelli M, Locardi B, Rosato N. Fibroblast growth and polymorphonuclear granulocyte activation in the presence of a new biologically active sol-gel glass. J Mater Sci Mater Med. 1997;8(7):417–21. doi: 10.1023/a:1018553505001. [DOI] [PubMed] [Google Scholar]
- 25.Alvarez SD, Derfus AM, Schwartz MP, Bhatia SN, Sailor MJ. The compatibility of hepatocytes with chemically modified porous silicon with reference to in vitro biosensors. Biomaterials. 2009;30(1):26–34. doi: 10.1016/j.biomaterials.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chin V, Collins BE, Sailor MJ, Bhatia SN. Compatibility of Primary Hepatocytes with Oxidized Nanoporous Silicon. Advanced Materials. 2001;13(24):1877–1880. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.