

Abstract
Purpose
Patients with mixed lineage leukemia (MLL)–rearranged B-lymphoblastic leukemias (B-ALL) have an unfavorable prognosis and require intensified treatment. Multiple MLL fusion partners have been identified, complicating the diagnostic evaluation of MLL rearrangements. We analyzed molecular markers of MLL rearrangement for use in rapid diagnostic assays and found the immunomodulatory protein, Galectin-1 (Gal-1), to be selectively expressed in MLL-rearranged B-ALL.
Experimental Design
Transcriptional profiling of ALL subtypes revealed selective overexpression of Gal-1 in MLL-rearranged ALLs. For this reason, we analyzed Gal-1 protein expression in MLL-germline and MLL-rearranged adult and infant pediatric B-ALLs and cell lines by immunoblotting, immunohistochemistry, and intracellular flow cytometry of viable tumor cell suspensions. Because deregulated gene expression in MLL-rearranged leukemias may be related to the altered histone methyltransferase activity of the MLL fusion protein complex, we also analyzed histone H3 lysine 79 (H3K79) dimethylation in the LGALS1 promoter region using chromatin immunoprecipitation.
Results
Gal-1 transcripts were significantly more abundant in MLL-rearranged B-ALLs. All 32 primary MLL-rearranged B-ALLs exhibited abundant Gal-1 immunostaining, regardless of the translocation partner, whereas only 2 of 81 germline-MLL B-ALLs expressed Gal-1. In addition, Gal-1 was selectively detected in newly diagnosed MLL-rearranged B-ALLs by intracellular flow cytometry. The LGALS1 promoter H3K79 was significantly hypermethylated in MLL-rearranged B-ALLs compared with MLL-germline B-ALLs and normal pre-B cells.
Conclusion
In B-ALL, Gal-1 is a highly sensitive and specific biomarker of MLL rearrangement that is likely induced by a MLL-dependent epigenetic modification.
B-lymphoblastic leukemia (B-ALL) is the most common malignancy of childhood and a disease with a poor prognosis among adults (1). There are several cytogenetic abnormalities characteristic of B-ALL that largely determine the biology of the disease, affect prognosis, and guide therapy (2, 3). Leukemias with rearrangements of the mixed lineage leukemia (MLL) gene on chromosome 11q23 exhibit unique clinical and biological features (4–7). MLL rearrangements occur in over 70% of infant B-ALLs and are less frequent in older patients (4–7). MLL aberrations are largely restricted to immature CD10-negative blasts that often coexpress myeloid markers (5–7). In both adults and children, MLL rearrangements are frequently associated with a particularly poor outcome (1, 8–11).
MLL translocations generate a new chimeric gene, in which the NH2-terminal portion of MLL is fused to the COOH-terminal sequence from multiple different partners (4–7). The common result of many of these rearrangements is the expression of a DNA-binding protein that recruits additional histone methyltransferases such as DOT1L and leads to ectopic histone H3 lysine 79 dimethylation (H3K79diMe; refs. 5, 12, 13). This epigenetic modification is associated with the deregulated transcription of multiple genes including HOXA cluster (12). The H3K79diMe histone modification mark and associated gene expression signature are distinguishing features of human and murine MLL-rearranged leukemias (12).
Current time-consuming diagnostic techniques such as fluorescence in situ hybridization or Southern blotting are not always available at initial diagnosis and are not able to detect all genetic abnormalities involving MLL (6, 7). Because the early identification of MLL rearrangements may guide therapy or clinical trial enrollment (8, 9, 14), it would be useful to have a more efficient method of identifying MLL-rearranged B-ALLs at diagnosis.
Galectins are a conserved family of carbohydrate-binding proteins that regulate innate and adaptive immune responses and promote tumor immune escape (15–17). Galectin-1 (Gal-1), a prototype member of this family, is a potent anti-inflammatory factor and a suppressive agent for T-cell responses (15–18). Through the selective recognition of multiple Gal β1,4 GlcNAc (N-acetyllactosamine) units on the branches of N- or O-linked glycans, Gal-1 controls T-cell homeostasis by inducing the selective apoptosis of TH1 and TH-17 and cytotoxic T effector cells, promoting the tolerogenic function of dendritic cells, and controlling T regulatory cell function (16–20). In solid tumor models, Gal-1 also promotes escape from T-cell–dependent immunity and confers immune privilege to tumor cells (16, 17, 21).
Recently, we showed that the malignant Hodgkin Reed-Sternberg cells and variants of classical Hodgkin lymphoma express and secrete high levels of Gal-1 in an activator protein (AP-1)–dependent manner, and that the expression of Gal-1 promotes the skewed, ineffective TH2-type T-cell infiltrate that is characteristic of this disease (22). Among other large cell lymphomas, only anaplastic large cell lymphoma exhibits constitutive AP-1 signaling and Gal-1 expression—a finding that suggests a common mechanism of Gal-1 transcriptional regulation by these tumors (23). The expression and regulation of Gal-1 in other hematopoietic neoplasms remains unknown.
Herein, we report that Gal-1 is a highly sensitive and specific marker of MLL-rearranged B-ALL. In addition, we show that the specific overexpression of Gal-1 in MLL-rearranged B-ALLs is associated with MLL-mediated modifications of the LGALS1 locus.
Materials and Methods
Case selection
One series of adult primary B-ALLs was derived from the files of Brigham & Women’s Hospital with Institutional Review Board approval (series 1). All diagnoses were established at the time of the original biopsy evaluation and based on the criteria established by the current WHO classification system (24). A second independent series of 60 infant/pediatric B-ALLs was derived from the Department of Pediatrics, Hematology, Oncology & Diabetology, Medical University of Lodz, Lodz, Poland and the centers of the Polish Pediatric Leukemia/Lymphoma Study Group with appropriate Institutional Review Board approvals (series 2). Rearrangements of the MLL locus (chromosome 11q23) in series 1 were identified in diagnostic bone marrow aspirates by either routine karyotypic analysis (G-banding) or fluorescent in situ hybridization analysis with a break-apart probe targeting the MLL locus (Vysis probes/Abbott Molecular). Rearrangements of the MLL locus in series 2 were identified using MLL11q23 split probe DC (Qbiogene), in accordance with the manufacturer’s instructions. In series 2, the partner genes for MLL rearrangements were identified using a long-distance inverse PCR approach as previously described (25).
Immunohistochemistry
Immunohistochemistry of series 1 primary B-ALLs was performed using 5-μm-thick Zenker’s-fixed, paraffin-embedded tissue sections on individual slides as previously described (23). Briefly, slides were soaked in xylene, passed through graded alcohols, and then pretreated with 10-mmol/L citrate (pH 6.0; Zymed) in a steam pressure cooker (Decloaking Chamber, BioCare Medical) as per the manufacturer’s instructions. All further steps were performed at room temperature in a hydrated chamber. Slides were then treated with Peroxidase Block (DAKO USA) for 5 min to quench endogenous peroxidase activity. A primary rabbit polyclonal anti–Gal-1 antiserum (1:10,000 dilution; generated in the laboratory of M.A.S.) was applied in the DAKO diluent (DAKO) for 1 h at room temperature. Slides were washed in 50 mmol/L Tris-Cl (pH 7.4) and anti-rabbit horseradish peroxidase–conjugated antibody solution (Envision+ detection kit, DAKO) was applied for 30 min. After further washing, immunoperoxidase staining was developed using a diaminobenzidine chromogen kit (DAKO) per the manufacturer’s instruction and counterstained with Harris hematoxylin (Polyscientific).
Immunocytochemistry of series 2 primary B-ALLs was performed using formalin-fixed bone marrow aspirate smears. First, endogenous peroxidase activity was quenched by treatment of the slides with a 3% perhydrol solution in methanol for 5 min. Subsequently, slides were treated with Target Retrieval Solution (pH 9.0; DAKO) in a water bath at 95°C for 45 min. Staining with antibodies was performed as described above. After further washing, immunoperoxidase staining was developed using a diaminobenzidine chromogen kit (DAKO) and was counterstained with Mayer hematoxylin. Following a brief wash in water, slides were dehydratated with a series of alcohol and xylene solutions, mounted in Histokit medium, and coverslipped.
Immunohistochemical evaluation
Reactivity for Gal-1 in series 1 B-ALLs was scored independently by two hematopathologists (S.J.R and J.L.K). Gal-1 staining intensity was scored as follows: 0, no staining detected; 1+, weak staining; 2+, moderate staining; and 3+, strong staining of the tumor cells. Positive staining for a case was defined as 2+ or 3+ cytoplasmic staining in >25% of the tumor cells. Zero or 1+ staining in >25% of tumor cells or only focal reactivity of 2+ or 3+ in <25% of the tumor cells was considered negative. Positive staining of endothelial cells and macrophages served as positive internal controls. All cases were photographed at ×1,000 original magnification with an Olympus BX41 microscope with the objective lens of ×100/0.75 Olympus UPlanFL (Olympus). The pictures were taken using Olympus QColor3 and analyzed with the acquisition software QCapture v2.60 (QImaging) and Adobe Photoshop 6.0 (Adobe).
Gal-1 reactivity in series 2 B-ALLs was assessed independently by two hematopathologists (K.M. and W.M.). Gal-1 staining intensity was scored using the above-mentioned criteria. The cases were photographed in ×400 magnification with the Nikon Microphot FXA (Nikon). The pictures were analyzed using the MultiScanBase v 8.08 Image Analysis System (Computer Scanning Systems).
Immunoblotting
MLL-rearranged (SEM-K2, RS4;11) and MLL-germline (REH and NALM6) B-ALL cell lines were maintained in RPMI 1640 supplemented with 10% fetal bovine serum (Cellgro Mediatech), 10 mmol/L HEPES buffer, 4 mmol/L L-glutamine, 50 U/mL penicillin, and 50 U/mL streptomycin. For immunoblotting, cells were washed with ice-cold PBS and lysed with 1% NP40 buffer. Lysates were size fractionated on NuPAGE Novex 4% to 12% Bis-Tris Gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Millipore). Blots were incubated with primary antibodies (αGal-1 or α-β-actin) and appropriate horseradish peroxidase–labeled secondary antibodies and developed by enhanced chemiluminescence as previously described (22).
Intracellular flow cytometry
Intracellular flow cytometry was performed on NALM6 (MLL germline) and SEM-K2 (MLL rearranged) cell lines and viable primary tumor specimens from four B-ALL patients with known MLL translocation status (2 with MLL rearrangements and 2 with MLL-germline). After thawing the previously cryopre-served primary B-ALL specimens, viable tumor cells were isolated by Ficoll-Hypaque (GE Healthcare) gradient centrifugation and washed. Thereafter, 1 × 106 cells were fixed and permeabilized with the Cytofix/Cytoperm Fixation/Permeabilization kit (BD Biosciences) according to the manufacturer’s instructions. Cells were then stained sequentially with the affinity-purified anti-human Gal-1 (diluted 1:5,000; ref. 22) at 4°C for 30 min and anti-rabbit FITC– or Cy5-conjugated AffiniPure Fab Fragment (Jackson ImmunoResearch Laboratories, Inc.). For background fluorescence control, cells were stained with normal rabbit IgG and anti-rabbit FITC– or Cy5-conjugated Fab fragment alone. Intracellular Gal-1 expression was analyzed with the BD FACS Canto II flow cytometer (BD Biosciences) and FlowJo software (Tree Star, Inc.).
Chromatin immunoprecipitation
RS4;11, SEM-K2, and NALM6 cells (50 × 106) were fixed in 1% formaldehyde for 10 min at room temperature. Reactions were subsequently quenched in 0.125 mol/L glycine for 5 min. Cells were then washed with 1×PBS and lysed in 1% SDS lysis buffer [1% SDS, 50 mmol/L Tris (pH 8.0), and 10 mmol/L EDTA] containing protease inhibitors (Complete protease inhibitor cocktail; Roche Applied Science) and were sonicated. Lysates were diluted 10× with the dilution buffer [0.01% SDS, 1.1% Triton-X100, 1.2 mmol/L EDTA, 16.7 mmol/L Tris (pH 8.0), and 167 mmol/L NaCl, supplemented with protease inhibitor cocktail], precleared and subsequently incubated with rabbit polyclonal anti-H3K79diMe antibody (Abcam) or with normal rabbit IgG antibody (Santa Cruz Biotechnology) for 4 h. Immunocomplexes were captured with protein A/G plus agarose preblocked with salmon sperm DNA (Abcam) and washed twice with radioimmunoprecipitation assay buffer, twice with high salt wash buffer [0.1% SDS, 1% Triton-X100, 2 mmol/L EDTA, 20 mmol/L Tris (pH 8.0), and 500 mmol/L NaCl], twice with LiCl wash buffer [0.25 mol/L LiCl, 1% NP40, 1% sodium deoxycholate, 1 mmol/L EDTA, and 10 mmol/L Tris (pH 8.0)], and once with TE buffer. Thereafter, immune complexes were eluted with 1% SDS in 100 mmol/L NaHCO3 and cross-links were reversed by incubating samples for 8 h at 65°C. DNA fragments enriched by chromatin immunoprecipitation were recovered by standard phenol-chloroform extraction followed by ethanol precipitation and quantified by real-time PCR using LGALS1 promoter and control region primers (LGALS1 promoter amplicon 1, F: GGGTGGAGTCTTCTGACAGCTG, R: CCTGCCCTATCCCCTGGAC; LGALS1 promoter amplicon 2, F: TGGACTCAATCATGGCTTGTG, R: GGGCTAGAATCT-GCTCCCGAT; control region 1, F: ATGAGCCACAGTGCT-TGGC, R: GCCGCAGTGCTCTGTGGTAT; Control region 2, F: CTGATTGCTGGGCAGAGAGAA, R: TTTGCCTCCATCT-CAAAGCC), the PowerSYBR green kit (Applied Biosystems), and an ABI 7700 thermal cycler (Applied Biosystems). Relative enrichment in H3K79 dimethylation in Gal-1 locus and control regions versus input in H3K79diMe- and IgG-immunoprecipitated samples was calculated by using the 2−(ΔCT H3K79diMe − ΔCT IgG) method. SDs were calculated from triplicate ΔΔCT values.
H3K79 dimethylation in primary tumors
Normal bone marrow samples and diagnostic primary leukemia samples (peripheral blood or bone marrow) were obtained with informed consent from individuals treated according to protocols approved by the Institutional Review Board at the Dana-Farber Cancer Institute between 2000 and 2007. Samples were immunoprecipitated with anti–dimethyl H3K79 antibody (Abcam 3594) and hybridized to an Affymetrix-GeneChip Human Promoter 1.0R Array as described (12). The raw CEL files were processed with MAT (26) to obtain the signal strength at each probe. To analyze LGALS1 gene promoter methylation in normal pre-B cells, MLL-germline, and MLL-rearranged tumors, the signal strength of all of the probes in the promoter region (7.5 kb upstream to 2.5 kb downstream of TSS) was added and compared using the Kruskal-Wallis test. The MAT library, mapping files, and TSS definitions are based on the National Center for Biotechnology Information Build 36.1 human reference sequence.
Results
Gal-1 is overexpressed in MLL-rearranged B-ALLs
To identify molecular markers of MLL translocation that might be used in rapid diagnostic assays, we first compared the transcriptional profiles of primary B-ALLs with and without MLL translocations. Gal-1 transcripts were significantly more abundant in MLL-rearranged B-ALLs from two large independent data sets (Supplementary Data; refs. 27, 28). Consistent with these findings, Gal-1 protein was also more abundant in MLL-rearranged B-ALL cell lines and primary tumors than in MLL-germline B-ALL lines and tumors by Western blotting (Fig. 1A).
Fig. 1.
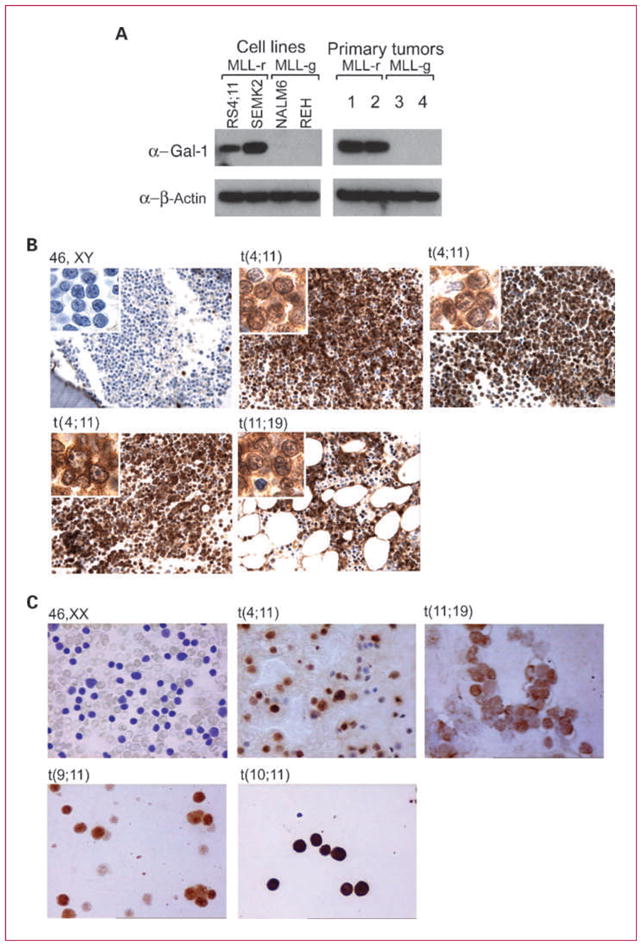
Gal-1 is overexpressed in MLL-rearranged B-ALL cell lines and primary tumors. A, Gal-1 protein expression in B-ALL cell lines (left) and primary tumors (right) with or without MLL-rearrangements [MLL-r, MLL-g (MLL-germline), respectively]. B and C, immunohistochemical analyses of Gal-1 in representative primary B-ALLs with known MLL status from two independent series. B, series 1 bone marrow biopsies were analyzed; C, series 2 bone marrow aspirates were assessed. B and C, representative primary B-ALLs with specific MLL translocations t(4;11), t(11;19), t(9;11), or t(10;11) or germline MLL are shown.
To assess the diagnostic utility of Gal-1 expression in identifying the MLL-rearranged B-ALL subtype, we performed Gal-1 immunostaining on an initial series of adult primary B-ALLs with known MLL status (series 1; Table 1). All 11 MLL-rearranged B-ALLs exhibited Gal-1 expression (11 of 11, 100%); in 10 of 11 cases, strong Gal-1 staining was present in >50% of the tumor cells. Gal-1+ tumors included seven MLL-rearranged B-ALLs with t(4;11) and four MLL-rearranged B-ALLs with t(11;19; Table 1; Fig. 1B). In marked contrast, only 1 of 40 B-ALLs without a cytogenetically detectable MLL translocation expressed Gal-1 (2.5%; P < 0.001, Fisher exact test; Table 1; Fig. 1B).
Table 1.
Gal-1 expression and MLL status in primary B-ALLs
| Karyotypic abnormality | No. of Gal-1-positive cases | No. of total cases | % positive for Gal-1 |
|---|---|---|---|
| Series 1* | |||
| MLL-rearranged | |||
| t(4;11)(q21;q23) | 7 | 7 | 100 |
| t(11;19)(q23;p13) | 4 | 4 | 100 |
| Total | 11 | 11 | 100 |
| MLL-germline | |||
| 46 XX or XY | 1 | 8 | 13 |
| t(9;22)/Ph+ | 0 | 16 | 0 |
| Hyperdiploid | 0 | 2 | 0 |
| Hypodiploid | 0 | 2 | 0 |
| Simple abnormal | 0 | 4 | 0 |
| Complex abnormal | 0 | 8 | 0 |
| Total | 1 | 40 | 3 |
| Series 2†‡ | |||
| MLL-rearranged | |||
| t(4;11)(q21;q23) | 14 | 14 | 100 |
| t(11;19)(q23;p13) | 2 | 2 | 100 |
| t(9;11) | 3 | 3 | 100 |
| t(10;11) | 1 | 1 | 100 |
| t(11;?) | 1 | 1 | 100 |
| Total | 21 | 21 | 100 |
| MLL-germline | |||
| 46 XX or XY | 1 | 24 | 4 |
| t(9;22)/Ph+ | 0 | 4 | 0 |
| t(1;19) | 0 | 1 | 0 |
| Hyperdiploid | 0 | 6 | 0 |
| Hypodiploid | 0 | 2 | 0 |
| Complex abnormal | 0 | 2 | 0 |
| Total | 1 | 39 | 3 |
In series 1, the median age at onset was 52 y (range, 18–73) for MLL-rearranged cases and 52.5 y (range, 24–84) for MLL-germline cases.
In series 2, the median age at onset was 0.4 y (range, 0–12.1) for MLL-rearranged cases and 5.75 y (range, 0.8–17.2) for MLL-germline cases.
In series 2, MLL fusion partner genes were confirmed by sequencing. Partner genes for the respective translocations included: t(4;11), MLL-AFF1; t(11;19), MLL-MLLT1; t(9;11), MLL-MLLT3; and t(10;11), MLL-MLLT10.
To further evaluate the utility of Gal-1 expression in the early diagnosis of primary MLL-rearranged B-ALL, we performed Gal-1 immunostaining of routine bone marrow smears from an independent infant/pediatric series of 21 MLL-rearranged B-ALLs and 39 B-ALLs without detectable MLL rearrangement (series 2). This series of MLL-rearranged B-ALLs included primary tumors with t(4;11), t(11;19), or additional MLL translocations that were not included in series 1, t(9;11) or t(10;11). All 21 MLL-rearranged B-ALLs exhibited strong Gal-1 expression (21 of 21, 100%), regardless of the specific MLL translocation and associated fusion partner (Table 1; Fig. 1C). In marked contrast, only 1 of 39 cases without a cytogenetically detectable MLL translocation expressed Gal-1 (2.6%; P < 0.001, Fisher exact test; Table 1).
Because intracellular flow cytometry is routinely used in the diagnostic evaluation of B-ALL, we next evaluated the utility of this approach for Gal-1 detection in B-ALL cell lines and primary B-ALLs with known MLL status. In fixed permeabilized cells, Gal-1 protein expression was high in MLL-rearranged B-ALL lines and primary tumors (Fig. 2A and B, left) and low/undetectable in MLL-germline B-ALL lines and primary tumors (Fig. 2A and B, right).
Fig. 2.

Gal-1 is detected by intracellular flow cytometry in MLL-rearranged B-ALL cell lines and primary tumors. Intracellular flow cytometry was performed on B-ALL cell lines (A) and viable primary tumor specimens from four B-ALL patients with known MLL translocation status (B). B, mean fluorescence intensity for control and anti–Gal-1 immunostaining was as follows: P1, 21.5 versus 108; P2, 24.2 versus 117; P3, 20.3 versus 35.5; and P4, 26.6 versus 63.3.
Mechanism of Gal-1 overexpression in MLL B-ALLs
We have previously shown that Gal-1 overexpression in classical Hodgkin lymphoma is mediated, in large part, by an AP-1–dependent enhancer (22). The AP-1–dependent Gal-1 enhancer did not increase the expression of a luciferase reporter in representative MLL-rearranged cell lines, suggesting an alternative mechanism of Gal-1 overexpression (data not shown). Because the Gal-1 promoter contains putative HOXA9 binding sites (data not shown), we next evaluated the consequences of HOXA9 overexpression on Gal-1 in MLL-rearranged B-ALLs. In two MLL-rearranged cell lines, shRNA-mediated depletion of HOXA9 did not decrease Gal-1 abundance, indicating that HOXA9 is not a major transcriptional activator of Gal-1 in MLL-rearranged ALL (data not shown).
Ectopic histone H3 lysine 79 (H3K79) dimethylation, a distinguishing feature of murine and human MLL B-ALLs, is related to the altered histone methyltransferase activity of MLL fusion protein complex (12). For this reason, we next analyzed H3K79 dimethylation in the LGALS1 promoter region using chromatin immunoprecipitation. LGALS1 promoter H3K79 dimethylation was ~5-fold higher in MLL-rearranged B-ALL cell lines (RS4;11 and SEM-K2) than in a MLL-germline B-ALL line (NALM-6; Fig. 3A). To determine whether similar abnormalities were found in primary MLL-rearranged B-ALLs, we next analyzed the H3K79 dimethylation of the LGALS1 locus in primary MLL-rearranged and germline B-ALLs and normal Lin− CD34+ CD19+ cells. Cumulative LGALS1 promoter region (−7.5 to + 2.5 kb) H3K79 dimethylation was significantly higher in primary MLL-rearranged B-ALLs than in MLL-germline BALLs and normal Lin− CD34+ CD19+ cell samples (Fig. 3B and C), suggesting that this epigenetic modification plays a role in the selective overexpression of Gal-1 in MLL-rearranged B-ALLs.
Fig. 3.

LGALS1 promoter exhibits enrichment of H3K79 dimethylation in MLL-rearranged B-ALL.
A, LGALS1 H3K79diMe in B-ALL cell lines with known MLL status. B, H3K79diMe ChiP-chip analysis of primary MLL-rearranged and MLL-germline B-ALLs and normal CD34/CD19+ cells. C, quantitative analysis of Gal-1 promoter H3K79 dimethylation in normal pre-B cells, MLL-germline (MLL-g), and MLL-rearranged (MLL-r) ALLs.
Discussion
Herein, we show that Gal-1 expression is a highly sensitive and specific marker of MLL-rearranged B-ALL in infant/pediatric and adult patients. Furthermore, Gal-1 expression can be evaluated in diagnostic clinical samples by immunohistochemistry of fixed, paraffin-embedded bone marrow biopsies or smears or by flow cytometry of permeabilized tumor cell suspensions. Gal-1 expression in MLL-rearranged B-ALL is likely driven by MLL-mediated chromatin modification.
Patients with MLL-rearranged B-ALLs have a particularly unfavorable prognosis, compared with patients with other types of B-ALL, and require intensified treatment (1, 8, 9). Therefore, it is important to identify MLL-rearranged B-ALLs at the earliest possible time point. To date, over 20 MLL fusion partners in B-ALL have been identified, making diagnostic evaluation of MLL rearrangements and follow-up monitoring a challenging and difficult task (6). Although several markers of MLL rearrangement have been proposed (7, 29–31), their specificity remains poor (7).
Several studies have shown that unique transcriptional profiles are characteristic of B-ALLs with distinct cytogenetic abnormalities (27, 28). The results suggest candidate tumor markers that may identify, with high certainty, an underlying genetic defect (27, 28). We verify this hypothesis by showing that the immunomodulatory carbohydrate-binding protein, Gal-1, is selectively expressed in B-ALLs harboring a MLL translocation.
Gal-1 expression is a highly sensitive, specific, and reproducible marker of MLL rearrangement, regardless of the fusion partner gene involved in the translocation. Fixed, paraffin-embedded biopsy specimens or routine bone marrow smears of B-ALL may be interrogated by rapid, standard immunohistochemical techniques to assess MLL translocation status. Furthermore, Gal-1 may be detected in MLL-rearranged B-ALLs by intracellular flow immunophenotyping at the time of diagnosis. Of note, the same high sensitivity and specificity of Gal-1 expression was observed in MLL-rearranged B-ALLs from adult and infant/pediatric patients. Finally, our results raise the possibility of monitoring minimal residual disease in patients with MLL-rearranged B-ALL (32, 33) by the detection of malignant Gal-1–positive B-lymphoblasts by flow cytometry, although additional studies are needed to verify this hypothesis.
In contrast to our previous studies in classical Hodgkin lymphoma and anaplastic large-cell lymphomas (22, 23), Gal-1 expression in MLL-rearranged B-ALLs was independent of constitutive AP-1 signaling. Instead, Gal-1 expression in MLL-rearranged B-ALLs is likely due to the aberrant H3K79 dimethylation of the LGALS1 promoter. The ectopic histone H3K79 dimethylation is a consequence of the MLL fusion protein complex–mediated activity and specifically correlates with unique gene expression signature in MLL-rearranged leukemias (12). The LGALS1 promoter had the same pattern of H3K79 diMe mark distribution as additional known MLL targets such as HOXA9. This epigenetic modification was significantly more abundant in MLL-rearranged primary B-ALLs than in MLL-germline B-ALLs or normal Lin− CD34+ CD19+ cells (12). Given that Gal-1 is not a transcriptional target of HOXA9, these observations suggest Gal-1 may be a direct target of the MLL fusion protein complex.
Although the mechanisms of Gal-1 overexpression differ in specific hematologic malignancies, this carbohydrate-binding protein plays a general role in limiting host anti-tumor immune responses (16, 17). In several tumor models, Gal-1 expression is associated with inefficient, Th2-skewed immune responses (16–18, 21, 22, 34). In one of the most extensively evaluated models, Gal-1 blockade resulted in tumor rejection that required intact CD4+ and CD8+ T-cell responses (17). Gal-1 may also promote the generation of tolerogenic dendritic cells that dampen tumor-specific T-cell–mediated immunity (19). Because spontaneous cytotoxic T-cell responses against leukemic cells are elicited in ALL and are used in experimental ALL immunotherapies (35–37), Gal-1 blockade may augment host antileukemia immune responses in MLL-rearranged B-ALLs.
Recent studies highlight the pathogenetic role of dynamic and bilateral interactions of leukemic blasts with bone marrow microenvironment (38–40). Specifically, leukemic cells modulate the architecture of the bone marrow microvasculature and rely upon protective, adhesion-mediated, and soluble signals from bone marrow stromal and endothelial cells. Given the additional roles of Gal-1 in the modulation of angiogenesis, adhesion, and cellular motility in tumor models (41–43), this protein may have additional pleiotropic effects in the pathogenesis of MLL-rearranged leukemias. More broadly, the identification of Gal-1 expression in MLL-rearranged B-ALLs represents a knowledge-based approach to biomarker discovery with both diagnostic and potential therapeutic implications.
Translational Relevance.
Patients with mixed lineage leukemia (MLL)–rearranged B-lymphoblastic leukemias (B-ALL) have an unfavorable prognosis and require intensified treatment. Multiple MLL fusion partners have been identified, complicating the diagnostic evaluation of MLL rearrangements and highlighting the need for a robust and rapidly detectable biomarker of MLL B-ALL. Herein, we show that MLL-rearranged B-ALLs selectively express the immunoregulatory protein, Galectin-1 (Gal-1), regardless of the specific MLL translocation and associated fusion partner. Gal-1 can be evaluated in diagnostic ALL samples using established techniques including intracellular flow cytometry or immunohistochemistry. The analysis of Gal-1 expression may accelerate the diagnosis, and guide therapy and clinical trial enrollment of patients with MLL-rearranged B-ALL. Furthermore, it may be possible to monitor minimal residual disease in patients with MLL-rearranged B-ALL by assessing malignant Gal-1–positive B-lymphoblasts by flow cytometry. Finally, because Gal-1 inhibits host anti-tumor immune responses and modulates tumor angiogenesis and adhesion, Gal-1 may also represent a rational therapeutic target.
Supplementary Material
Acknowledgments
We thank the Polish Pediatric Leukemia/Lymphoma Study Group for providing B-ALL cell samples, Drs. R. Marschalek and C. Meyer (Diagnostic Center of Acute Leukemia, Frankfurt/Main, Germany) for their assistance in the analysis of MLL partner genes, and Prof. Jozef Kobos and Dr. Elzbieta Los from the Medical University of Lodz for assistance in immunohistochemical evaluation.
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008;371:1030–43. doi: 10.1016/S0140-6736(08)60457-2. [DOI] [PubMed] [Google Scholar]
- 2.Teitell MA, Pandolfi PP. Molecular genetics of acute lymphoblastic leukemia. Annu Rev Pathol. 2009;4:175–98. doi: 10.1146/annurev.pathol.4.110807.092227. [DOI] [PubMed] [Google Scholar]
- 3.Meijerink JP, den Boer ML, Pieters R. New genetic abnormalities and treatment response in acute lymphoblastic leukemia. Semin Hematol. 2009;46:16–23. doi: 10.1053/j.seminhematol.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 4.Harper DP, Aplan PD. Chromosomal rearrangements leading to MLL gene fusions: clinical and biological aspects. Cancer Res. 2008;68:10024–7. doi: 10.1158/0008-5472.CAN-08-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krivtsov AV, Armstrong SA. MLL translocations, histone modifications and leukaemia stem-cell development. Nat Rev Cancer. 2007;7:823–33. doi: 10.1038/nrc2253. [DOI] [PubMed] [Google Scholar]
- 6.Meyer C, Kowarz E, Hofmann J, et al. New insights to the MLL recombinome of acute leukemias. Leukemia. 2009;23:1490–9. doi: 10.1038/leu.2009.33. [DOI] [PubMed] [Google Scholar]
- 7.Burmeister T, Meyer C, Schwartz S, et al. The MLL recombinome of adult CD10-negative B-cell precursor acute lymphoblastic leukemia—results from the GMALL study group. Blood. 2009;113:4011–5. doi: 10.1182/blood-2008-10-183483. [DOI] [PubMed] [Google Scholar]
- 8.Pieters R, Schrappe M, De Lorenzo P, et al. A treatment protocol for infants younger than 1 year with acute lymphoblastic leukaemia (Interfant-99): an observational study and a multicentre randomised trial. Lancet. 2007;370:240–50. doi: 10.1016/S0140-6736(07)61126-X. [DOI] [PubMed] [Google Scholar]
- 9.van der Linden MH, Valsecchi MG, De Lorenzo P, et al. Outcome of congenital acute lymphoblastic leukemia treated on the Interfant-99 protocol. Blood. 2009;114:3764–8. doi: 10.1182/blood-2009-02-204214. [DOI] [PubMed] [Google Scholar]
- 10.Pullarkat V, Slovak ML, Kopecky KJ, Forman SJ, Appelbaum FR. Impact of cytogenetics on the outcome of adult acute lymphoblastic leukemia: results of Southwest Oncology Group 9400 study. Blood. 2008;111:2563–72. doi: 10.1182/blood-2007-10-116186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hilden JM, Dinndorf PA, Meerbaum SO, et al. Analysis of prognostic factors of acute lymphoblastic leukemia in infants: report on CCG 1953 from the Children’s Oncology Group. Blood. 2006;108:441–51. doi: 10.1182/blood-2005-07-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krivtsov AV, Feng Z, Lemieux ME, et al. H3K79 methylation profiles define murine and human MLL-AF4 leukemias. Cancer Cell. 2008;14:355–68. doi: 10.1016/j.ccr.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okada Y, Feng Q, Lin Y, et al. hDOT1L links histone methylation to leukemogenesis. Cell. 2005;121:167–78. doi: 10.1016/j.cell.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 14.Tomizawa D, Koh K, Sato T, et al. Outcome of risk-based therapy for infant acute lymphoblastic leukemia with or without an MLL gene rearrangement, with emphasis on late effects: a final report of two consecutive studies, MLL96 and MLL98, of the Japan Infant Leukemia Study Group. Leukemia. 2007;21:2258–63. doi: 10.1038/sj.leu.2404903. [DOI] [PubMed] [Google Scholar]
- 15.van Kooyk Y, Rabinovich GA. Proteinglycan interactions in the control of innate and adaptive immune responses. Nat Immunol. 2008;9:593–601. doi: 10.1038/ni.f.203. [DOI] [PubMed] [Google Scholar]
- 16.Liu FT, Rabinovich GA. Galectins as modulators of tumour progression. Nat Rev Cancer. 2005;5:29–41. doi: 10.1038/nrc1527. [DOI] [PubMed] [Google Scholar]
- 17.Rubinstein N, Alvarez M, Zwirner NW, et al. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection; a potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5:241–51. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 18.Toscano MA, Bianco GA, Ilarregui JM, et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat Immunol. 2007;8:825–34. doi: 10.1038/ni1482. [DOI] [PubMed] [Google Scholar]
- 19.Ilarregui JM, Croci DO, Bianco GA, et al. Tolerogenic signals delivered by dendritic cells to T cells through a galectin-1-driven immunoregulatory circuit involving interleukin 27 and interleukin 10. Nat Immunol. 2009;10:981–91. doi: 10.1038/ni.1772. [DOI] [PubMed] [Google Scholar]
- 20.Garin MI, Chu CC, Golshayan D, Cernuda-Morollon E, Wait R, Lechler RI. Galectin-1: a key effector of regulation mediated by CD4+CD25+ T cells. Blood. 2006;109:2058–65. doi: 10.1182/blood-2006-04-016451. [DOI] [PubMed] [Google Scholar]
- 21.Le QT, Shi G, Cao H, et al. Galectin-1: a link between tumor hypoxia and tumor immune privilege. J Clin Oncol. 2005;23:8932–41. doi: 10.1200/JCO.2005.02.0206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Juszczynski P, Ouyang J, Monti S, et al. The AP1-dependent secretion of galectin-1 by Reed Sternberg cells fosters immune privilege in classial Hodgkin lymphoma. Proc Natl Acad Sci U S A. 2007;104:13134–9. doi: 10.1073/pnas.0706017104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodig SJ, Ouyang J, Juszczynski P, et al. AP1-dependent galectin-1 expression delineates classical hodgkin and anaplastic large cell lymphomas from other lymphoid malignancies with shared molecular features. Clin Cancer Res. 2008;14:3338–44. doi: 10.1158/1078-0432.CCR-07-4709. [DOI] [PubMed] [Google Scholar]
- 24.Jaffe ES, Harris NL, Stein H, Vardiman JW. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues. 3. IARC; 2001. pp. 111–4. [Google Scholar]
- 25.Meyer C, Schneider B, Reichel M, et al. Diagnostic tool for the identification of MLL rearrangements including unknown partner genes. Proc Natl Acad Sci U S A. 2005;102:449–54. doi: 10.1073/pnas.0406994102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson WE, Li W, Meyer CA, et al. Model-based analysis of tiling-arrays for ChIP-chip. Proc Natl Acad Sci U S A. 2006;103:12457–62. doi: 10.1073/pnas.0601180103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Armstrong SA, Staunton JE, Silverman LB, et al. MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat Genet. 2002;30:41–7. doi: 10.1038/ng765. [DOI] [PubMed] [Google Scholar]
- 28.Yeoh EJ, Ross ME, Shurtleff SA, et al. Classification, subtype discovery, and prediction of outcome in pediatric acute lymphoblastic leukemia by gene expression profiling. Cancer Cell. 2002;1:133–43. doi: 10.1016/s1535-6108(02)00032-6. [DOI] [PubMed] [Google Scholar]
- 29.Hilden JM, Smith FO, Frestedt JL, et al. MLL gene rearrangement, cytogenetic 11q23 abnormalities, and expression of the NG2 molecule in infant acute myeloid leukemia. Blood. 1997;89:3801–5. [PubMed] [Google Scholar]
- 30.Behm FG, Smith FO, Raimondi SC, Pui CH, Bernstein ID. Human homologue of the rat chondroitin sulfate proteoglycan, NG2, detected by monoclonal antibody 7.1, identifies childhood acute lymphoblastic leukemias with t(4;11)(q21;q23) or t(11;19)(q23;p13) and MLL gene rearrangements. Blood. 1996;87:1134–9. [PubMed] [Google Scholar]
- 31.Pui CH, Frankel LS, Carroll AJ, et al. Clinical characteristics and treatment outcome of childhood acute lymphoblastic leukemia with the t(4;11)(q21;q23): a collaborative study of 40 cases. Blood. 1991;77:440–7. [PubMed] [Google Scholar]
- 32.Van der Velden VH, Corral L, Valsecchi MG, et al. Prognostic significance of minimal residual disease in infants with acute lymphoblastic leukemia treated within the Interfant-99 protocol. Leukemia. 2009;23:1073–9. doi: 10.1038/leu.2009.17. [DOI] [PubMed] [Google Scholar]
- 33.Szczepanski T. Why and how to quantify minimal residual disease in acute lymphoblastic leukemia? Leukemia. 2007;21:622–6. doi: 10.1038/sj.leu.2404603. [DOI] [PubMed] [Google Scholar]
- 34.Salatino M, Croci DO, Bianco GA, Ilarregui JM, Toscano MA, Rabinovich GA. Galectin-1 as a potential therapeutic target in auto-immune disorders and cancer. Expert Opin Biol Ther. 2008;8:45–57. doi: 10.1517/14712598.8.1.45. [DOI] [PubMed] [Google Scholar]
- 35.Yotnda P, Garcia F, Peuchmaur M, et al. Cytotoxic T cell response against the chimeric ETV6-1 protein in childhood acute lympho blastic leukemia. J Clin Invest. 1998;102:455–62. doi: 10.1172/JCI3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fujii H, Trudeau JD, Teachey DT, et al. In vivo control of acute lymphoblastic leukemia by immunostimulatory CpG oligonucleotides. Blood. 2007;109:2008–13. doi: 10.1182/blood-2006-02-002055. [DOI] [PubMed] [Google Scholar]
- 37.Nijmeijer BA, Willemze R, Falkenburg JH. An animal model for human cellular immunotherapy: specific eradication of human acute lymphoblastic leukemia by cytotoxic T lymphocytes in NOD/scid mice. Blood. 2002;100:654–60. doi: 10.1182/blood.v100.2.654. [DOI] [PubMed] [Google Scholar]
- 38.Costa LF, Balcells M, Edelman ER, Nadler LM, Cardoso AA. Proangiogenic stimulation of bone marrow endothelium engages mTOR and is inhibited by simultaneous blockade of mTOR and NF-κB. Blood. 2006;107:285–92. doi: 10.1182/blood-2005-06-2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Veiga JP, Costa LF, Sallan SE, Nadler LM, Cardoso AA. Leukemia-stimulated bone marrow endothelium promotes leukemia cell survival. Exp Hematol. 2006;34:610–21. doi: 10.1016/j.exphem.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 40.Fragoso R, Pereira T, Wu Y, Zhu Z, Cabecadas J, Dias S. VEGFR-1 (FLT-1) activation modulates acute lymphoblastic leukemia localization and survival within the bone marrow, determining the onset of extramedullary disease. Blood. 2006;107:1608–16. doi: 10.1182/blood-2005-06-2530. [DOI] [PubMed] [Google Scholar]
- 41.van den Brule F, Califice S, Garnier F, Fernandez PL, Berchuck A, Castronovo V. Galectin-1 accumulation in the ovary carcinoma peritumoral stroma is induced by ovary carcinoma cells and affects both cancer cell proliferation and adhesion to laminin-1 and fibronectin. Lab Invest. 2003;83:377–86. doi: 10.1097/01.lab.0000059949.01480.40. [DOI] [PubMed] [Google Scholar]
- 42.Thijssen VL, Postel R, Brandwijk RJ, et al. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci U S A. 2006;103:15975–80. doi: 10.1073/pnas.0603883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh SH, Ying NW, Wu MH, et al. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene. 2008;27:3746–53. doi: 10.1038/sj.onc.1211029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.