

Abstract
Structure-activity correlations were investigated for substituted peptide conjugates that function as dual receptor site antagonists of HIV-1 gp120. A series of peptide conjugates were constructed via click reaction of both aryl and alkyl acetylenes with an internally-incorporated azidoproline 6 derived from the parent peptide 1 (12p1, RINNIPWSEAMM). Compared to 1, many of these conjugates were found to exhibit several orders of magnitude increase in both affinity for HIV-1 gp120 and inhibition potencies at both the CD4 and co-receptor binding sites of gp120. We sought to determine structural factors in the added triazole grouping responsible for the increased binding affinity and antiviral activity of the dual inhibitor conjugates. We measured peptide conjugate potencies in both kinetic and cell infection assays. High affinity was sterically specific, being exhibited by the cis but not the trans triazole. The results demonstrate that aromatic, hydrophobic and steric features in the residue 6 side-chain are important for increased affinity and inhibition. Optimizing these features provides a basis for developing gp120 dual inhibitors into peptidomimetic and increasingly smaller molecular weight entry antagonist leads.
Introduction
Human immunodeficiency virus type 1 (HIV-1) has infected over 60 million and killed over 20 million individuals worldwide since the beginning of the AIDS pandemic.1 The primary targets for HIV-1 infection in vivo are CD4+ T cells and cells of the monocyte/macrophage lineage.2, 3 Major advances have been made over the past decade in understanding the molecular machinery of HIV-1 entry into host cells. The functional HIV-1 envelope complex, which binds specifically to the host cell surface, is a trimer of three gp120 surface glycoproteins, each noncovalently attached to one of three subunits of the gp41 transmembrane glycoproteins.4–6 Crystal structures of gp120-CD4 with co-receptor surrogate antibody complexes have provided insights into the formation of protein-protein interactions in viral entry.7–10 An initial step in the entry process is the binding of the external viral spike trimer with the T-cell CD4 receptor molecule. This binding event promotes conformational structuring in the envelope gp120 that stabilizes a binding site for a co-receptor, most commonly CCR5 or CXCR4.11–13 The interaction of virus envelope gp120-CD4 complex with co-receptor is believed to promote further conformational rearrangements in HIV-1 envelope that drive exposure of gp41 and ultimately fusion of the viral and host cell membranes. In the absence of a vaccine, one of the most effective approaches to prevent and inhibit viral infections could be to block binding of virus envelope gp120 protein to either or both CD4 and CCR5 cell surface receptors. The efficacies of the fusion inhibitor T-20 and CCR5 inhibitors14, 15 have provided encouragement for the pursuit of entry inhibitors as clinically realistic strategies for AIDS treatment.
Currently, the development of effective HIV-1 gp120 inhibitors has focused mainly on natural ligands,16, 17 monoclonal antibodies,18–20 small synthetic compounds obtained either by high-throughput screening of large compound libraries21–23 or compounds derived by structure-guided rational design to interfere with the gp120 interaction with CD4 and co-receptor.24, 25 Recently, we reported a peptide conjugate, generated by click chemistry, that has the ability to inhibit interactions of gp120 with both CD4 and the co-receptor surrogate mAb 17b.26 Using initial screening of click conjugated peptides constructed from both aryl and alkyl acetylenes on an internally-incorporated azidoproline of the parent peptide 1 (12p1, RINNIPWSEAMM, entry 1 in Table 1), we synthesized a conjugated peptide through reaction with phenylacetylene that exhibited high affinity to gp120.26 The modified peptide, 2 (denoted HNG-105, entry 2 in Table 1) showed in vitro inhibition of the interactions of viral gp120s from clades A, B, C, D and CRF AE with soluble CD4 (sCD4). 27 Similarly, 2 also inhibited infection by pseudoviruses from HIV-1 subtypes A, B and C.27 In sum, formation of a phenyl triazole at residue 6 led to a two-order magnitude increase in gp120 affinity and close-to-nanomolar inhibitory potency.
Table 1.
Binding constants of click chemistry-modified conjugates of 1 determined using SPR, by direct interaction with surface-immobilized YU2 gp120.
| Compound Code | Peptide Designationa | R1 | Mass (Da) | ka(1/Ms) | kd(1/s) | KDa (nM) |
|---|---|---|---|---|---|---|
| 1 | 12p1 | Parent Pro6 | 1362.8 | 1.4 × 104 | 0.07 | 5 × 103 |
| 2 | 105 | 1603.8 | 3.4(±0.2) × 105 | 7.8 (±3.2) × 10−3 | 23 | |
| 3 | 107 | 1607.6 | 6.5(±2.1) × 104 | 5.9(±0.1) × 10−3 | 91 | |
| 4 | 113 | 1617.6 | 2.4(±0.8) × 105 | 2.8(±0.2) × 10−3 | 12 | |
| 5 | 115 | 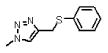 |
1536.5 | 7.1(± 1.7) 104 | 8.2(±0.2) × 10−3 | 115 |
| 6 | 116 | 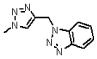 |
1658.6 | 5.4(±0.1) × 104 | 2.1 ×10−2 | 388 |
| 7 | 118 | 1624.6 | NB | NB | NB | |
| 8 | 124 | 1631.7 | 4.3(±0.5) × 105 | 3.8(±0.4) × 10−3 | 9 | |
| 9 | 125 | 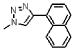 |
1653.7 | 1.6(±0.7) × 105 | 8.6(±0.6) × 10−3 | 54 |
| 10 | 128 | 1618.7 | 2.1(±1.6) ×104 | 9.18(±0.4) × 10−3 | 437 | |
| 11 | 131 | 1653.4 | 1.5(±0.2) × 105 | 5.7 × 10−3 | 38 | |
| 12 | 132 | 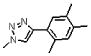 |
1645.7 | 7.5(±1.0) × 104 | 1.2(±0.1) × 10−2 | 160 |
| 13 | 134 | 1579.8 | NS | NS | NS | |
| 14 | 135 | 1633.7 | 5.7(±1.3) × 104 | 8.8(±0.2) × 10−3 | 154 | |
| 15 | 137 | 1631.7 | 2.3(±0.8) × 105 | 2.9(±0.2) × 10−3 | 13 | |
| 16 | 145 | 1630.4 | 8.6(±1.3) × 104 | 1.1(±0.2) × 10−2 | 128 | |
| 17 | 146 | 1620.0 | 8.9(±0.3) × 104 | 7.4(±0.4) × 10−2 | 83 | |
| 18 | 147 | 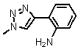 |
1619.2 | ND | ND | 25 μM(SS) |
| 19 | 149 | 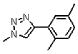 |
1631.7 | 2.7(±0.2) × 104 | 2.0(±0.2) × 10−2 | 741 |
| 20 | 150 | 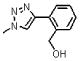 |
1764.8 | 1.5(±0.3) × 103 | 2.9(±0.4) × 10−3 | 1933 |
| 21 | 151 | 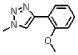 |
1632.6 | ND | ND | 14 μM(SS)) |
| 22 | 152 | 1621.7 | 1.0 × 105 | 9.1(±0.1) × 10−3 | 91 |
All the experiments were performed in duplicate and the average values are given in the table. Numbers in parentheses represent one standard deviation. The equilibrium constant KD values were derived from the average ka and kd.
Designations are HNG-XXX except parent 12p1, RINNIPWSEAMM, which contains the non-triazole conjugated proline at position 6.
NB, no detectable binding up to 1 mM analyte; NS, non-specific; ND, not determined; SS, determined by steady state analysis.
The impressive affinity increase achieved with 2 (22.9 nM KD, vs. 5.27 μM KD for 12p1), and its potential as an entry inhibitor functioning through a unique dual antagonism mechanism, led us in the present study to determine what structural elements in the added phenyl triazole group of 2 were most responsible for the affinity increase. We derived a family of 4-aryl 1, 2, 3-1H –triazole antagonists from 12p1 constructed around the phenyl ring of 2 formed by copper catalyzed 1, 3-dipolar cycloadditions at the internally incorporated azidoproline 6 position. We measured direct binding of conjugates to gp120, inhibition of CD4 binding and inhibition of the interactions of envelope gp120 with neutralizing and non-neutralizing antibodies (CD4bs and CD4i) and with co-receptor surrogate antibody mAb17b. These results demonstrate the importance of aromatic, hydrophobic and steric features in the residue 6 side chain for the increased affinity of the triazole-conjugated 12p1 peptides.
Results
Click Conjugates of cis-4-Azidoproline-12p1 and their gp120 Binding Activities
In our study of the novel peptide entry inhibitor 1 (Figure 1), we identified the opportunity to produce substantially higher affinity variants of the parent peptide by chemical modification in the Pro 6 position.23, 28 Initially, we found that the Trp 7 side chain was greatly suppressed by substitution as judged from Ala scanning experiments,28 aromatic side-chain functionalization of cysteine conjugates29 at position 7 of 12p1 and non-natural amino acid replacements. We speculated that Trp 7 was part of a possible hot-spot for stabilizing interactions with gp120. We further reasoned that introducing a more hydrophobic environment around Trp 7 without disturbing the core sequence and nature of the peptide might increase the binding affinity of the peptide to gp120. Since it is synthetically challenging to introduce more hydrophobic alkyl or aryl substitutions on the indole ring of Trp7, we chose to modify the neighboring Pro 6 to introduce hydrophobic structural elements. To achieve this, we adopted copper catalyzed 1, 3-dipolar cycloaddition to generate a small library of conjugates of 1 by using commercially available alkynes (Figure 1).
Figure 1.

Scheme for synthesis of triazole-based covalently modified various HNG peptide derivatives of 1 on solid phase using click chemistry strategy.
We replaced Pro 6 of 1 with cis-4-azidoproline and constructed a series of triazole conjugates using a previously-reported on-resin methodology.26 We used a Biacore® 3000 surface plasmon resonance (SPR) optical biosensor to screen the direct interactions of compound 1 conjugate library peptides to YU2 gp120. In the direct binding experiments, YU2 gp120 was covalently immobilized on a CM5 biosensor chip using standard amine coupling chemistry (5000 RU). Interactions were monitored in real time after injecting various concentrations of peptide analytes in PBS buffer. The kinetic binding constants were calculated using Biaevaluation 4.1 software. The kinetic binding parameters and equilibrium constants for the peptides derived from the click conjugation and determined by SPR are given in Table 1. The conjugates 4, 8 and 15 showed affinities for gp120 equivalent to or greater than the previously-reported values for 2, with KD values of 12, 8.8 and 12.9 nM, respectively. Sensorgrams for direct binding of 4, 8 and 15 are shown in Figure 2.
Figure 2.

Sensorgrams for the direct binding of triazole conjugates of 1 to immobilized YU-2 gp120. (A) 4; (B) 8 and (C) 15, each at concentrations of 10, 50, 100, 250 and 500 nM. Black lines are experimental data; red lines are fits to a 1:1 Langmuir binding model with a parameter included for mass transport.
The values for the binding affinities to gp120 were validated by isothermal titration calorimetric analysis for the parental peptide 1 and peptide conjugate 2. Figure 3 shows ITC results where gp120 was titrated with either 1 or 2. The ITC experiments yielded KD’s of 2.6 μM and 33 nM, respectively, in excellent agreement with the values determined by SPR. According to the ITC results, the binding of both compounds is characterized by favorable enthalpies (−24.9 and −16.5 kcal/mol) and unfavorable entropies (−58 and −21 cal/K×mol). ITC experiments at different temperatures yielded heat capacity change of −750 cal/K×mol for both compounds. A negative heat capacity is indicative of the burial of non-polar groups from the solvent, a process associated with a favorable entropy change. The fact that the measured overall entropy is unfavorable indicates that the favorable desolvation entropy is not large enough to overcome the unfavorable conformational entropy associated with a structuring of gp120. gp120 is a largely unstructured protein and the binding of some ligands can be associated with structuring processes.30, 31 Based upon the magnitude of the thermodynamic parameters, it can be concluded that the binding of this class of compounds induces a local structuring involving anywhere between 50 and 60 residues.
Figure 3.

Calorimetric titrations of gp120 with the parent peptide 1 (left) and conjugate 2 (right) at 25 °C in PBS, pH 7.4. The concentration of gp120 was 3 μM whereas the concentrations of 1 and 2 in the titration syringe were 60 and 30 μM, respectively. The affinity and enthalpy change for the binding of 1 to gp120 were 2.6 μM and −24.9 kcal/mol, respectively, whereas 2 bound to gp120 with an affinity of 33 nM and an enthalpy change of −16.5 kcal/mol.
Comparing SPR-derived binding parameters of peptides in Table 1, we observed that conjugate peptides 2, 4, 8 and 15 were approximately 2–3 orders of magnitude more potent than 1. By contrast, conjugates derived from azidohomoalanine at residue position 6 were either weak binders to gp120 or showed no detectable binding to gp120 (data not shown). Similarly, we replaced Trp 7 of 1 with azidohomoalanine and constructed 4-aryl substituted 1, 2, 3-1H-triazole derivatives. Triazole conjugates at position 7 of 1 showed either low affinity to gp120 or no detectable binding. The gp120 binding sensorgrams of the low affinity naphthyl derivative formed by click conjugation of azidohomoalanine at position 7 are shown in Supporting Information. No inhibition of gp120 binding to CD4 was observed with azidohomoalanine-derived conjugates (data not shown).
Peptide Conjugate Inhibition of gp120 Binding to sCD4 and to CD4 Binding Site (CD4bs) and CD4-induced (CD4i) Antibodies
Previously, we reported that 2 inhibited the interactions of YU2 gp120 to CD4 and mAb 17b. Here, we evaluated whether the higher-affinity peptides 4, 8 and 15 (based on direct-binding data as shown in Table 1) had similar dual antagonist activities, and also evaluated the inhibition activity for a set of CD4bs and CD4i antibodies. Antibodies that bind to the CD4-binding site (CD4bs) recognize HIV-1 gp120 epitopes that overlap the binding site for CD4 but are believed to interact with conformations of gp120 that are distinct from that recognized by CD4.5, 32, 33 CD4bs antibodies include both potent (e.g., IgG b12, herein designated b12) and less potent neutralizing agents (e.g., F105). The CD4i antibodies such as 17b recognize gp120 epitopes that overlap the chemokine receptor-binding site and that are stabilized upon exposure to CD4.34–36 We used SPR-based inhibition analysis to confirm whether or not the high-affinity conjugate peptides as a group inhibited the interactions of envelope gp120 with CD4 or with CD4bs and CD4i antibodies. Soluble CD4 and both types of antibodies were covalently immobilized on a CM5 biosensor chip (2000 RU), a concentration series of gp120 then was passed over the surfaces and SPR responses were recorded. Kinetic responses were evaluated using Biaevaluation software. The kinetic parameters of YU2 gp120 binding to CD4, CD4bs antibodies and CD4i antibodies are shown in Table 2. SPR sensorgrams for the direct binding of YU2 gp120 to CD4bs antibodies, and its inhibition by 4, are shown in Supporting Information. Based on the affinities determined for gp120-antibody interactions, fixed concentrations of gp120 were identified for competition experiments. These were mixed with increasing concentrations of conjugate and the resulting mixtures injected over immobilized antibody/receptor on the SPR chip surface. The inhibition profiles of YU2 gp120 binding to CD4 and the CD4i antibody 17b by 4 (representative case) are shown in Figure 4. All high-affinity conjugate peptides inhibited the interactions of YU2 gp120 with CD4 as well as with CD4bs and CD4i antibodies. Biacore-derived IC50 values for the inhibition of the gp120 binding to CD4, as well as to CD4bs and CD4i antibodies, by high affinity conjugate peptides are given in Table 2. Conjugate peptides did not inhibit the binding of gp120 to another broadly neutralizing antibody, IgG 2G12, which binds to carbohydrate sites and is not sensitive to conformational changes in gp120 induced by CD4 (data not shown).
Table 2.
IC50 values (in nM units) for the inhibition of both virus infection and the binding of gp120 to CD4, CD4bs antibodies and CD4i antibodiesa
| Compound | b6 (CD4bs) | b12 (CD4bs) | F105 (CD4bs) | 17b (CD4i) | CD4 | HIV-1 Infection |
|---|---|---|---|---|---|---|
| 1 | 10±1×103 | 8.8±0.6 ×103 | 6.4±0.2 ×103 | 7.2 ±0.1×103 | 5.4±0.4 ×103 | 48±1.2 × 103 |
| 2 | 106±10 | 162±15 | 109±9 | 172±42 | 154±37 | 1.4±1.3 × 103 |
| 4 | 136± 11 | 117± 13 | 76 ± 11 | 94± 2 | 67±6 | 156±1 |
| 8 | 235±19 | 191±18 | 137±22 | 177± 42 | 146± 6 | 418±2 |
| 15 | 85±18 | 65±14 | 23± 1 | 129± 3 | 99± 2 | 610±1 |
YU2 gp120 concentrations used in competition experiments were selected based upon KD values from isotherms for direct binding of gp120 to immobilized antibodies. These KD values for b6, b12, F105, 17b and CD4 were, respectively, 0.56±0.1, 27±4, 11±2.2, and 0.6±0.15 and 7.0±0.5 nM. IC50 values (obtained from fitting data to sigmoidal dose response curve) for the viral inhibition assay were derived from data illustrated in Figure 6 using GraphPad Prism software.
Figure 4.

Binding of YU2 gp120 to CD4 and CD4i antibody 17b in the presence of increasing concentrations of 4. (Left) Direct binding of YU2 gp120 to immobilized CD4 and antibodies. Approximately 2000 RUs of either sCD4 or mAb17b were immobilized on separate flow cells of biosensor chip and exposed to increasing concentrations (1 – 200 nM) of YU-2 gp120. Black lines indicate experimental data, whereas red lines indicate fitting to a 1:1 Langmuir binding model. (Right) Inhibition of the binding of YU2 gp120 (100 nM) to CD4 and 17b with increasing concentration of peptide 4. Data are presented as percent of the binding observed in the absence of 4.
Effect of Stereochemistry of 4-Substituted Triazole at Pro 6
The specific pattern of efficacy observed in the structure-activity relationship (SAR) analyses of click conjugates, combined with prior determination of 1:1 stoichiometry of interaction, led us to deduce that the efficacy of particular conjugates is dependent on interaction with a specific binding site in gp120. We examined the stereochemical requirements in the peptide for this interaction by variations in the side chain triazole. The trans-4-azidoproline ((2S, 4R)-4-azidopyrrolidine-2-carboperoxoic acid) was synthesized starting from commercially available cis-4-hydroxy-proline ((2S, 4S)-4-hydroxypyrrolidine-2-carboperoxoic acid). Strikingly, the click conjugation of phenyl acetylene to internally incorporated trans-4-azidoproline (peptide 23 in Figure 5c) leads to a peptide with a weaker affinity (KD 2.7 μM), in contrast to the two order of magnitude greater affinity of the other cis-isomer ((2S, 4S)-4-azidopyrrolidine-2-carboperoxoic acid), 2 (KD 22.9 nM) (Figure 5b). Peptide 23 also showed poor inhibition of binding of gp120 to sCD4 and 17b. Indeed, the trans triazole peptide 23 binds with similar affinity as the parent compound 1.26 These results provide a strong argument that affinity-enhanced gp120 binding and inhibition of triazole conjugates is driven by a clearcut stereospecificity.
Figure 5.
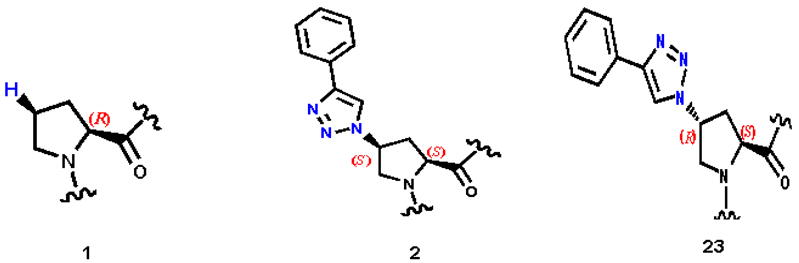
Side chain structures at position 6 in conjugates that the highlight importance of stereochemistry. Shown are (Left) Proline 6 in 1; (Center) 2, with cis-4–triazole ((2S,4S)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboperoxoic acid); (Right) 23(Mass=1630.6 Da), with trans-4-triazole ((2S,4R)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboperoxoic acid). Compound 1, parent peptide, binds to gp120 with a Kd of 5 μM. Compound 2 with cis, and 23 with trans–triazole bind to gp120 with Kd values of 22.9 nM and 2.7 μM respectively.
Inhibition of HIV-1 Infection by Conjugate Peptides
Recently we reported the effective inhibition of isolates from HIV-1 subtypes A, B, and C by 2. 27 Here, we used cell assays with fully infectious subtype B strain HIV-1BaL virus (R5 phenotype) to compare the ability of high-affinity conjugate peptides (2, 4, 8 and 15) to exert an inhibitory effect on the HIV-1 envelope in the context of an intact viral spike. Viral inhibition data are shown in Figure 6, and the resultant IC50 values obtained from these data are given in Table 2. In the fully infectious virus assay, 4 (IC50 = 156±1.2 nM) was more effective than the other conjugate peptides. 8 and 15 showed similar IC50 values, slightly better than 2. The conjugate peptides used in the cell infection assays did not exhibit appreciable cell toxicity (Figure 6 legend and Supporting Information). These results demonstrated the antiviral activity of conjugate peptides and the substantially increased efficacy of a subset of these peptides vs. the parental 1.
Figure 6.
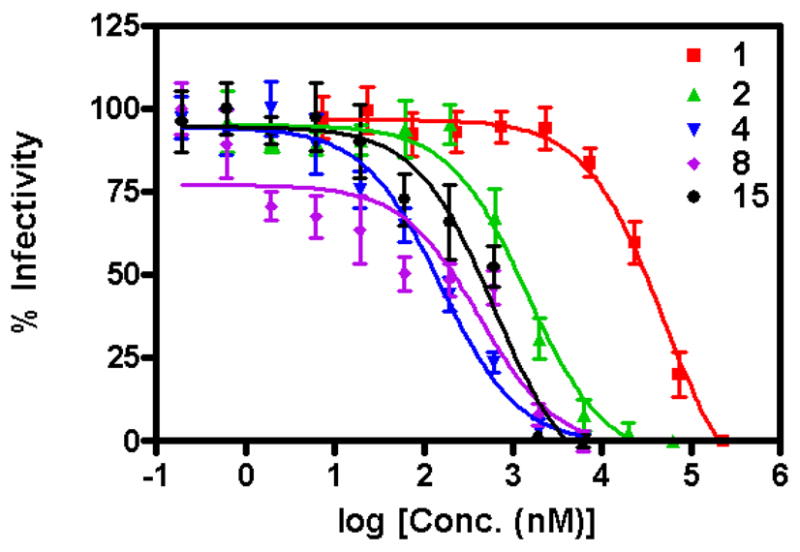
Inhibition of HIV-1 BaL infection by high affinity conjugate peptides. Cell-free HIV-1 BaL and P4-CCR5 indicator cells were incubated for 2 h with with 1, 2,4, 8 or 15. Infectivity remaining was calculated with respect to the level of infection determined in cells infected in the absence of compound. Graph depicts the combined results of two independent experiments. In assays of in vitro cytotoxicity using P4-CCR5 cells (2 h exposure), peptides 1, 2, 4, 8 and 15 had no or minimal impact on cell viability at concentrations corresponding to IC50 values (data in Supporting Information).
Importantly, the hierarchy of potencies of inhibition of cell infection was similar to that measured for inhibition of gp120 binding to CD4 and antibodies. In contrast, the absolute IC50 values for inhibition of viral infection were about an order of magnitude greater than those for gp120 inhibition in SPR assays. These differences are discussed further below.
SAR Evaluation of 12p1 Conjugates
As shown by the data of Figure 5, a conjugate peptide 23 synthesized from the trans-4-azidoproline ((2S,4R)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboperoxoic acid) exhibited a large reduction in binding affinity compared to the cis diastereoisomer. This suggests a strong stereo-specific requirement in the gp120 binding site for these peptides. In the absence of direct structural information of the gp120-peptide interface, the epitope in gp120 that recognizes peptide groups stereospecifically remains unknown. Nonetheless, as a group, the cis-4-azidoproline ((2S,4S)-4-(4-phenyl-1H-1,2,3-triazol-1-yl)pyrrolidine-2-carboperoxoic acid) derivatized compounds demonstrated a broad range of binding affinity. We evaluated SAR relationships of the azidoproline derivatized portions of these peptides based on various molecular descriptors for the Pro 6 triazole conjugated portion of the compounds (Table 1). The most correlated three-dimensional descriptor was the hydrophobic solvent accessible surface area (SASA). Descriptors of positive charge or polarity exhibited poor correlations with gp120 binding affinities.
The relationship between potency and molecular properties for the peptide conjugates is illustrated by the plot of hydrophobic SASA vs. binding affinity in Figure 7. The data points on the plot were color-coded by the number of aromatic atoms in the conjugates. There was a clear trend of increased potency as the number of aromatic atoms increased and as the hydrophobicity increased. We analyzed the effect of ortho, meta or para substitution on the phenyl-triazole of 2. Para substitutions produced the most potent compounds, followed by meta substitutions. Ortho substitutions (depicted as diamonds in Figure 7) produced the most negative affects on potency. For instance, substitution of the phenyl triazole with a hydroxymethyl at the ortho position (20, Table 1, Figure 7) produced a weaker binding peptide conjugate. The methoxy ortho substitution (21, Table 1) further decreased binding.
Figure 7.

Correlation of hydrophobic solvent accessible surface area (SASA) plotted vs. the pKD for peptide conjugates (R=0.81 and R2 =0.66). Data points are colored to denote the number of aromatic atoms conjugated to proline 6: 0, red; 5, yellow; 11, cyan.
Discussion
Antagonism of Multiple Molecular and Cellular Interactions of HIV-1 Envelope Protein by High-Affinity Peptide Conjugates
In this work, we found that formation of 4-aryl-substituted triazoles at the gamma position of Pro 6 in the phage-library derived compound 1 generated high-affinity inhibitors of the HIV-1 envelope protein gp120. Screening by direct binding to immobilized gp120 using SPR (Table 1) identified the affinity range for the conjugates. Binding of this class of peptides was found to be 1:1 as judged by the magnitude of SPR response. Peptide binding disrupts multiple gp120 interactions as observed in both SPR and cell infection assays. SPR showed disruption of interaction to both CD4 and co-receptor binding site ligands. Moreover, even though the sCD4 and antibody ligands range in affinity over 3–4 orders of magnitudes, the inhibition potencies for the different ligands were equivalent for a particular triazole conjugate, indicating that a single binding site is likely responsible for all inhibitory effects. Of the conjugates examined, 2, 4, 8 and 15, showed IC50 values in a relatively narrow range from 23 to 235 nM respectively in SPR analyses with CD4 and CD4bs and CD4i antibodies. The conjugates also inhibited cell infection by the infectious HIV-1BaL virus, though the IC50 values varied more widely in this set and were generally higher than those measured in SPR competition (see below).
Importance of Aromatic Substitutions on Triazole
Survey of the direct binding (Table 1) and competition potencies (Table 2, 1Figures 4, 6) of peptide conjugates with the similar sequence background (Figure 2), in addition to the hydrophobic solvent accessible surface area at position 6 (Figure 7), argue that the primary structural change leading to increased potency (vs. the initial 1) is the addition of a substituted aromatic ring substituent on the triazole at Pro 6. Incorporation of small alkyl groups at the para-position of the phenyl group (for example 4, 8) led to some of the most potent variants examined. In contrast, introducing a charged amine (10) or polar ether (14) group at the same position decreased the affinity of the peptide. Extension of the alkyl chain from methyl (4) to ethyl (8) preserved increased binding affinity, while extending the alkyl chain from ethyl to pentyl (13) led to nonspecific binding. Two of the most potent peptides 8 and 15 bind with similar affinity to gp120 even though these have reverse par-substitution of an ethyl group. Assessment of the solvent accessible surface area of 8 and 15 indicate that their molecular volumes are similar (data not shown). From the viewpoint of molecular surface area or volume, it is plausible that the incorporation of a sulfur atom in the alkyl chain (in compound 5) alters the shape of the molecule and decreases the affinity 10-fold.
Meta substitution is less tolerated than para substitution as demonstrated by the decreased affinity of 17 (methyl at the meta-position). The binding affinity appears even more sensitive to ortho substitution on the phenyl ring. Incorporation of an ortho-methyl group (16) decreases the affinity 5-fold from 2. Addition of an ortho-hydroxymethyl group (20) decreases the affinity another 15 fold, whereas incorporation of an amine (18) or methoxy group (21) at the ortho-position abolishes the affinity of the peptide. The deleterious affects of ortho substitution are also exhibited in the relative affinities of the ortho- and meta-dimethyl substituted peptide (19) and the ortho-, meta- and para-trimethyl substituted peptide (12). In both cases, the ortho-substituted compounds bind more poorly than their meta-(17) and meta-, para-(11) analogues. Ortho-substituted 4-phenyl-triazoles as a sub-class have reduced binding affinities. Overall, the results of different substitutions on the 4-substituted phenyl group of 1, 2, 3-1H-triazole suggest that para substitution with alkyl groups is favorable up to the limit of the pentyl substitution, as shown in Figure 7. We conclude that the 4-phenyl-triazole binding site on gp120 imposes a restricted steric environment in the span of the peptide molecule between the ortho phenyl carbon and the triazole.
The napthyl triazole (9) has a solvent accessible surface area comparable to 15, but incorporation of a ring system fused to the primary ring may decrease affinity in a manner similar to the ortho-substituted phenyl derivatives. Comparison of the two bicyclic substitutions, napthyl (9) and benzotriazole (6), shows a loss of approximately 100 Å2 in SASA and the associated loss of potency for the benzotriazole. The decreased affinity for the benzotriazole vs the napthyl further emphasizes the importance of the phenyl group. To confirm the relative preference for the phenyl component on the triazole, we replaced the 4-substituted phenyl group by a cyclohexenyl group (3, Table 1). This peptide binds to gp120 with less affinity (98 nM) than 2, though still more strongly than the initial 1. The importance of an aromatic ring in this region of the peptide was also examined with Trp 7 variations. We replaced Trp 7 in the original peptide 1 by azidohomoalanine and constructed a conjugate library at the site of Trp 7. The conjugates derived from azidohomoalanine showed either poor or no binding to gp120. No inhibition of gp120 binding to CD4 or 17b was observed in the case of homoalanine-derived conjugates at position 7.
The hydrophobic SASA and presence of a phenyl group are not the only factors driving high-affinity binding. We examined the importance of stereochemistry of the triazole itself. Strikingly, 4-phenyl substituted trans triazole (23, Figure 5c) decreased the affinity of the peptide to the level of that for 1. We would have expected similar affinity between these two conjugates if the increasing affinity were due only to the hydrophobic interaction of the side chain. This difference argues for the importance of steric factors in the position 6 side chain. This and the limitation imposed by ortho-substitution (Figure 7) point to a clearcut steric specificity of the high-affinity peptides.
Inhibition Potencies of Triazole Conjugates in Molecular and Cellular Assays
As shown in Table 2, key compounds in Table 1 with the highest inhibition potencies in SPR show significant inhibition potencies in live virus cell infection assays. The overall hierarchy of inhibition potencies of high-affinity triazole conjugates of 1 was found to be similar in both the SPR and cell infection assays. Consistent virus-neutralizing effects by the conjugates reinforce the notion that the conjugates can lead to useful antagonist leads for AIDS treatment. Furthermore, the generally parallel hierarchies of affinity and infection neutralization argue that peptide conjugate binding is the main driving event in neutralization. The relatedness of binding and neutralization has been found for neutralizing antibodies, 37, 38 though the correlation for antibodies is most evident with gp120 trimers but not monomers. In contrast to the above, the absolute potencies of peptide conjugates in the cell-based assays were lower by about one order of magnitude than those in the molecular assays. Lower potencies also were observed in single round cell infection assays for 2 (753 nM IC50 for JR-CSF pseudovirus). This suggests that additional factors may limit neutralizing activity of the conjugates in the viral context, such as constrained access to the conjugate binding site in the viral trimer spike, the need for a threshold of multiple ligand binding to effect neutralization or degradation of peptide in the virus-cell environment. Nonetheless, the sub-micromolar efficacies of conjugates such as 2,4, 8 and 15, and the underlying steric specificity of these inhibitors, strongly argue that the peptide conjugates identified here provide realistic molecular tools to design further potent entry inhibitors for HIV-1 envelope gp120.
MATERIALS AND METHODS
Materials
All Fmoc-protected amino acids, HBTU, HOBt and Hyp(OMe).HCl were purchased from Novabiochem. Rink amide resin was obtained from AppliedBiosystem. Solvents and other chemicals were purchased from Aldrich or Fisher and used without further purification. Fmoc-cis-4-azidoproline was synthesized starting with commercially available trans-Hyp-(OMe).HCl. Fmoc-trans-4-azidoproline was synthesized starting from methyl ester of cis-4-hydroxyproline hydrochloride. Fmoc-azido homoalanine was synthesized according to the reported procedures.39
Peptide Synthesis and Click Conjugation
Peptides were synthesized by manual solid phase synthesis using Fmoc chemistry on Rink amide resin at 0.1 mmol scale. The [3+2] cycloaddition reaction of azide and terminal alkynes was carried out by an on-resin method.26 All peptides were purified using reverse-phase HPLC, and their intended masses validated by mass spectrometry. The peptides were cleaved from the resin using a cocktail mixture of 95:2:2:1 trifluoroacetic acid/1–2-ethanedithiol/water/thioanisole. The crude peptides were purified by fractionation on a C18 column on HPLC (Beckmann Coulter), with a gradient between 95:5:0.1 and 5:95:0.1 water/acetonitrile/trifluoroacetic acid. All peptides were ≥ 98% homogeneous chromatographically when re-run on an analytical C18 HPLC column with the above gradient. The chemical structures of purified peptides were confirmed by MALDI-TOF. Chromatograms for key compounds are given in Supporting Information.
Protein Reagents
YU2-gp120 was produced in Drosophila S2 cells.40 Cells were spun down and supernatant sterile filtered. Supernatant was purified over an F105-antibody column (NHS-activated Sepharose, Amersham; F105 antibody coupled according to manufacturer’s instructions). YU2-gp120 was eluted from the column with glycine buffer, pH 2.4, dialysed against PBS and frozen at −80ºC. sCD4 was expressed in CHO cells in a hollow fiber bioreactor. Supernatant from the hollow fiber bioreactor was fractionated on an SP-column, and bound fractions were run over a Q-column. Unbound material was concentrated and analyzed by SDS-PAGE. The monoclonal antibodies mAb, b12, b6, F105 and 2G12 were obtained from the NIH AIDS Research and Reference Reagent Program. Antibody Fab X5 was obtained from Dimiter Dimitrov, NIH. 17b was purchased from Applied Biosystems.
Optical Biosensor Binding Assays
All surface plasmon resonance experiments (SPR) were performed on a BIA3000 optical biosensor (Biacore, Inc., Uppsala, Sweden). A CM5 sensor chip was derivatized by amine coupling using EDC.HCl/HOSu with either YU2 gp120, soluble CD4, mAb 17b Fab, or, for a control surface, an anti-IL5 receptor α antibody 2B6R.41 For direct binding experiments, YU2 gp120 was immobilized on the surface (~4000 RU); peptide analytes in PBS buffer were passed over the surface at a flow rate of 15 μL/min with 5 min association and 5 min dissociation phases. For competition experiments, ligands (sCD4, 17b mAb, b6, b12, and F105) were immobilized on a surface with a density of approximately 2000 RU. The indicated analytes were passed over the surfaces at a flow rate of 50 μL/min with 2.5 min association and 2.5 min dissociation phases. Surfaces were regenerated using 35 mM NaOH and 1.3M NaCl for sCD4 and YU2 gp120 surfaces, and 10 mM HCl for 17b surface. Buffer injections and control surface binding were subtracted in all of the experiments.
Data analysis was performed using BIAEvaluation® 4.0 software (Biacore Inc., NJ). The responses of a buffer injection and responses from the control flow cell (2B6R) were subtracted to account for nonspecific binding. Experimental data were fitted to a simple 1:1 binding model with a parameter included for mass transport. The average kinetic parameters (association [ka] and dissociation [kd] rates) generated from a minimum of 4 data sets were used to define equilibrium association (KA) and dissociation constants (KD).
The evaluation method for SPR inhibition data included a calculation of the inhibitor concentrations at 50% of the maximal response, IC50.42 The inhibition curve was converted into a calibration curve by the use of a fitting function. The fitting was done using the 4-parameter equation in BIAevaluation software,
where Rhigh is the response value at high inhibitor concentrations and Rlow is response at low inhibitor concentrations. Conc is the concentration of inhibitor and A1 and A2 are fitting parameters. At the concentration corresponding to IC50,
which means that A1 has to equal Conc and is therefore the desired parameter.
Isothermal Titration Calorimetry (ITC)
Isothermal titration calorimetric experiments were performed using a high-precision VP-ITC titration calorimetric system from MicroCal Inc. (Northampton, MA). The calorimetric cell (~1.4 mL) contained gp120 at a concentration of about 3 μM, and the concentration of inhibitor in the injection syringe was about 60 μM for 1 (12P1) and 30 μM for 2. Both gp120 and the inhibitors were dissolved in PBS, pH 7.4 (Roche Diagnostics GmbH). The inhibitor solution was added in steps of 10 μL. All solutions were degassed to avoid any formation of bubbles in the calorimeter during stirring. The experiments were performed at 25 °C.
Inhibition of HIV-1 Infection by Live Virus
P4-CCR5 cells (AIDS Reagent Program #3580) were maintained in DMEM supplemented with 0.1 mg/ml puromycin, 10% fetal bovine serum (FBS), L-glutamine (0.3 mg/ml), antibiotics (penicillin, streptomycin, and kanamycin at 0.04 mg/ml each) and 0.05% sodium bicarbonate. 43 The P4-CCR5 cells were seeded at a density of 1.2 × 104 cells/well in a 96 well plate approximately 18 h prior to experiment. The cells were then incubated for 2 h with HIV-1 strain BaL (AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH) 44, 45 in the presence of 1, 2, 4, 8 and 15. Dextran sulfate was used as a positive control. After the 2 h incubation, cells were washed, cultured for an additional 46 h, and subsequently assayed for HIV-1 infection using the Galacto-Star ®-Galactosidase Reporter Gene Assay System for Mammalian Cells as per manufacturer’s instructions (Applied Biosystems, Bedford, MA). Infectivity remaining is expressed relative to mock-treated, HIV-1-infected cells. Data were fit with GraphPad Prism software. From the fit curves, the concentration at which exposure to the compound resulted in a 50% decrease in infectivity relative to mock-treated, HIV-1-infected cells is referred to as the IC50 value.
Cytotoxicity Assays
P4-CCR5 cells were seeded at a density of 4×104 cells/well in a 96 well plate approximately 18 h prior to experiment. Cells were then exposed to varying concentrations of 1, 2, 4, 8, 15, and dextran sulfate for 2 h. The cells were subsequently washed and assessed for viability using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay of viability (as previously described46). Concentrations were tested in triplicate in two independent assays.
Inhibition of Single-round Luciferase Viral Infection
293T human embryonic kidney and Cf2Th canine thymocytes (ATCC) were grown at 37ºC and 5% CO2 in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum and 100 μg/ml penicillin-streptomycin. Cf2Th cells stably expressing human CD4 and CCR5 or CXCR4 (21) were grown in medium supplemented with 0.4 mg/ml G418 and 0.15 mg/ml hygromycin B.
293T human embryonic kidney cells were co-transfected with vectors expressing the pCMVΔP1Δenv HIV Gag-Pol packaging construct 47, the envelope glycoprotein of HIV-1 JR-CSF isolate or of amphotropic murine leukemia virus (AMLV) or vesicular stomatitis virus (VSV) as a control and a firefly luciferase reporter gene at a DNA ratio of 1:1:3 μg using Effectene transfection reagent. Co-transfection produced single-round, replication-defective viruses.The virus-containing supernatants were harvested 24–30 h after transfection, filtered (0.45 μm), aliquoted, and frozen at −80°C until further use.The reverse transcriptase (RT) activities of all viruses were measured as described previously 48.
Cf2Th-CD4-CCR5/CXCR4 target cells were seeded at a density of 6 × 103 cells/well in 96-well luminometer-compatible tissue culture plates 24 h before infection. On the day of infection, peptide 2 (0–10 μM) was added to recombinant viruses (10,000 RT units) to a final volume of 50 μl and incubated at 37°C for 30 min. The medium was then removed from the target cells, and the cells incubated with virus-peptide mixture for 48 h at 37°C. After this time, the medium was removed from each well, and the cells were lysed with 30 μl of passive lysis buffer and by three freeze-thaw cycles. An EG&G Berthold Microplate Luminometer LB 96V was used to measure luciferase activity of each well after the addition of 100 μl of luciferin buffer (15 mM MgSO4, 15 mM KPO4, pH 7.8, 1 mM ATP, and 1 mM dithiothreitol) and 50 μl of 1 mM D-luciferin potassium salt.
Computational Methods
Small Molecule Preparation
The individual peptides were reduced to the Proline 6 triazole portion of the peptide and constructed in MOE.49 The Pro-6 amino and carboxy termini were capped with a methyl group, and the small molecules were ionized using MOE’s WashMDB function before adding hydrogens. The small molecule analogues were minimized to a gradient of 0.01 in the MMFF94 force field 50, 51 using a distance-dependent dielectric constant of 2.
Calculation of Molecular Descriptors
Using the MOE QSAR module, 56 2-dimensional and 3-dimensional molecular descriptors were calculated for the 34 compounds, including 7 weaker binding compounds not reported in this manuscript. Individual correlation plots were analyzed for each descriptor and the measured binding affinity pKd for the compounds. The 3-dimensional descriptor with the best correlation coefficient (R=0.78, R2=0.61) was the hydrophobic solvent accessible surface area. We choose to analyze the relative potency of compounds in Table 1 by plotting the hydrophobic solvent accessible surface area versus pKd as shown in Figure 6 to draw conclusions concerning SAR.
Supplementary Material
Acknowledgments
We acknowledge Dr. Srivats Rajagopal for proofreading this manuscript. This research was supported by National Institutes of Health (NIH) grants P01 GM 56550 and R21 AI071965.
Abbreviations
- AIDS
acquired immune deficiency syndrome
- Azp
cis-4S-Azido-L-proline
- EDC
1-ethyl-3-[3 (dimethylamino) propyl] carbodiimide
- Fmoc-
9-fluorenylmethyloxycarbonyl
- gp120
glycoprotein 120
- HBTU
2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- HIV-1
human immunodeficiency virus type 1
- HOBt
1-Hydroxybenzotriazole
- Hyp
trans- 4R-Hydroxy-L-proline
- ITC
isothermal titration calorimetry
- sCD4
soluble CD4
- SPR
surface plasmon resonance
Footnotes
Supporting Information Available
Material presented are the following. I. Reverse phase liquid chromatographic analyses of key position 6 triazole conjugates 2, 4, 8 and 15, as well as parent compound 1. II. Direct binding of YU2gp120 to immobilized CD4 site binding antibodies (CD4bs and the inhibition of binding of YU2 gp120 to CD4bs by 4 (HNG-113). III. Profiles of inhibition of gp120 binding by residue 6 triazole conjugates 2, 8 and 15. IV. Inhibition of binding of gp120 to CD4bs by 1 (12p1). V. Cytotoxicity of triazole conjugates. VI. Low-Affinity Peptides Included in SAR Analysis. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.WHO/UNAIDS. AIDS Epidemic Update. Dec, 2005. [Google Scholar]
- 2.Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, Gluckman JC, Montagnier L. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312(5996):767–8. doi: 10.1038/312767a0. [DOI] [PubMed] [Google Scholar]
- 3.Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312(5996):763–7. doi: 10.1038/312763a0. [DOI] [PubMed] [Google Scholar]
- 4.Chan DC, Fass D, Berger JM, Kim PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell. 1997;89(2):263–73. doi: 10.1016/s0092-8674(00)80205-6. [DOI] [PubMed] [Google Scholar]
- 5.Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280(5371):1884–8. doi: 10.1126/science.280.5371.1884. [DOI] [PubMed] [Google Scholar]
- 6.Tan K, Liu J, Wang J, Shen S, Lu M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci U S A. 1997;94(23):12303–8. doi: 10.1073/pnas.94.23.12303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kwong PD, Wyatt R, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393(6686):648–59. doi: 10.1038/31405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang CC, Stricher F, Martin L, Decker JM, Majeed S, Barthe P, Hendrickson WA, Robinson J, Roumestand C, Sodroski J, Wyatt R, Shaw GM, Vita C, Kwong PD. Scorpion-toxin mimics of CD4 in complex with human immunodeficiency virus gp120 crystal structures, molecular mimicry, and neutralization breadth. Structure. 2005;13(5):755–68. doi: 10.1016/j.str.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Huang CC, Tang M, Zhang MY, Majeed S, Montabana E, Stanfield RL, Dimitrov DS, Korber B, Sodroski J, Wilson IA, Wyatt R, Kwong PD. Structure of a V3-containing HIV-1 gp120 core. Science. 2005;310(5750):1025–8. doi: 10.1126/science.1118398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kwong PD, Wyatt R, Majeed S, Robinson J, Sweet RW, Sodroski J, Hendrickson WA. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure. 2000;8(12):1329–39. doi: 10.1016/s0969-2126(00)00547-5. [DOI] [PubMed] [Google Scholar]
- 11.Wu L, Gerard NP, Wyatt R, Choe H, Parolin C, Ruffing N, Borsetti A, Cardoso AA, Desjardin E, Newman W, Gerard C, Sodroski J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature. 1996;384(6605):179–83. doi: 10.1038/384179a0. [DOI] [PubMed] [Google Scholar]
- 12.Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, Cayanan C, Maddon PJ, Koup RA, Moore JP, Paxton WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature. 1996;381(6584):667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- 13.Doranz BJ, Berson JF, Rucker J, Doms RW. Chemokine receptors as fusion cofactors for human immunodeficiency virus type 1 (HIV-1) Immunol Res. 1997;16(1):15–28. doi: 10.1007/BF02786321. [DOI] [PubMed] [Google Scholar]
- 14.Moore JP, Doms RW. The entry of entry inhibitors: a fusion of science and medicine. Proc Natl Acad Sci U S A. 2003;100(19):10598–602. doi: 10.1073/pnas.1932511100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsibris AM, Kuritzkes DR. Chemokine antagonists as therapeutics: focus on HIV-1. Annu Rev Med. 2007;58:445–59. doi: 10.1146/annurev.med.58.080105.102908. [DOI] [PubMed] [Google Scholar]
- 16.Munk C, Wei G, Yang OO, Waring AJ, Wang W, Hong T, Lehrer RI, Landau NR, Cole AM. The theta-defensin, retrocyclin, inhibits HIV-1 entry. AIDS Res Hum Retroviruses. 2003;19(10):875–81. doi: 10.1089/088922203322493049. [DOI] [PubMed] [Google Scholar]
- 17.Gallo SA, Wang W, Rawat SS, Jung G, Waring AJ, Cole AM, Lu H, Yan X, Daly NL, Craik DJ, Jiang S, Lehrer RI, Blumenthal R. Theta-defensins prevent HIV-1 Env-mediated fusion by binding gp41 and blocking 6-helix bundle formation. J Biol Chem. 2006;281(27):18787–92. doi: 10.1074/jbc.M602422200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang MY, Dimitrov DS. Novel approaches for identification of broadly cross-reactive HIV-1 neutralizing human monoclonal antibodies and improvement of their potency. Curr Pharm Des. 2007;13(2):203–12. doi: 10.2174/138161207779313669. [DOI] [PubMed] [Google Scholar]
- 19.Cardoso RM, Zwick MB, Stanfield RL, Kunert R, Binley JM, Katinger H, Burton DR, Wilson IA. Broadly neutralizing anti-HIV antibody 4E10 recognizes a helical conformation of a highly conserved fusion-associated motif in gp41. Immunity. 2005;22(2):163–73. doi: 10.1016/j.immuni.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhang MY, Shu Y, Phogat S, Xiao X, Cham F, Bouma P, Choudhary A, Feng YR, Sanz I, Rybak S, Broder CC, Quinnan GV, Evans T, Dimitrov DS. Broadly cross-reactive HIV neutralizing human monoclonal antibody Fab selected by sequential antigen panning of a phage display library. J Immunol Methods. 2003;283(1–2):17–25. doi: 10.1016/j.jim.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 21.Lin PF, Blair W, Wang T, Spicer T, Guo Q, Zhou N, Gong YF, Wang HG, Rose R, Yamanaka G, Robinson B, Li CB, Fridell R, Deminie C, Demers G, Yang Z, Zadjura L, Meanwell N, Colonno R. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc Natl Acad Sci U S A. 2003;100(19):11013–8. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhao Q, Ma L, Jiang S, Lu H, Liu S, He Y, Strick N, Neamati N, Debnath AK. Identification of N-phenyl-N′-(2,2,6,6-tetramethyl-piperidin-4-yl)-oxalamides as a new class of HIV-1 entry inhibitors that prevent gp120 binding to CD4. Virology. 2005;339(2):213–25. doi: 10.1016/j.virol.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 23.Ferrer M, Harrison SC. Peptide ligands to human immunodeficiency virus type 1 gp120 identified from phage display libraries. J Virol. 1999;73(7):5795–802. doi: 10.1128/jvi.73.7.5795-5802.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vita C, Drakopoulou E, Vizzavona J, Rochette S, Martin L, Menez A, Roumestand C, Yang YS, Ylisastigui L, Benjouad A, Gluckman JC. Rational engineering of a miniprotein that reproduces the core of the CD4 site interacting with HIV-1 envelope glycoprotein. Proc Natl Acad Sci U S A. 1999;96(23):13091–6. doi: 10.1073/pnas.96.23.13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DeMarco SJ, Henze H, Lederer A, Moehle K, Mukherjee R, Romagnoli B, Robinson JA, Brianza F, Gombert FO, Lociuro S, Ludin C, Vrijbloed JW, Zumbrunn J, Obrecht JP, Obrecht D, Brondani V, Hamy F, Klimkait T. Discovery of novel, highly potent and selective beta-hairpin mimetic CXCR4 inhibitors with excellent anti-HIV activity and pharmacokinetic profiles. Bioorg Med Chem. 2006;14(24):8396–404. doi: 10.1016/j.bmc.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 26.Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM. Click chemistry on azidoproline: high-affinity dual antagonist for HIV-1 envelope glycoprotein gp120. ChemMedChem. 2006;1(1):54–7. doi: 10.1002/cmdc.200500037. [DOI] [PubMed] [Google Scholar]
- 27.Cocklin S, Gopi H, Querido B, Nimmagadda M, Kuriakose S, Cicala C, Ajith S, Baxter S, Arthos J, Martin-Garcia J, Chaiken IM. Broad-spectrum anti-HIV Potential of a Peptide HIV-1 Entry Inhibitor. J Virol. 2007 doi: 10.1128/JVI.01778-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Biorn AC, Cocklin S, Madani N, Si Z, Ivanovic T, Samanen J, Van Ryk DI, Pantophlet R, Burton DR, Freire E, Sodroski J, Chaiken IM. Mode of action for linear peptide inhibitors of HIV-1 gp120 interactions. Biochemistry. 2004;43(7):1928–38. doi: 10.1021/bi035088i. [DOI] [PubMed] [Google Scholar]
- 29.Smith AB, 3rd, Savinov SN, Manjappara UV, Chaiken IM. Peptide-small molecule hybrids via orthogonal deprotection-chemoselective conjugation to cysteine-anchored scaffolds. A model study. Org Lett. 2002;4(23):4041–4. doi: 10.1021/ol026736d. [DOI] [PubMed] [Google Scholar]
- 30.Leavitt SA, SchOn A, Klein JC, Manjappara U, Chaiken IM, Freire E. Interactions of HIV-1 proteins gp120 and Nef with cellular partners define a novel allosteric paradigm. Curr Protein Pept Sci. 2004;5(1):1–8. doi: 10.2174/1389203043486955. [DOI] [PubMed] [Google Scholar]
- 31.Schon A, Madani N, Klein JC, Hubicki A, Ng D, Yang X, Smith AB, 3rd, Sodroski J, Freire E. Thermodynamics of Binding of a Low-Molecular-Weight CD4 Mimetic to HIV-1 gp120. Biochemistry. 2006;45(36):10973–80. doi: 10.1021/bi061193r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyatt R, Kwong PD, Desjardins E, Sweet RW, Robinson J, Hendrickson WA, Sodroski JG. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393(6686):705–11. doi: 10.1038/31514. [DOI] [PubMed] [Google Scholar]
- 33.Xiang SH, Kwong PD, Gupta R, Rizzuto CD, Casper DJ, Wyatt R, Wang L, Hendrickson WA, Doyle ML, Sodroski J. Mutagenic stabilization and/or disruption of a CD4-bound state reveals distinct conformations of the human immunodeficiency virus type 1 gp120 envelope glycoprotein. J Virol. 2002;76(19):9888–99. doi: 10.1128/JVI.76.19.9888-9899.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rizzuto CD, Wyatt R, Hernandez-Ramos N, Sun Y, Kwong PD, Hendrickson WA, Sodroski J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science. 1998;280(5371):1949–53. doi: 10.1126/science.280.5371.1949. [DOI] [PubMed] [Google Scholar]
- 35.Thali M, Moore JP, Furman C, Charles M, Ho DD, Robinson J, Sodroski J. Characterization of conserved human immunodeficiency virus type 1 gp120 neutralization epitopes exposed upon gp120-CD4 binding. J Virol. 1993;67(7):3978–88. doi: 10.1128/jvi.67.7.3978-3988.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xiang SH, Wang L, Abreu M, Huang CC, Kwong PD, Rosenberg Robinson JE, Sodroski J. Epitope mapping and characterization of a novel CD4-induced human monoclonal antibody capable of neutralizing primary HIV-1 strains. Virology. 2003;315(1):124–34. doi: 10.1016/s0042-6822(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 37.Parren PW, Mondor I, Naniche D, Ditzel HJ, Klasse PJ, Burton DR, Sattentau QJ. Neutralization of human immunodeficiency virus type 1 by antibody to gp120 is determined primarily by occupancy of sites on the virion irrespective of epitope specificity. J Virol. 1998;72(5):3512–9. doi: 10.1128/jvi.72.5.3512-3519.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang X, Lipchina I, Cocklin S, Chaiken I, Sodroski J. Antibody binding is a dominant determinant of the efficiency of human immunodeficiency virus type 1 neutralization. J Virol. 2006;80(22):11404–8. doi: 10.1128/JVI.01102-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Link AJ, Vink MK, Tirrell DA. Presentation and detection of azide functionality in bacterial cell surface proteins. J Am Chem Soc. 2004;126(34):10598–602. doi: 10.1021/ja047629c. [DOI] [PubMed] [Google Scholar]
- 40.Culp JS, Johansen H, Hellmig B, Beck J, Matthews TJ, Delers A, Rosenberg M. Regulated expression allows high level production and secretion of HIV-1 gp120 envelope glycoprotein in Drosophila Schneider cells. Biotechnology (N Y) 1991;9(2):173–7. doi: 10.1038/nbt0291-173. [DOI] [PubMed] [Google Scholar]
- 41.Gopi HN, Tirupula KC, Baxter S, Ajith S, Chaiken IM. Click Chemistry on Azidoproline: High-Affinity Dual Antagonist for HIV-1 Envelope Glycoprotein gp120. ChemMedChem. 2006;1(1):54–57. doi: 10.1002/cmdc.200500037. [DOI] [PubMed] [Google Scholar]
- 42.Ishino T, Pillalamarri U, Panarello D, Bhattacharya M, Urbina C, Horvat S, Sarkhel S, Jameson B, Chaiken I. Asymmetric usage of antagonist chargedresidues drives interleukin-5 receptor recruitment but is insufficient for receptor activation. Biochemistry. 2006;45(4):1106–15. doi: 10.1021/bi0518038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Charneau P, Mirambeau G, Roux P, Paulous S, Buc H, Clavel F. HIV-1 reverse transcription. A termination step at the center of the genome. J Mol Biol. 1994;241(5):651–62. doi: 10.1006/jmbi.1994.1542. [DOI] [PubMed] [Google Scholar]
- 44.Gartner S, Markovits P, Markovitz DM, Kaplan MH, Gallo RC, Popovic M. The role of mononuclear phagocytes in HTLV-III/LAV infection. Science. 1986;233(4760):215–9. doi: 10.1126/science.3014648. [DOI] [PubMed] [Google Scholar]
- 45.Popovic M, Gartner S. Biology of human imunodeficiency virus: virus receptor and cell tropism. Curr Opin Immunol. 1989;1(3):516–20. doi: 10.1016/0952-7915(88)90036-2. [DOI] [PubMed] [Google Scholar]
- 46.Krebs FC, Miller SR, Malamud D, Howett MK, Wigdahl B. Inactivation of human immunodeficiency virus type 1 by nonoxynol-9, C31G, or an alkyl sulfate, sodium dodecyl sulfate. Antiviral Res. 1999;43(3):157–73. doi: 10.1016/s0166-3542(99)00044-3. [DOI] [PubMed] [Google Scholar]
- 47.Parolin C, Taddeo B, Palu G, Sodroski J. Use of cis- and trans-acting viral regulatory sequences to improve expression of human immunodeficiency virus vectors in human lymphocytes. Virology. 1996;222(2):415–22. doi: 10.1006/viro.1996.0438. [DOI] [PubMed] [Google Scholar]
- 48.Rho HM, Poiesz B, Ruscetti FW, Gallo RC. Characterization of the reverse transcriptase from a new retrovirus (HTLV) produced by a human cutaneous T-cell lymphoma cell line. Virology. 1981;112(1):355–60. doi: 10.1016/0042-6822(81)90642-5. [DOI] [PubMed] [Google Scholar]
- 49.MOE Molecular Operating Environment Chemical Computing Group. Canada: http//www.chemcomp.com/Montreal. [Google Scholar]
- 50.Halgren TA. MMFF VI. MMFF94s option for energy minimization studies. J Comput Chem. 1999;20:720–729. doi: 10.1002/(SICI)1096-987X(199905)20:7<720::AID-JCC7>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 51.Halgren TA. MMFF MMFF VII. Characterization of MMFF94, MMFF94s, other widely available force fields for conformational energies and for intermolecular-interaction energies and geometries. J Comput Chem. 1999;20:740–774. doi: 10.1002/(SICI)1096-987X(199905)20:7<730::AID-JCC8>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.