

Abstract
Proximal spinal muscular atrophy (SMA) is a neuromuscular disease caused by low levels of the survival motor neuron (SMN) protein. The reduced SMN levels are due to loss of the survival motor neuron-1 (SMN1) gene. Humans carry a nearly identical SMN2 gene that generates a truncated protein, due to a C to T nucleotide alteration in exon 7 that leads to inefficient RNA splicing of exon 7. This exclusion of SMN exon 7 is central to the onset of the SMA disease, however, this offers a unique therapeutic intervention in which corrective splicing of the SMN2 gene would restore SMN function. Exon 7 splicing is regulated by a number of exonic and intronic splicing regulatory sequences and trans-factors that bind them. A better understanding of the way SMN pre-mRNA is spliced has lead to the development of targeted therapies aimed at correcting SMN2 splicing. As therapeutics targeted toward correction of SMN2 splicing continue to be developed available SMA mouse models can be utilized in validating their potential in disease treatment.
Keywords: Spinal Muscular Atrophy (SMA), Survival Motor Neuron (SMN), pre-mRNA Splicing, Intronic Splicing Enhancer (ISE), Exonic Splicing Enhancer (ESE), RNA Therapy
2. INTRODUCTION
Proximal Spinal Muscular Atrophy (SMA) is a neuro-degenerative disease characterized by the loss of alpha-motor neurons. This loss of motor neurons results in progressive muscle atrophy that eventually leads to paralysis and death. This disease occurs in about 1 in 6000 to 10000 live births. Disease symptoms can occur at different times in life and can show varied intensities (1-6).
Children with type I SMA experience proximal muscle weakness and may never be able to sit unaided. Death occurs before age two. They may have decreased ability to move their heads and obtain adequate oxygen levels while sleeping. The onset of this disease occurs less than six month after birth. With type II SMA the proximal muscle weakness begins between 6 to 18 months. The patients can sit unaided and have limited movement. While they sleep these individuals often have difficulty taking adequately deep breaths. Finally, type III SMA patients are able to walk unaided and can have a normal lifespan with the onset of the disease occurring after 18 months of age. Once they begin walking the children may fall more frequently, have difficulty in righting themselves, and may be unable to run. However, even these patients develop progressive muscle weakness (7-9).
The decreased expression of the Survival Motor Neuron genes is central to the development of SMA. The SMN genes located in the inverted duplication were identified as the telomeric copy of SMN (SMN1) and the centromeric SMN (SMN2). It was revealed by analysis of patients with SMA that the SMN1 gene is deleted or interrupted in 98% of the patients analyzed while the other 2% of patients analyzed had missense, or splice site mutations in SMN1. All SMA patients in this study had the SMN2 gene present (10). More extensive analysis, based on a large study of 525 SMA patients, revealed the homozygous absence of SMN1 in 92% patients, subtle mutation in 3.4% patients, and no mutation in 4.6% patients (11, 12). The SMN2 gene is highly homologous to the SMN1 gene with a greater than 99% identity between the two (13). None of the nucleotide differences identified in SMN2 alter the amino acid sequence, but a single C to T nucleotide change in exon 7 has been shown to alter the processing of the SMN2 gene (10, 13, 14). The loss of the SMN1 gene compounded by the decreased processing of the SMN2 gene results in extremely low levels of SMN protein. It is this low level of SMN protein that leads to the SMA disease. Research continues to investigate the functions for the SMN protein, in both snRNP biogenesis and novel neuronal function, to understand which aspect of the SMN protein function leads to SMA (15). While a better understanding of the SMN protein function is important, understanding the regulation of the SMN genes will also provide insight into the genesis of the SMA disease. This review focuses on clearly outlining the currently identified SMN splicing regulators and understanding how RNA splicing is being investigated as a therapeutic point of intervention in SMA.
3. PRE-mRNA SPLICING AND SMA DISEASE
Pre-mRNA splicing involves the joining of exons, which contain the nucleotide sequence present in the mature RNA molecule and may provide the protein coding region of a gene, through the precise removal of the noncoding introns. Human genes typically contain multiple introns of variable size, however, nearly all introns start with nucleotide GU and ends with AG in the 5’ to 3’ direction and contain a larger, less highly conserved consensus region at each site. These exon intron boundaries are called the 5'splice site (splice donor site) and 3'splice site (splice acceptor site), respectively. The exact sequence present at the splice site modifies the capability of the U1 and U2 small nuclear ribonucleoproteins (snRNP) and other auxiliary factors such as U2AF to recognize this consensus region. Thus the relative strength of the splice site can directly affect the likelihood of the exon intron boundary being identified. To better define the exon, multiple interactions exist between regulatory elements in the pre-mRNA and trans-factors that recognize these regulatory elements in the pre-mRNA. Splicing elements that enhance exon inclusion are called the exonic or intronic splicing enhancers (ESE or ISE) depending upon their location within the gene. Additionally, elements can also interfere with inclusion of the exon and are likewise called exonic or intronic splicing silencers (ESS and ISS). It is the cooperative interaction between the splicing regulatory proteins that bind these regions that help define the exons (16). The eukaryotic pre-mRNA splicing, therefore, is a complex process involving several RNP complexes and an essential regulatory step in eukaryotic gene expression. When any of these steps becomes disrupted the results can lead to disruption in pre-mRNA splicing. The C to T transition in exon 7 of the SMN2 gene drastically reduces the recognition of exon 7 by the splicing machinery and therefore reduces the efficiency of exon 7 inclusion in the SMN2 transcript (13, 14, 17) resulting in a truncated protein with altered activity (10, 18, 19). The mechanism for the disrutpoin of exon 7 splicng has been extensively studied and is discussed below.
Additional alternative splice forms of SMN have been identified. It has also been reported that exon 5 of the SMN genes undergoes alternative splicing to produce transcripts lacking exon 5 (18). Exon 5 skipping can be coupled to exon 7 skipping however the function of the transcripts lacking exon 5 in not known. More recently axonal-SMN (a-SMN) has been identified (20). This isoform is preferentially encoded by SMN1 and expressed mainly in the axons of motor neurons (20). This isoform consists of all 9 exons of SMN but also retains intron 3 and the resulting protein has been implicated in motor neuron axonogenesis (20). The role of a-SMN, SMNdelta5, and SMNdelta5+7 in the SMA disease phenotype or normal SMN function has not been conclusively determined. While multiple SMN transcripts have been identified, exon 7 splicing continues to be the primary focus when studying the RNA splicing of the SMN genes. Because alerted splicing of the SMN2 exon 7 is central to decreased SMN levels and the SMA disease, correction of SMN2 splicing provides the possibility of therapeutic benefits for SMA patients. Indeed a SMA patient mutation has been identified in the SMN2 gene that can ameliorate the disease phenotype. In this individual a G to C change in SMN2 exon 7 has been shown to partially correct pre-mRNA splicing to increase full length transcript levels through the creation of a new ESE (21).
4. SMN SPLICING REGULATORS
4.1. The C to T nucleotide transition in exon 7
After the translationally silent C to T alteration in SMN2 exon 7 was identified as the causative change mediating the reduced incorporation of exon 7, the mechanism by which this alteration disrupted the natural splicing of SMN2 transcripts was further examined. Two competing models were proposed (Figure 1). In the first, the C to T alteration disrupts binding of the exonic splicing enhancer SF2/ASF (ESE model). Disrupting the binding of a positive regulator of splicing present in SMN2 leads to inefficient definition of exon 7 and results in predominantly skipped spliced product. In the second proposed model, the C to T alteration creates an exonic splicing silencer, which binds hnRNP A1 (ESS model). hnRPN A1 binding specifically to the SMN2 pre-mRNA transcript disrupts the binding of other splicing factors and leads to increased skipping of exon 7.
Figure 1.
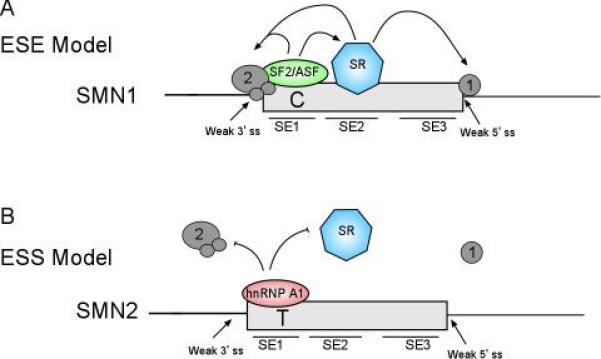
Exon 7 ESE and ESS model of pre-mRNA splicing. Schematic of the SMN genes with each consisting of a weak 3’ and 5’ splice site. The SMN1 gene contains an ESE where SF2/ASF binding occurs which promotes exonic inclusion. In the SMN2 gene where position 6 contains a T nucleotide the SF2/ASF ESE is disrupted while a hnRNP A1 site is formed creating a new ESS. This mutated site disrupts exon recognition of exon 7 and promotes exon skipping.
Three putative exonic splicing enhancer regions were identified in exon 7 and named SE1, SE2, and SE3 (17). In the ESE model, SF2/ASF had a predicted binding site in SE1 of exon 7 using the predictive ESE motif tool (ESE Finder) (17). Studies that support the ESE model have utilized SMN minigenes containing the SMN1 sequence, SMN1 sequence containing the SMN2 exon 7 nucleotide alteration, or compensatory mutations that restore the predicted SF2/ASF ESE motif. The C to T alteration generated in SMN1 was shown to mimic SMN2 exon 7 skipping. Inclusion of exon 7 is rescued by correcting the predicted SF2/ASF ESE in both in vivo and in vitro splicing assays. Most importantly, SF2/ASF was found to bind to SMN1 with a higher affinity than SMN2 in which the SF2/ASF binding site was reduced due to the C to T alteration. These data verified the role of SF2/ASF in mediating exon 7 inclusion (17, 22).
In evaluating hnRNP A1, the factor proposed to bind exon 7 of SMN2 but not SMN1 as proposed in the ESS model, it was shown that hnRNP A1 bound with equal intensity to SMN1 and SMN2 (22). Depletion of hnRNP A1 was shown to relieve exon 7 skipping in vivo whereas over expression led to an increase of exon 7 skipping in vitro and in vivo for both SMN1 and SMN2, suggesting hnRNP A1 may function via a mechanism that is independent of the C to T transition (22). Finally, minigenes containing multiple point mutations designed to either disrupt the proposed SF2/ASF site and/or the hnRNP A1 site found a strong correlation between SF2/ASF and exon 7 inclusion, whereas no correlation was found between the hnRNP A1 motif and exon 7 inclusion using a Spearman rank correlation coefficient (22). In agreement with this model, a bifunctional oligonucleotide was capable of stimulating exon 7 inclusion of SMN2 by providing SF2/ASF binding motifs through binding to the exonic 3’ splice site (23).
In the ESS model, hnRNP A1 was proposed to bind to a UAG motif in SMN2, which had been previously characterized in other hnRNP A1 dependent ESSs. The hnRNP A1 binding site is created by the C to T transition and leads to the exclusion of exon 7 specifically in SMN2 (24). However, when the UAG motif was disrupted, as in SMN1, hnRNP A1 binding was abolished and the exon skipping was not observed. Point mutations in the UAG motif, which is essential for hnRNP A1 binding, promoted exon 7 inclusion in SMN2. SMN1 and SMN2 minigenes cotransfected with siRNA to hnRNP A1 were capable of increasing full length transcripts from SMN2 minigenes while depletion of SF2/ASF (the factor proposed to bind SMN1 but not SMN2 exon 7 in the ESE model) resulted in no change in splicing in both SMN1 and SMN2 minigenes (25). Additionally, over expression of SF2/ASF showed no change in SMN1 or SMN2 minigene exon 7 splicing (24). Finally, hnRNP A1 was found to bind to SMN2 RNA, and to a lesser extent SMN1, while SF2/ASF showed little to no binding (24, 25).
Clearly, both models have experimental evidence that support and/or refute the SF2/ASF ESE and the hnRNP A1 ESS models in SMN exon 7 splicing. Variations in results from these two groups are suggested to be due to differences in experimental techniques and/or reagents used. However, it is possible that these models are not mutually exclusive, where the loss of SF2/ASF and gain of hnRNP A1 binding contribute to the exclusion of exon 7 in SMN2 transcripts.
The function of splicing regulatory elements are often utilized to mediate recognition of exon/intron boundaries by the recruitment of the spliceosome to splice sites, however, it is possible that the C to T alteration also reduces splice site strength. Using two-exon minigene systems it was determined that the C to T alteration decreased the strength of the exon 7 3'ss by 2-fold, whereas the 5'ss was unaffected (26). Further evidence of reduced recognition of the SMN2 exon 7 3'ss was shown to be dependent on the U2 accessory factor (U2AF), a spliceosomal factor that binds the 3'ss and mediates recruitment of the U2 snRNP, where U2AF bound differentially to SMN1 and SMN2 (27). Using RNAs encoding exon 7 and the surrounding intronic sequences of SMN1 and SMN2, U2AF binding was nearly 2-fold higher on SMN1, consistent with the Hertel groups data on splice site strength. This difference in U2AF occupancy of SMN1 and SMN2 RNAs also correlated to differential levels of U2 assembly. The addition of recombinant SF2/ASF increased U2 assembly whereas the addition of hnRNP A1 reduced U2 assembly. However, these results held true for both SMN1 and SMN2 RNAs and were not dependent on U2AF binding. Thus, the role of SF2/ASF and hnRNP A1 likely function in other steps of U2 assembly other than U2AF binding (27).
While the loss of a critical ESE or creation of an ESS may tip the balance toward exon 7 skipping in SMN2, other necessary factors mediating exon 7 recognition are likely involved such as: a suboptimal polypyrimidine tract, weak 3’ splice site (26), and a weak 5’ splice site (28). It is the interplay between the recognition of the SMN exon/intron boundaries and the recruitment of regulatory proteins that determine the amount of full length and skipped transcripts generated from the SMN1 and SMN2 genes.
4.2. Exonic splicing regulation
Once differential splicing of exon 7 in the SMN genes was identified, researchers began the methodical task of better characterizing the complex interactions that are involved in the regulated splicing of SMN1 and SMN2. Attention was placed first on the sequence of exon 7. The role of many splicing and regulatory factors has been examined in an attempt to understanding the role these factors may play in altering the splicing of exon 7 (Table 1 and Figure 2).
Table 1.
The effect of Trans-splicing factor over-expression on SMN pre-mRNA splicing
| Factor | FL/Delta7 Transcript Ratioa | Endogenouse and/or Minigene Assay | Reference |
|---|---|---|---|
| 9G8 | No Change | Minigene | 24, 30 |
| CLK2 | None Reported | Minigene | 29 |
| CLK2-KR | None Reported | Minigene | 29 |
| hnRNP A1 | Decrease | Endogenous | 22 |
| hnRNP G | Increase | Minigene | 31 |
| hnRNP Q1 | Increase | Minigene and Endogenous | 32 |
| hnRNP Q2 | Decrease | Minigene | 32 |
| hnRNP Q3 | Decrease | Minigene | 32 |
| hTra2alpha | Increase | Minigene | 24, 29 |
| hTra2beta1 | Increase | Minigene | 24, 29 |
| hTra2beta3 | None Reported | Minigene | 29 |
| HuD | No Change | Minigene | 24 |
| PTB | None Reported | Minigene | 29 |
| SAF-B | None Reported | Minigene | 29 |
| SC35 | No Change | Minigene | 24, 30 |
| SF1 | None Reported | Minigene | 29 |
| SF2/ASF | No Change | Minigene | 22, 24, 29 |
| SLM-2 | None Reported | Minigene | 29 |
| SRp20 | No Change | Minigene | 30 |
| SRp30c | Increase | Minigene | 29, 30 |
| SRp40 | No Change | Minigene | 30 |
| SRp55 | No Change | Minigene | 30 |
| U2AF65 | None Reported | Minigene | 29 |
| YT521B | None Reported | Minigene | 29 |
Expression constructs encoding RNA binding proteins were evaluated for their effect on SMN splicing in transfection studies. Those evaluated but where results were not shown or explicitly communicated have been labeled as none reported. The effect of factors that were tested with the results shown or clearly stated have been listed according to their published findings.
Figure 2.
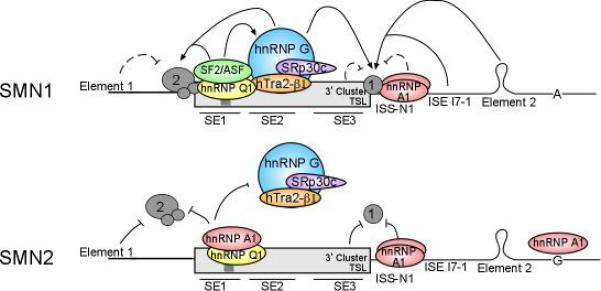
SMN exon 7 is Regulated my multiple exonic and intronic regulators. Schematic of the SMN genes are shown with previously identified cis elements and any known proteins that bind them. Inclusion of exon 7 is regulated by the interaction of multiple factors.
hTra2beta1, an SR-like protein which functions through ESEs containing AG-rich motifs showed a ~2.7 fold increase of full length transcript when overexpressed in cell culture and was shown to bind the SE2 AG-rich motif of SMN exon 7 (29). In addition to hTra2beta1, over expression of the splicing factor SRp30c is capable of increasing the levels of exon 7 containing transcript and also found to intact at the SE2 element. Using RNA affinity chromatography and biomolecular interaction analysis (BIA), it was demonstrated that SRp30c has the ability to stimulate SMN exon 7 inclusion through direct interaction with hTra2beta1 (30). In similar studies hnRNP G was also shown to promote exon 7 inclusion via a direct interaction with hTra2beta1 (31). Taken together this suggests an hTra2beta1/hnRNP G/SRp30c complex that binds the SE2 region of the SMN genes and help in exon 7 pre-mRNA recognition by the splicing machinery.
Investigating the splicing of the SMN transgene in SMA transgenic mice has also been used to determine regulators of SMN splicing. Using RNA affinity chromatography from the testes of mice, where SMN exon 7 including transcript levels were increased, several RNA associated proteins were pulled down and identified by mass spectrometry. From the list of proteins identified hnRNP Q1 was shown to bound directly to SMN1 and SMN2 exon 7 near in SE1 regions in the +6 nucleotide region of the exon using human HEK293 cell extract. Additional analysis revealed that over expression of hnRNP Q1 promoted inclusion of exon 7 of the SMN2. Interestingly, over expression of the other hnRNP Q isoforms (hnRNP Q2 and hnRNP Q3) resulted in suppression of exon 7 inclusion suggesting a coordinated function between the opposing isoforms (32). This is the first example of a splicing factor that affects splicing being identified by looking at tissue specific splicing of the SMN genes.
At the 3’ end of the SMN exon 7 two cis-elements were discovered and characterized. The 3’ cluster, an inhibitory element, was discovered using an in vivo selection technique to identify nucleotides important in regulating SMN exon 7 (28). The 3’ cluster comprises a 7 nucleotide sequence toward the 3’ end of exon 7. Deletion or alteration of any part of the 3’ cluster promoted exon 7 inclusion in an SMN2 minigene system. Additionally this region of exon 7 was also shown to contain terminal stem-loop 2 (TSL2) (33). TLS2 acted as an inhibitor to SMN exon 7 splicing and was predicted to form a stem-loop using the MFOLD program, a program used to predict RNA secondary structure, and then confirmed in human SMN minigene constructs using RNA structure probing (Singh et al 2006). This loop, when stabilized, prevents the 5’ splice site of intron 7 from being available for use by the U1 snRNP. In SMN1 where the ESE is still intact, this inhibitory complex has little effect on constitutive splicing. However, in SMN2 where the exon 7 splicing elements have been disrupted, this stem loop is sufficient to cause exon 7 skipping. Mutational disruptions of this loop, along with compensatory mutations, support the role of this structural element in the splicing of the SMN genes (34). The 3’ cluster overlaps the nucleotides making up the top of the stem and the loop of TSL2. While this overlap was not explored it could be that the TSL2 and 3’ cluster interact in the regulation of the already weak 5’ splice site. The TSL2 element highlights the importance of RNA structure on the regulation of pre-mRNA splicing of the SMN genes.
4.3. Intronic splicing regulation
The role of intronic sequences in pre-mRNA splicing is less understood than exonic regulators making it more challenging to identify intronic elements that play a role in SMN splicing. However, the introns flanking exon 7 of the SMN genes began to be examined for any potential role in pre-mRNA splicing. The first investigations of the intron were carried out by the Imaizumi lab using a modified exon trapping vector used to examine both exonic and intronic splicing regulators. An exon trapping vector typically has two exons that are normally spliced together to form a mature transcript. However, a DNA fragment can be inserted between these two exons whereby its incorporation in the final transcript can be evaluated to identify functional exons. The trapping vector utilized in this study contained either the wildtype SMN1 or a mutated SMN1 exon 7 containing the C>T alteration and flanking regions of intron 6 and 7. To examine the effects of intronic regions in SMN exon 7 splicing, deletions were then made in either intron 6 or intron 7 containing regions. This technique led to the discovery of two intronic elements that altered SMN splicing, element 1 and element 2.
Element 1, a pyrimidine rich sequence, is located in intron 6 near the branch point and polyprymidine tract proceeding the 3’ splice site of exon 7. Element 1 consists of 45 nucleotides that, when deleted, was capable of increasing inclusion of full length SMN (35, 36). Additionally, when this region was targeted by antisense oligonucleotides, expression of the SMN exon 7 containing transcript was increased. This led to the conclusion that element 1 was a silencer element (36). Additionally, using RNA affinity chromatography, the proteins FUSE-BP (aka FUBP1 and FBP1) and PTB were found to bind element 1; however, their role in modifying SMN splicing has not yet been elucidated (35).
The study performed by the Imaizumi lab also uncovered element 2, an intronic splicing enhancer. This element was located in intron 7 downstream of exon 7. Using the MFOLD program element 2 was predicted to form a stem loop secondary structure. Using their SMN exon trapping vectors, point mutations designed to disrupt the secondary structure of element 2 were shown to disrupt exon 7 inclusions, while compensatory mutations were capable of restoring exon 7 ratios. Additionally, tandem repeats of element 2 were capable of increasing exon 7 levels. A genome wide search using The Basic Local Alignment Search Tool (BLAST) Internet based search tool found that the general stem loop structure appeared in a number of other genes, however its general role in splicing was never addressed. This data would suggest that element 2 is a structural element located in intron 7 that is acting as a splicing enhancer and again underscores the importance of RNA secondary structure as a regulator of RNA splicing (37).
Further analysis of intron 7 uncovered a unique silencer element termed ISS-N1. ISS-N1, located immediately downstream of the exon 7 5’ splice site, was found to profoundly enhance SMN exon 7 containing transcripts when deleted or mutated in an SMN minigene system (33). When inserted into a heterologous Casp3 minigene system it was likewise sufficient in increasing exon skipping. Finally, when antisense oligonucleotides were developed against the ISS-N1 nucleotide sequence, exon 7 containing transcripts were increased (33, 38). These experiments lead to the conclusion that ISS-N1 is a strong intronic splicing silencer. Later experiments also found that ISS-N1 contains two hnRNP A1 binding sites and that hnRNP A1 binds this region (39).
Located between ISS-N1 and element 2 is a region that was examined by the Chandler lab. This sequence, termed ISE I7-1, was observed to increase the skipping of SMN exon 7 after deletion or mutation. When the 3’ portion of this element was repeated in tandem in an SMN minigene system complete restoration of exon 7 containing transcripts was observed. When ISE I7-1 was placed into a heterlogouse Casp3 minigene system downstream of the alternatively spliced exon it was capable of increasing exon inclusion. Furthermore, a genomic search for sequence matches to this element identified the presence of partial ISE I7-1 sequence in a number of other genes both in exons and introns indicating that this element could play a role in regulating the splicing of other genes as well (40).
Finally, an SMN2 specific intronic splicing silencer was investigated by the Manley laboratory to better characterize the differences between SMN1 and SMN2. This group found that, like the SMN2 exon 7 C>T alteration that forms an hnRNP A1 binding site, there is a specific nucleotide alteration located in intron 7 of SMN2 which also creates a high affinity hnRNP A1 binding site. The SMN2 specific A>G transition at position 100 results in increased exclusion of exon 7. Using a SMN2 minigene containing reduced intronic sequences they were able to show that altering this SMN2 specific G to an SMN1 A resulted in an increase of exon 7 containing transcripts. Furthermore, this region was found to contain an hnRNP A1 binding motif. When the SMN minigene was UV crosslinked in HeLa nuclear extract and probed for hnRNP A1 there was preferential binding to intron 7 of the SMN2 minigene over the intron of the SMN1 gene which would contain the hnRNP A1 binding motif (41).
ISS-N1, ISE I7-1 and element 2 and the SMN2 A>G comprises the first 100 nucleotides of the 444 nucleotide intron 7. Their close proximity and competing functions (ISS-N1 acts as a silencer while ISE I7-1 and element 2 functions as enhancers and the SMN2 A>G site acts as a silencer) suggest that intron 7 plays a critical role in regulating SMN exon 7 splicing. Additionally these elements or the proteins that bind them may be interacting in a cooperative or antagonistic fashion to ultimately determine the levels of skipping in the SMN2 gene. Further analysis of the factors that bind these regions and their interactions with each other may provide a better understanding of how SMN is regulated. Additionally, this type of analysis can lead to an understanding of how other alternatively spliced RNAs are regulated making the SMN gene a powerful tool in understanding RNA splicing as well as the therapeutic treatment of SMA.
5. SMN SPLICING THERAPIES, TOOLS AND TECHNIQUES
Spinal muscular atrophy is a unique genetic disorder in which correcting the inefficient splicing of the modifier gene SMN2, which is present in all SMA patients, can rescue for the loss of the SMN1 gene. Furthermore, as SMN2 is expressed is similar to that of SMN1 and as the correction of SMN2 splicing would not alter the expression pattern Correction of SMN2 splicing can be achieved by several strategies to increase the levels of SMN2 exon 7 inclusion and thus SMN protein: (1) antisense oligonucleotides, (2) trans-splicing, and (3) drugs (Figure 3). We will introduce which mouse models are available to address the use of these approaches and how each of these approaches has been used in SMN2 splicing correction.
Figure 3.
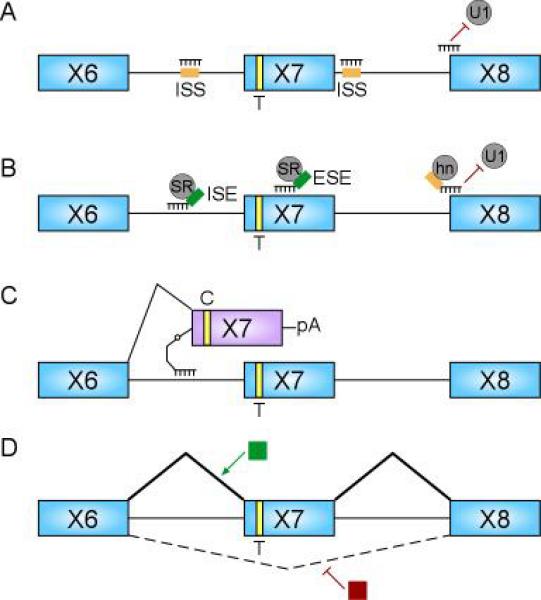
Therapeutic strategies targeting splicing correction in SMN2. A. Standard ASOs targeting intron splicing silencers (ISS) and the exon 8 3'ss to promote exon 7 inclusion. B. Bifunctional ASOs linked to binding sites for positively acting splicing factors (SR proteins) to increase exon 7 inclusion. A bifunctional ASO linked to binding sites for inhibitory splicing factors (hnRNP proteins) to block the exon 8 3'ss and increase exon 7 inclusion by inhibiting splicing of exon 6 to 8. C. Trans splicing of an RNA molecule that can be used instead of SMN2 exon 7 to produce full length SMN transcripts. D. Drugs that promote the incorporation of exon 7 (green box) or inhibit the splicing of exon 6 to 8 (red box).
5.1. SMA mouse models for therapy testing
Several genetic models of SMA are available that mimic the genetic basis and phenotype of SMA. In this review we are focusing on mouse models that contain a human SMN2 transgene that requires splicing for expression of SMN (Table 2). Initially the mouse Smn gene was inactivated by insertion of a lac Z cassette in the region of exon 2. This null allele when homozygous led to early embryonic lethality (42). The addition of a bacterial artificial chromosome (bac) transgene carrying the human SMN2 gene in low copy number (1 copy per allele) rescued the embryonic lethality and generated mice exhibiting a severe SMA phenotype (average lifespan of 5.3-6 days). However, eight copies of the SMN2 bac transgene were capable of correcting for the loss of Smn (5). A second null allele was generated by the replacement of Smn exon 7 by an HPRT cassette, and the addition of an SMN2 bac transgene produced mice with severe to mild SMA (10 day lifespan to normal lifespan associated with a short tail or necrotic tail and hind limbs). These variations were again due to variable copy number of the SMN2 transgene with the low copy mouse carrying 2 copies of the SMN2 transgene per allele (43). The addition of a transgene encoding SMN cDNA lacking exon 7 (SMNdelta7) to the severe SMA mouse (5) produced a less severe SMA mouse model that extended the average lifespan of these SMA mice from 5.3 to 13.3 days (3). Interestingly, the lifespan of the Hsieh-Li mouse and the SMAdelta7 mice are comparable which is likely due to similar genetics. Both of these mice have 2 copies of an SMN2 transgene and 2 copies of alleles that produce transcripts lacking exon 7, either in the Smn gene or by the SMNdelta7 transgene (44). Additional transgenic mice harboring 1 and 2 copies of an SMN2 transgene per allele were generated(45) and crossed to the Smn null mice (42). When these transgenic lines are crossed together they generate a 3 copy SMN2 mouse on the Smn null background and a severe SMA mouse model that has an average lifespan of 15.2 days. This SMA mouse model lacks the SMA phenotype at early postnatal days but develops a progressive SMA phenotype and lifespan similar to the SMA delta7 mice (45). The SMN2 transgenes used for the generation of these mouse models contain the promoter for SMN2 and as such are considered under normal transcriptional control; however, due to random insertion of the transgenes, varied epigenetic control of SMN2 expression may not truly mimic endogenous Smn expression. Nonetheless, all of these mice validate SMN2 as a modifier of the SMA disease in vivo, and as they carry the SMN2 gene they are good genetic models for SMN2 splicing correction in SMA therapeutic intervention.
Table 2.
Available SMA mouse models for SMN2 splicing correction
| Mouse Model | SMA I Monani et al. 2000 (5) | SMA I-III Hsieh-Li et al. 2000 (43) | SMAdelta7 Le et al. 2005 (3) | SMA I Michaud et al. 2010 (45) |
|---|---|---|---|---|
| Genetics |
aSmn -/- cSMN2 bac +/+ |
bSmn -/- dSMN2 bac +/+ |
aSmn -/- cSMN2 bac +/+ SMN delta7 +/+ |
aSmn -/- eSMN2 +/-; fSMN2 +/- |
| Disease Onset | PND1 | PND1 | PND1-2 | PND5 |
| Survival | 5.3-6 days | Type I: 10 days Type II: 2-4 weeks Type III: normal/ necrotic tail and hind limbs |
13.3 days | 15.2 days |
| Advantages | The carrier Smn +/- mice are long-lived and can be used for testing SMN2 splicing correction. Affected SMA mice are amenable to rescue of SMN protein levels and SMA phenotype. | The less severe mice are longer lived and have a mild phenotype of tail/hind limb necrosis. These mice can be used for testing SMN2 splicing and SMA phenotype correction. | Extension of lifespan over SMA I pups for ease in treatment of older pups. One can observe increases in SMN protein and rescue of the SMA phenotype. | Extension of lifespan over SMA I without the SMNdelta7 transgene. These mice can be used to evaluate SMN2 splicing and treatment of older pups |
| Disadvantages | The SMA affected mice have a very short lifespan and would require treatment when animals are neonates. | Variability in disease severity and short lifespan of SMA type I mice. | The SMNdelta7 transgene perturbs the observation of SMN2 splicing by RT-PCR. There are three loci to follow. | There are three loci to follow, and the SMN2 transgene loci need to be heterozygous to obtain the SMA mice. |
Smn -/- allele due to exon 2 disruption by lacZ
Smn -/- allele due to exon 7 replacement with HPRT
28 kb human bac transgene with the SMN2 promoter and SMN2 gene (single copy of SMN2 at the transgene locus)
115 kb human bac transgene with SMN2, SERF1, and part of NAIP genes (two copies of SMN2 at the trangsgene locus)
35.5 kb human pac transgene with 1 copy of SMN2 at the transgene locus
35.5 kb human pac transgene with 2 copies of SMN2 at the transgene locus.
The available SMA mouse models offer several ways to evaluate the effectiveness of a given therapeutic for SMN2 splicing correction: by observing in vivo correction of the SMN2 splicing pattern, an associated increase in SMN protein levels, and correction of the SMA neurodegenerative phenotype. Several major considerations when selecting an appropriate SMA mouse model include the complexity of the genetics of the SMA model, the lifespan of the mouse, and the consideration of the downstream assay to be preformed (RT-PCR, western blot, Kaplan-Meyer survival, etc.). When first evaluating the potential of a given therapeutic to function in SMN2 splicing correction it is best to use a mouse model that has a normal lifespan and carries the SMN2 transgene. However, long-lived SMA models correlate with a higher basal level of SMN protein, thereby making identification of increased SMN protein levels difficult. Correction of the SMA phenotype would be more pronounced in more severe SMA models where SMN levels are the lowest, however this makes administration of the treatment more technically difficult as the animals are much younger when treatment would be required for rescue. Therefore, the use of several SMA mouse models may be required to evaluate the viability of a given therapeutic for in vivo correction of SMN2 splicing and SMA phenotype correction.
5.2. Therapeutic strategies
A greater understanding of how SMN exon 7 is regulated has lead to targeted approaches for the correction of SMN2 splicing. Several approaches have been tested to increase exon 7 inclusion: preventing the function of inhibitory elements, introducing positively acting elements to exon 7, modulating the availability of competing splice sites, or introduction of corrected exons by trans-splicing.
5.2.1. Antisense oligonucleotides
Correction of SMN2 splicing by the use of anti-sense oligonucleotides (ASOs) can be achieved by either masking the effect of an inhibitory element or introducing an enhancer to the effected exon. ASOs target sequences in the messenger RNA by Watson-Crick base pairing to the mRNA (Figure 3A). In an effort to introduce positively acting elements to promote exon 7 inclusion the Krainer group utilized exon-specific splicing enhancement by small chimeric effectors (ESSENCE) (Figure 3B). This technology combines the use of an antisense moiety linked to a minimal RS domain targeted to the affected exon. The minimal RS domain is meant to mimic the function of SR proteins in mediating protein-protein interactions that increase the association of the spliceosome to an exon. Using an in vitro competent splicing SMN2 minigene they showed the addition of the ESSENCE moiety in splicing reactions was capable of increasing exon 7 inclusion in a manner consistent with SR protein mediated exon inclusion (46). Though validated in vitro, neither cell based assays nor in vivo tests have been performed. It was observed in this study that the ASO alone was capable of promoting exon 7 inclusion, and therefore ASOs alone may be functional in SMN2 splicing correction. This was tested utilizing an ASO-tiling method of SMN2 exon 7, from which a central region of splicing enhancement and flanking regions of inhibition were identified. The use of ASOs targeting the inhibitory regions in exon 7 were successful in increasing SMN2 exon 7 inclusion in in vitro splicing reactions, transfection experiments, and endogenous SMN2 splicing. The ASOs were further capable of increasing SMN protein in SMA fibroblasts (47).
While targeting splicing regulatory elements in exon 7 can correct SMN2 splicing it is also possible to block intronic inhibitory elements, thereby increasing exon 7 inclusion (Figure 3A). A similar ASO-tiling approach as conducted for exon 7 was used to indentifying cis-regulatory elements in intron 6 and 7 proximal to exon 7. This approach identified a mild intron 6 inhibitory region and further validated ISS-N1 in intron 7 as a strong ISS and a target of hnRNPA1/A2 binding (39). A 2'-O methyl ASO with a phosphorothioate backbone targeting this inhibitory element was administered by tail vein injection and found to significantly increase SMN2 exon 7 inclusion in the liver of mice carrying the human SMN2 transgene (39). This inhibitory element was further analyzed by the Singh group where it was determined that an 8 nucleotide ASO was sufficient to promote the correction of SMN2 splicing thereby reducing the potential of off target effects (38). Targeting ISS-N1 in the central nervous system (CNS) by the intracranial ventricular (ICV) injection of ASOs in neonatal SMA mice led to increased SMN2 exon 7 inclusion and SMN protein in the brain and spinal cord. An improvement in the SMA phenotype was observed although an increase in lifespan was not shown (48).
In an effort to increase the potential of an ASO to promote exon 7 inclusion bi-functional ASOs that target regulatory sequences as well as recruit positively acting splicing regulatory proteins such as SR proteins may offer an alternative treatment for increased exon 7 inclusion (Figure 3B). RNA oligonucletides targeting SMN2 exon 7 with additional ESE sequences were capable of increasing exon 7 inclusion and SMN protein in SMA fibroblasts (23). The addition of a U7 snRNA sequence can be used to mediate the incorporation of an ASO into the splicing machinery and therefore increase the likelihood of correcting splicing. Virally transducible vectors expressing U7 snRNA linked to bi-functional ASOs with ESE sequences were targeted to exon 7 in SMA patient fibroblasts and shown to increase exon 7 inclusion and SMN protein levels (49, 50). To develop second generation bi-functional ASOs the intronic suppressor of exon 7 inclusion, element 1, was targeted using bi-functional ASOs linked to binding sites for SF2/ASF or hTra2beta. As expected the use of the ASO alone was capable of correcting SMN2 splicing through inhibiting element 1, however, the use of the bi-functional ASO was more effective. The hTra2beta bi-functional RNA was administered to SMA mouse models by intracranial ventricular (ICV) injection and was capable of increasing SMN protein in brain and spinal cord (35). Transgenic mice carrying the U7 ASO generated by lentiviral transgenesis were crossed to severe SMA mice and were capable of rescuing the early lethality and SMA phenotype (51).
An alternative approach to increasing exon 7 inclusion is to prevent the incorporation of exon 8, and therefore increase the availability of exon 7 for splicing (Figure 3A). The use of U7 ASOs targeting the exon 8 3'ss were capable of increasing SMN2 exon 7 inclusion and SMN protein levels in cell culture (52, 53). Inhibition of exon 8 was also achieved by the use of a bi-functional ASO targeting the exon 8 3'ss linked to hnRNP A1 binding sites. This ASO was capable of correcting SMN2 splicing and increasing SMN protein both in SMA fibroblasts and in an SMA mouse model (54).
5.2.2. Trans-splicing
Trans-splicing is an approach in which two different RNA molecules are used in the splicing reaction in order to replace a mutant exon with the desired correct RNA sequence (Figure 3C). This approach was used to introduce the SMN1 exon 7 sequence into the SMN2 mRNA. A trans-splicing RNA encoding the correct SMN1 exon 7 sequence along with necessary splicing regulatory sequences was tethered to intron 6 by an ASO upstream of the SMN2 branch point and 3'ss. This allows the spliceosome to utilize the trans-splicing RNA instead of the endogenous SMN2 exon 7. This approach was validated for correction of SMN2 splicing, increasing SMN protein levels, and correction of snRNP assembly (55). To observe SMN2 splicing correction in vivo the authors coupled the trans-splicing approach with the blocking of the exon 8 3'ss by an ASO. This approach made use of a single AAV vector encoding both RNA molecules under independent promoters to increase the likelihood that both RNAs were present for increased trans-splicing (56). Trans-splicing provides an alternative approach for correction of SMN2 splicing and can be utilized in combination with ASOs to promote exon 7 inclusion.
5.2.3. Drugs
The use of small molecules or drugs is a common treatment option for a variety of diseases and as such is an alternative approach by which SMN2 splicing correction may be achieved. Though small molecules can be utilized to increase the level of SMN2 transcripts, stabilize SMN protein, or increase SMN protein activity, in this review we are focusing on drugs shown to increase the ratio of full length to skipped SMN2 transcripts and as such are potentially mediating splicing correction (Table 3). However, an increase in full length transcripts may be produced by multiple mechanisms involved in RNA metabolism including altered transcriptional activity, 5’ capping and nuclear export, or RNA stability. In order to identify drugs that increase the inclusion of exon 7 often reporter minigenes encoding SMN2 exons 6 to 8 were used in high throughput screens (HTS) or in evaluating candidate drugs, from which novel drugs and those that are FDA approved have been identified. However, the mechanism of increased exon 7 inclusion is often not well understood.
Table 3.
Drugs that alter the ratio of full length to skipped SMN2 transcripts
| Drug | Drug type | Effect on SMN2 Transcription | Animal tested | Drug Trial | Selected Citations |
|---|---|---|---|---|---|
| 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) | Na+/H+ exchange inhibitor | Not evaluated | No | No | 72 |
| Aclarubicin | Anthracycline antibiotic | Increased | No | No | 66, 67 |
| Curcumin | Polyphenol | Not evaluated | No | No | 71 |
| Epigallocatechin galate (EGCG) | Polyphenol | Not evaluated | No | No | 71 |
| Hydroxyurea (HU) | ribonucleotide reductase inhibitor | No increase in SMN2 transcripts | Yes | Yes | 65, 76, 84 |
| LBH589 | Hydroxamic acid derived HDAC inhibitor | Increased | Yes | No | 63 |
| M344 | Benzamide, HDAC inhibitor | Increased | No | No | 64 |
| Phenylbutyrate (PB) | Aromatic fatty acid, HDAC inhibitor | Increased | No | Yes | 57, 77, 78, 79 |
| PTK-SMA1 | Tetracycline derivative | N/A | Yes | No | 70 |
| Resveratrol | Polyphenol | Not evaluated | No | No | 71 |
| Salbutamol | Beta2-adrenoceptor agonist | Increased | No | Yes | 69, 85 |
| Sodium butyrate (SB) | HDAC inhibitor | Increased SMN2 promoter activity | Yes | No | 60, 62 |
| Sodium vanadate | Protein phosphatase inhibitor | Increased | No | No | 67, 68 |
| Suberoylanilide hydroxamic acid (SAHA) | HDAC inhibitor | Increased | No | No | 61 |
| Trichostatin A (TSA) | HDAC inhibitor | Increased SMN2 promoter activity | Yes | No | 60, 74, 75 |
| Valproic acid (VPA) | Branched chain fatty acid, HDAC inhibitor | Increased | Yes | Yes | 58, 59, 73, 80, 81, 82, 83 |
For this review we have provided a table of drugs that have be shown to increase the ratio of full length to skipped SMN2 transcripts (Table 3). Many of the drugs identified to increase exon 7 inclusion are of the class of drugs that alter transcriptional activity by inhibition of histone deacetylases (HDAC)s. HDAC inhibitors phenylbutyrate (PB) (57), valproic acid (VPA) (58, 59), trichoistatin A (TSA) (60), suberoylanilide hydroxamic acid (SAHA) (61), sodium butyrate (60, 62), LBH589 (63), and M344 (64) were capable of increasing SMN2 transcriptional activity and increase the ratio of SMN2 full length to skipped transcripts. Other compounds shown to increase this ratio of SMN2 transcripts include the ribonucleotide reductase inhibitor hydroxyurea (HU) (65), anthracyclin antibiotic aclarubicin (66, 67), phosphatase inhibitor sodium vanadate (67, 68), b2-adrenoceptor agonist salbutamol (69), tetracycline derivative PTKSMA1 (70), polyphenols Epigallocatechin galate (EGCG), curcumin, and resveratrol (71), and Na+/H+ exchange inhibitor 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) (72). Of the drugs listed above valproic acid (73), trichostatin A (74, 75), Sodium butyrate (62), LBH589 (63), hydroxyurea (76), and PTK-SMA1 (70) and have been tested in SMA mouse models. Finally, human drug trials have been performed for the potential SMA therapeutics phenylbutyrate (77-79), valproic acid (80-83), hydroxyurea (84), and salbutamol (85).
Recently the drug PTK-SMA1, a compound in the family of tetracyclines, was shown to increase SMN2 exon 7 inclusion by specifically altering splicing. Using the in vitro cell free splicing assay they were able to evaluate the effect of SMA drugs on the splicing reaction by eliminating the role of transcription, mRNA export, and mRNA turnover from the system. This group evaluated several drugs currently in use in SMA treatment that had previously shown to increase the levels of exon 7 inclusion or SMN protein. Only PTK-SMA1 was capable of increasing SMN2 exon 7 inclusion, and this effect was mediated by increasing exon 6 to 7 splicing as well as inhibiting exon 8 incorporation. Furthermore, administration of this drug to two SMA mouse models showed increased SMN2 exon 7 inclusion and SMN protein levels in the liver of these mice, as this drug does not pass the blood brain barrier (BBB) (70). Additionally, a novel HDAC inhibitor, LBH589, was found to markedly increase SMN levels by multiple mechanisms including transcriptional activation of the SMN2 promoter, increased protein stability, and increased exon 7 inclusion. LBH589 treatment increased levels of hTra2beta, which in turn increased the levels of exon 7 inclusion and SMN. LBH589 was further shown to pass the BBB in SMA mice where an increase in SMN2 transcripts and SMN protein was observed in the spinal cord and brain (63).
In the treatment of SMA a wide variety of therapeutic options targeting splicing correction are under development. The limitations common to all of the described therapeutic approaches are method of delivery, dosage, and specificity. Reaching the affected cell type, the motor neurons in the spinal cord, limits the application of therapies due to the BBB. However, ICV injections of ASOs have been shown to correct SMN2 splicing and SMA phenotype in SMA mouse models. As more becomes known about the regulatory elements governing SMN2 exon 7 splicing there exists an increasing availability of therapeutic targets. The use of therapies specifically targeting SMN2 such as ASOs and trans-splicing reduces the likelihood of off target effects often associated with drug treatments while retaining the transcriptional regulation of the endogenous SMN2 gene. However, the best option may be the application of a combinatorial approach: specific treatment of the CNS in conjunction with systemic treatment, and this could be achieved through the use of ASOs and drugs. The correction of SMN2 splicing for the rescue of SMN function is underscored by the benefit of targeting SMN2, a gene that is under the same expression control of SMN1, thereby increasing SMN in the appropriate temporal and spatial context.
6. SUMMARY AND PERSPECTIVES
Though the focus of this review is primarily on splicing in SMA and treatment options that target splicing correction, approaches to increase the levels or stability of the SMN protein may serve as alternative options for SMA treatment. As SMN2 does produce some full length SMN, either stabilization of the transcript and/or protein are options currently under development through approaches such as drug treatment or by gene replacement therapy. As more drugs are identified it will be important to understand their effect on RNA splicing in general. Techniques such as splicing microarrays and deep sequencing will help identify whether the observed change in SMN2 splicing is gene specific or more likely a global alteration in RNA processing. Understanding the mechanism of action for these drugs will increase our understanding of how the SMN genes are regulated. A further consideration in the treatment of SMA and a large focus in the field is the timing of SMN replacement in the correction of the disease. As most SMA patients are diagnosed following appreciable loss of motor function, and thus motor neurons, the treatments may be limited by a low number of available motor neurons. SMN is developmentally required as well, and though low levels are sufficient to proceed past embryonic development the requirement of SMN throughout development is not fully understood. Early detection by newborn screening would allow for earlier treatment and potentially increased rescue of motor neurons prior to disease progression. For SMA patients more progressed in the disease, neuron replacement therapies may be a requirement for correction of the SMA disease phenotype. As we continue to discover the regulatory paradigms directing SMN splicing, there is great hope for therapeutic benefit for SMA patients by the correction of SMN2 splicing.
7. ACKNOWLEDGMENTS
Authors JTG and TWB equally contributed to this review. This work was generously supported by The Research Institute at Nationwide Children's Hospital, and the National Institute of Neurological Disorders and Stroke (NINDS) Grant, 1R21NS054690, to DSC, and the National Institute of General Medical Sciences (NIGMS) Grant, 1F31GM080151-01A1, to JTG.
Abbreviations
- SMN
Survival Motor Neuron
- SMA
Spinal Muscular Atrophy
- ESE
Exonic Splicing Enhancer
- ESS
Exonic Splicing Silencer
- ISE
Intronic Splicing Enhancer
- ISS
Intronic Splicing Silencer
- 3'ss
Three Prime Splice Site
- 5'ss
Five Prime Splice Site
- BBB
Blood Brain Barrier
- ICV
Intracranial Ventricular
- HDAC
Histone Deacetylase
- ASO
Antisense oligonucleotide
8. REFERENCES
- 1.Coovert DD, Le TT, McAndrew PE, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy EJ, Prior TW, Burghes AH. The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet. 1997;6(8):1205–14. doi: 10.1093/hmg/6.8.1205. [DOI] [PubMed] [Google Scholar]
- 2.Le TT, Coovert DD, Monani UR, Morris GE, Burghes AH. The survival motor neuron (SMN) protein: effect of exon loss and mutation on protein localization. Neurogenetics. 2000;3(1):7–16. doi: 10.1007/s100480000090. [DOI] [PubMed] [Google Scholar]
- 3.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14(6):845–57. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 4.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16(3):265–9. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- 5.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossol W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(-/-) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9(3):333–9. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- 6.Gavrilov DK, Shi X, Das K, Gilliam TC, Wang CH. Differential SMN2 expression associated with SMA severity. Nat Genet. 1998;20(3):230–1. doi: 10.1038/3030. [DOI] [PubMed] [Google Scholar]
- 7.Zerres K, Rudnik-Schoneborn S. Natural history in proximal spinal muscular atrophy. Clinical analysis of 445 patients and suggestions for a modification of existing classifications. Arch Neurol. 1995;52(5):518–23. doi: 10.1001/archneur.1995.00540290108025. [DOI] [PubMed] [Google Scholar]
- 8.Zerres K, Rudnik-Schoneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. J Neurol Sci. 1997;146(1):67–72. doi: 10.1016/s0022-510x(96)00284-5. [DOI] [PubMed] [Google Scholar]
- 9.Zerres K, Wirth B, Rudnik-Schoneborn S. Spinal muscular atrophy--clinical and genetic correlations. Neuromuscul Disord. 1997;7(3):202–7. doi: 10.1016/s0960-8966(97)00459-8. [DOI] [PubMed] [Google Scholar]
- 10.Lefebvre S, Burglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80(1):155–65. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 11.Wirth B, Hahnen E, Morgan K, DiDonato CJ, Dadze A, Rudnik-Schoneborn S, Simard LR, Zerres K, Burghes AH. Allelic association and deletions in autosomal recessive proximal spinal muscular atrophy: association of marker genotype with disease severity and candidate cDNAs. Hum Mol Genet. 1995;4(8):1273–84. doi: 10.1093/hmg/4.8.1273. [DOI] [PubMed] [Google Scholar]
- 12.Sun Y, Grimmler M, Schwarzer V, Schoenen F, Fischer U, Wirth B. Molecular and functional analysis of intragenic SMN1 mutations in patients with spinal muscular atrophy. Hum Mutat. 2005;25(1):64–71. doi: 10.1002/humu.20111. doi:10.1002/humu.20111. [DOI] [PubMed] [Google Scholar]
- 13.Monani UR, Lorson CL, Parsons DW, Prior TW, Androphy EJ, Burghes AH, McPherson JD. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum Mol Genet. 1999;8(7):1177–83. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 14.Lorson CL, Hahnen E, Androphy EJ, Wirth B. A single nucleotide in the SMN gene regulates splicing and is responsible for spinal muscular atrophy. Proc Natl Acad Sci U S A. 1999;96(11):6307–11. doi: 10.1073/pnas.96.11.6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10(8):597–609. doi: 10.1038/nrn2670. doi:nrn2670 [pii] 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291–336. doi: 10.1146/annurev.biochem.72.121801.161720. doi:10.1146/annurev.biochem.72.121801.161720 121801.161720 [pii] [DOI] [PubMed] [Google Scholar]
- 17.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet. 2002;30(4):377–84. doi: 10.1038/ng854. [DOI] [PubMed] [Google Scholar]
- 18.Gennarelli M, Lucarelli M, Capon F, Pizzuti A, Merlini L, Angelini C, Novelli G, Dallapiccola B. Survival motor neuron gene transcript analysis in muscles from spinal muscular atrophy patients. Biochem Biophys Res Commun. 1995;213(1):342–8. doi: 10.1006/bbrc.1995.2135. doi:S0006-291X(85)72135-3 [pii] 10.1006/bbrc.1995.2135. [DOI] [PubMed] [Google Scholar]
- 19.Parsons DW, McAndrew PE, Monani UR, Mendell JR, Burghes AH, Prior TW. An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet. 1996;5(11):1727–32. doi: 10.1093/hmg/5.11.1727. [DOI] [PubMed] [Google Scholar]
- 20.Setola V, Terao M, Locatelli D, Bassanini S, Garattini E, Battaglia G. Axonal-SMN (a-SMN), a protein isoform of the survival motor neuron gene, is specifically involved in axonogenesis. Proc Natl Acad Sci U S A. 2007;104(6):1959–64. doi: 10.1073/pnas.0610660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Prior TW, Krainer AR, Hua Y, Swoboda KJ, Snyder PC, Bridgeman SJ, Burghes AH, Kissel JT. A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet. 2009;85(3):408–13. doi: 10.1016/j.ajhg.2009.08.002. doi:S0002-9297(09)00345-0 [pii] 10.1016/j.ajhg.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cartegni L, Hastings ML, Calarco JA, Stanchina E, Krainer AR. Determinants of Exon 7 Splicing in the Spinal Muscular Atrophy Genes, SMN1 and SMN2. Am J Hum Genet. 2006;78(1):63–77. doi: 10.1086/498853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Skordis LA, Dunckley MG, Yue B, Eperon IC, Muntoni F. Bifunctional antisense oligonucleotides provide a trans-acting splicing enhancer that stimulates SMN2 gene expression in patient fibroblasts. Proc Natl Acad Sci U S A. 2003;100(7):4114–9. doi: 10.1073/pnas.0633863100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kashima T, Manley JL. A negative element in SMN2 exon 7 inhibits splicing in spinal muscular atrophy. Nat Genet. 2003;34(4):460–3. doi: 10.1038/ng1207. [DOI] [PubMed] [Google Scholar]
- 25.Kashima T, Rao N, David CJ, Manley JL. hnRNP A1 functions with specificity in repression of SMN2 exon 7 splicing. Hum Mol Genet. 2007;16(24):3149–59. doi: 10.1093/hmg/ddm276. doi:ddm276 [pii] 10.1093/hmg/ddm276. [DOI] [PubMed] [Google Scholar]
- 26.Lim SR, Hertel KJ. Modulation of survival motor neuron pre-mRNA splicing by inhibition of alternative 3' splice site pairing. J Biol Chem. 2001;276(48):45476–83. doi: 10.1074/jbc.M107632200. [DOI] [PubMed] [Google Scholar]
- 27.Martins de Araujo M, Bonnal S, Hastings ML, Krainer AR, Valcarcel J. Differential 3' splice site recognition of SMN1 and SMN2 transcripts by U2AF and U2 snRNP. RNA. 2009;15(4):515–23. doi: 10.1261/rna.1273209. doi:rna.1273209 [pii] 10.1261/rna.1273209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singh NN, Androphy EJ, Singh RN. In vivo selection reveals combinatorial controls that define a critical exon in the spinal muscular atrophy genes. Rna. 2004;10(8):1291–305. doi: 10.1261/rna.7580704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hofmann Y, Lorson CL, Stamm S, Androphy EJ, Wirth B. Htra2-beta 1 stimulates an exonic splicing enhancer and can restore full-length SMN expression to survival motor neuron 2 (SMN2). Proc Natl Acad Sci U S A. 2000;97(17):9618–23. doi: 10.1073/pnas.160181697. doi:10.1073/pnas.160181697 160181697 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young PJ, DiDonato CJ, Hu D, Kothary R, Androphy EJ, Lorson CL. SRp30c-dependent stimulation of survival motor neuron (SMN) exon 7 inclusion is facilitated by a direct interaction with hTra2 beta 1. Hum Mol Genet. 2002;11(5):577–87. doi: 10.1093/hmg/11.5.577. [DOI] [PubMed] [Google Scholar]
- 31.Hofmann Y, Wirth B. hnRNP-G promotes exon 7 inclusion of survival motor neuron (SMN) via direct interaction with Htra2-beta1. Hum Mol Genet. 2002;11(17):2037–49. doi: 10.1093/hmg/11.17.2037. [DOI] [PubMed] [Google Scholar]
- 32.Chen HH, Chang JG, Lu RM, Peng TY, Tarn WY. The RNA binding protein hnRNP Q modulates the utilization of exon 7 in the survival motor neuron 2 (SMN2) gene. Mol Cell Biol. 2008;28(22):6929–38. doi: 10.1128/MCB.01332-08. doi:MCB.01332-08 [pii] 10.1128/MCB.01332-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh NK, Singh NN, Androphy EJ, Singh RN. Splicing of a critical exon of human survival motor neuron is regulated by a unique silencer element located in the last intron. Mol Cell Biol. 2006;26(4):1333–46. doi: 10.1128/MCB.26.4.1333-1346.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh NN, Singh RN, Androphy EJ. Modulating role of RNA structure in alternative splicing of a critical exon in the spinal muscular atrophy genes. Nucleic Acids Res. 2007;35(2):371–89. doi: 10.1093/nar/gkl1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baughan TD, Dickson A, Osman EY, Lorson CL. Delivery of bifunctional RNAs that target an intronic repressor and increase SMN levels in an animal model of spinal muscular atrophy. Hum Mol Genet. 2009 doi: 10.1093/hmg/ddp076. doi:ddp076 [pii] 10.1093/hmg/ddp076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miyajima H, Miyaso H, Okumura M, Kurisu J, Imaizumi K. Identification of a cis-acting element for the regulation of SMN exon 7 splicing. J Biol Chem. 2002;277(26):23271–7. doi: 10.1074/jbc.M200851200. doi:10.1074/jbc.M200851200 M200851200 [pii] [DOI] [PubMed] [Google Scholar]
- 37.Miyaso H, Okumura M, Kondo S, Higashide S, Miyajima H, Imaizumi K. An intronic splicing enhancer element in survival motor neuron (SMN) pre-mRNA. J Biol Chem. 2003;278(18):15825–31. doi: 10.1074/jbc.M209271200. doi:10.1074/jbc.M209271200 M209271200 [pii] [DOI] [PubMed] [Google Scholar]
- 38.Singh NN, Shishimorova M, Cao LC, Gangwani L, Singh RN. A short antisense oligonucleotide masking a unique intronic motif prevents skipping of a critical exon in spinal muscular atrophy. RNA Biol. 2009;6(3):341–50. doi: 10.4161/rna.6.3.8723. doi:8723 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82(4):834–48. doi: 10.1016/j.ajhg.2008.01.014. doi:S0002-9297(08)00163-8 [pii] 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gladman JT, Chandler DS. Intron 7 conserved sequence elements regulate the splicing of the SMN genes. Hum Genet. 2009 doi: 10.1007/s00439-009-0733-7. doi:10.1007/s00439-009-0733-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kashima T, Rao N, Manley JL. An intronic element contributes to splicing repression in spinal muscular atrophy. Proc Natl Acad Sci U S A. 2007;104(9):3426–31. doi: 10.1073/pnas.0700343104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schrank B, Gotz R, Gunnersen JM, Ure JM, Toyka KV, Smith AG, Sendtner M. Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci U S A. 1997;94(18):9920–5. doi: 10.1073/pnas.94.18.9920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hsieh-Li HM, Chang JG, Jong YJ, Wu MH, Wang NM, Tsai CH, Li H. A mouse model for spinal muscular atrophy. Nat Genet. 2000;24(1):66–70. doi: 10.1038/71709. [DOI] [PubMed] [Google Scholar]
- 44.Gogliotti RG, Hammond SM, Lutz C, Didonato CJ. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2009 doi: 10.1016/j.bbrc.2009.11.090. doi:S0006-291X(09)02267-0 [pii] 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michaud M, Arnoux T, Bielli S, Durand E, Rotrou Y, Jablonka S, Robert F, Giraudon-Paoli M, Riessland M, Mattei MG, Andriambeloson E, Wirth B, Sendtner M, Gallego J, Pruss RM, Bordet T. Neuromuscular defects and breathing disorders in a new mouse model of spinal muscular atrophy. Neurobiol Dis. 2010 doi: 10.1016/j.nbd.2010.01.006. doi:S0969-9961(10)00008-2 [pii] 10.1016/j.nbd.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 46.Cartegni L, Krainer AR. Correction of disease-associated exon skipping by synthetic exon-specific activators. Nat Struct Biol. 2003;10(2):120–5. doi: 10.1038/nsb887. [DOI] [PubMed] [Google Scholar]
- 47.Hua Y, Vickers TA, Baker BF, Bennett CF, Krainer AR. Enhancement of SMN2 exon 7 inclusion by antisense oligonucleotides targeting the exon. PLoS Biol. 2007;5(4):e73. doi: 10.1371/journal.pbio.0050073. doi:06-PLBI-RA-1492R3 [pii] 10.1371/journal.pbio.0050073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams JH, Schray RC, Patterson CA, Ayitey SO, Tallent MK, Lutz GJ. Oligonucleotide-mediated survival of motor neuron protein expression in CNS improves phenotype in a mouse model of spinal muscular atrophy. J Neurosci. 2009;29(24):7633–8. doi: 10.1523/JNEUROSCI.0950-09.2009. doi:29/24/7633 [pii] 10.1523/JNEUROSCI.0950-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baughan T, Shababi M, Coady TH, Dickson AM, Tullis GE, Lorson CL. Stimulating full-length SMN2 expression by delivering bifunctional RNAs via a viral vector. Mol Ther. 2006;14(1):54–62. doi: 10.1016/j.ymthe.2006.01.012. doi:S1525-0016(06)00055-4 [pii] 10.1016/j.ymthe.2006.01.012. [DOI] [PubMed] [Google Scholar]
- 50.Marquis J, Meyer K, Angehrn L, Kampfer SS, Rothen-Rutishauser B, Schumperli D. Spinal muscular atrophy: SMN2 pre-mRNA splicing corrected by a U7 snRNA derivative carrying a splicing enhancer sequence. Mol Ther. 2007;15(8):1479–86. doi: 10.1038/sj.mt.6300200. doi:6300200 [pii] 10.1038/sj.mt.6300200. [DOI] [PubMed] [Google Scholar]
- 51.Meyer K, Marquis J, Trub J, Nlend Nlend R, Verp S, Ruepp MD, Imboden H, Barde I, Trono D, Schumperli D. Rescue of a severe mouse model for spinal muscular atrophy by U7 snRNA-mediated splicing modulation. Hum Mol Genet. 2009;18(3):546–55. doi: 10.1093/hmg/ddn382. doi:ddn382 [pii] 10.1093/hmg/ddn382. [DOI] [PubMed] [Google Scholar]
- 52.Madocsai C, Lim SR, Geib T, Lam BJ, Hertel KJ. Correction of SMN2 Pre-mRNA splicing by antisense U7 small nuclear RNAs. Mol Ther. 2005;12(6):1013–22. doi: 10.1016/j.ymthe.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 53.Geib T, Hertel KJ. Restoration of full-length SMN promoted by adenoviral vectors expressing RNA antisense oligonucleotides embedded in U7 snRNAs. PLoS One. 2009;4(12):e8204. doi: 10.1371/journal.pone.0008204. doi:10.1371/journal.pone.0008204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dickson A, Osman E, Lorson CL. A negatively acting bifunctional RNA increases survival motor neuron both in vitro and in vivo. Hum Gene Ther. 2008;19(11):1307–15. doi: 10.1089/hum.2008.067. doi:10.1089/hgt.2008.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coady TH, Shababi M, Tullis GE, Lorson CL. Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol Ther. 2007;15(8):1471–8. doi: 10.1038/sj.mt.6300222. doi:6300222 [pii] 10.1038/sj.mt.6300222. [DOI] [PubMed] [Google Scholar]
- 56.Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS One. 2008;3(10):e3468. doi: 10.1371/journal.pone.0003468. doi:10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Andreassi C, Angelozzi C, Tiziano FD, Vitali T, De Vincenzi E, Boninsegna A, Villanova M, Bertini E, Pini A, Neri G, Brahe C. Phenylbutyrate increases SMN expression in vitro: relevance for treatment of spinal muscular atrophy. Eur J Hum Genet. 2004;12(1):59–65. doi: 10.1038/sj.ejhg.5201102. doi:10.1038/sj.ejhg.5201102 5201102 [pii] [DOI] [PubMed] [Google Scholar]
- 58.Sumner CJ, Huynh TN, Markowitz JA, Perhac JS, Hill B, Coovert DD, Schussler K, Chen X, Jarecki J, Burghes AH, Taylor JP, Fischbeck KH. Valproic acid increases SMN levels in spinal muscular atrophy patient cells. Ann Neurol. 2003;54(5):647–54. doi: 10.1002/ana.10743. doi:10.1002/ana.10743. [DOI] [PubMed] [Google Scholar]
- 59.Brichta L, Hofmann Y, Hahnen E, Siebzehnrubl FA, Raschke H, Blumcke I, Eyupoglu IY, Wirth B. Valproic acid increases the SMN2 protein level: a well-known drug as a potential therapy for spinal muscular atrophy. Hum Mol Genet. 2003;12(19):2481–9. doi: 10.1093/hmg/ddg256. doi:10.1093/hmg/ddg256 ddg256 [pii] [DOI] [PubMed] [Google Scholar]
- 60.Jarecki J, Chen X, Bernardino A, Coovert DD, Whitney M, Burghes A, Stack J, Pollok BA. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum Mol Genet. 2005;14(14):2003–18. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 61.Hahnen E, Eyupoglu IY, Brichta L, Haastert K, Trankle C, Siebzehnrubl FA, Riessland M, Holker I, Claus P, Romstock J, Buslei R, Wirth B, Blumcke I. In vitro and ex vivo evaluation of second-generation histone deacetylase inhibitors for the treatment of spinal muscular atrophy. J Neurochem. 2006;98(1):193–202. doi: 10.1111/j.1471-4159.2006.03868.x. doi:JNC3868 [pii] 10.1111/j.1471-4159.2006.03868.x. [DOI] [PubMed] [Google Scholar]
- 62.Chang JG, Hsieh-Li HM, Jong YJ, Wang NM, Tsai CH, Li H. Treatment of spinal muscular atrophy by sodium butyrate. Proc Natl Acad Sci U S A. 2001;98(17):9808–13. doi: 10.1073/pnas.171105098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garbes L, Riessland M, Holker I, Heller R, Hauke J, Trankle C, Coras R, Blumcke I, Hahnen E, Wirth B. LBH589 induces up to 10-fold SMN protein levels by several independent mechanisms and is effective even in cells from SMA patients non-responsive to valproate. Hum Mol Genet. 2009;18(19):3645–58. doi: 10.1093/hmg/ddp313. doi:ddp313 [pii] 10.1093/hmg/ddp313. [DOI] [PubMed] [Google Scholar]
- 64.Riessland M, Brichta L, Hahnen E, Wirth B. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum Genet. 2006;120(1):101–10. doi: 10.1007/s00439-006-0186-1. doi:10.1007/s00439-006-0186-1. [DOI] [PubMed] [Google Scholar]
- 65.Grzeschik SM, Ganta M, Prior TW, Heavlin WD, Wang CH. Hydroxyurea enhances SMN2 gene expression in spinal muscular atrophy cells. Ann Neurol. 2005;58(2):194–202. doi: 10.1002/ana.20548. [DOI] [PubMed] [Google Scholar]
- 66.Andreassi C, Jarecki J, Zhou J, Coovert DD, Monani UR, Chen X, Whitney M, Pollok B, Zhang M, Androphy E, Burghes AH. Aclarubicin treatment restores SMN levels to cells derived from type I spinal muscular atrophy patients. Hum Mol Genet. 2001;10(24):2841–9. doi: 10.1093/hmg/10.24.2841. [DOI] [PubMed] [Google Scholar]
- 67.Ting CH, Lin CW, Wen SL, Hsieh-Li HM, Li H. Stat5 constitutive activation rescues defects in spinal muscular atrophy. Hum Mol Genet. 2007;16(5):499–514. doi: 10.1093/hmg/ddl482. doi:ddl482 [pii] 10.1093/hmg/ddl482. [DOI] [PubMed] [Google Scholar]
- 68.Zhang ML, Lorson CL, Androphy EJ, Zhou J. An in vivo reporter system for measuring increased inclusion of exon 7 in SMN2 mRNA: potential therapy of SMA. Gene Ther. 2001;8(20):1532–8. doi: 10.1038/sj.gt.3301550. doi:10.1038/sj.gt.3301550. [DOI] [PubMed] [Google Scholar]
- 69.Angelozzi C, Borgo F, Tiziano FD, Martella A, Neri G, Brahe C. Salbutamol increases SMN mRNA and protein levels in spinal muscular atrophy cells. J Med Genet. 2008;45(1):29–31. doi: 10.1136/jmg.2007.051177. doi:jmg.2007.051177 [pii] 10.1136/jmg.2007.051177. [DOI] [PubMed] [Google Scholar]
- 70.Hastings ML, Berniac J, Liu YH, Abato P, Jodelka FM, Barthel L, Kumar S, Dudley C, Nelson M, Larson K, Edmonds J, Bowser T, Draper M, Higgins P, Krainer AR. Tetracyclines That Promote SMN2 Exon 7 Splicing as Therapeutics for Spinal Muscular Atrophy. Science Translational Medicine. 2009;1(5):5ra12–5ra12. doi: 10.1126/scitranslmed.3000208. doi:10.1126/scitranslmed.3000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakla MS, Lorson CL. Induction of full-length survival motor neuron by polyphenol botanical compounds. Hum Genet. 2008;122(6):635–43. doi: 10.1007/s00439-007-0441-0. doi:10.1007/s00439-007-0441-0. [DOI] [PubMed] [Google Scholar]
- 72.Yuo CY, Lin HH, Chang YS, Yang WK, Chang JG. 5-(N-ethyl-N-isopropyl)-amiloride enhances SMN2 exon 7 inclusion and protein expression in spinal muscular atrophy cells. Ann Neurol. 2008;63(1):26–34. doi: 10.1002/ana.21241. doi:10.1002/ana.21241. [DOI] [PubMed] [Google Scholar]
- 73.Tsai LK, Tsai MS, Ting CH, Li H. Multiple therapeutic effects of valproic acid in spinal muscular atrophy model mice. J Mol Med. 2008;86(11):1243–54. doi: 10.1007/s00109-008-0388-1. doi:10.1007/s00109-008-0388-1. [DOI] [PubMed] [Google Scholar]
- 74.Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117(3):659–71. doi: 10.1172/JCI29562. doi:10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marce M, Taye AA, Eckhaus MA, Sumner CJ. Sustained improvement of spinal muscular atrophy mice treated with trichostatin A plus nutrition. Ann Neurol. 2008;64(4):465–70. doi: 10.1002/ana.21449. doi:10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mattis VB, Butchbach ME, Lorson CL. Detection of human survival motor neuron (SMN) protein in mice containing the SMN2 transgene: applicability to preclinical therapy development for spinal muscular atrophy. J Neurosci Methods. 2008;175(1):36–43. doi: 10.1016/j.jneumeth.2008.07.024. doi:S0165-0270(08)00454-8 [pii] 10.1016/j.jneumeth.2008.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Brahe C, Vitali T, Tiziano FD, Angelozzi C, Pinto AM, Borgo F, Moscato U, Bertini E, Mercuri E, Neri G. Phenylbutyrate increases SMN gene expression in spinal muscular atrophy patients. Eur J Hum Genet. 2005;13(2):256–9. doi: 10.1038/sj.ejhg.5201320. doi:5201320 [pii] 10.1038/sj.ejhg.5201320. [DOI] [PubMed] [Google Scholar]
- 78.Mercuri E, Bertini E, Messina S, Pelliccioni M, D'Amico A, Colitto F, Mirabella M, Tiziano FD, Vitali T, Angelozzi C, Kinali M, Main M, Brahe C. Pilot trial of phenylbutyrate in spinal muscular atrophy. Neuromuscul Disord. 2004;14(2):130–5. doi: 10.1016/j.nmd.2003.11.006. doi:S0960896603002591 [pii] [DOI] [PubMed] [Google Scholar]
- 79.Mercuri E, Bertini E, Messina S, Solari A, D'Amico A, Angelozzi C, Battini R, Berardinelli A, Boffi P, Bruno C, Cini C, Colitto F, Kinali M, Minetti C, Mongini T, Morandi L, Neri G, Orcesi S, Pane M, Pelliccioni M, Pini A, Tiziano FD, Villanova M, Vita G, Brahe C. Randomized, double-blind, placebo-controlled trial of phenylbutyrate in spinal muscular atrophy. Neurology. 2007;68(1):51–5. doi: 10.1212/01.wnl.0000249142.82285.d6. doi:01.wnl.0000249142.82285.d6 [pii] 10.1212/01.wnl.0000249142.82285.d6. [DOI] [PubMed] [Google Scholar]
- 80.Swoboda KJ, Scott CB, Reyna SP, Prior TW, LaSalle B, Sorenson SL, Wood J, Acsadi G, Crawford TO, Kissel JT, Krosschell KJ, D'Anjou G, Bromberg MB, Schroth MK, Chan GM, Elsheikh B, Simard LR. Phase II open label study of valproic acid in spinal muscular atrophy. PLoS One. 2009;4(5):e5268. doi: 10.1371/journal.pone.0005268. doi:10.1371/journal.pone.0005268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tsai LK, Yang CC, Hwu WL, Li H. Valproic acid treatment in six patients with spinal muscular atrophy. Eur J Neurol. 2007;14(12):e8–9. doi: 10.1111/j.1468-1331.2007.01992.x. doi:ENE1992 [pii] 10.1111/j.1468-1331.2007.01992.x. [DOI] [PubMed] [Google Scholar]
- 82.Weihl CC, Connolly AM, Pestronk A. Valproate may improve strength and function in patients with type III/IV spinal muscle atrophy. Neurology. 2006;67(3):500–1. doi: 10.1212/01.wnl.0000231139.26253.d0. doi:01.wnl.0000231139.26253.d0 [pii] 10.1212/01.wnl.0000231139.26253.d0. [DOI] [PubMed] [Google Scholar]
- 83.Brichta L, Holker I, Haug K, Klockgether T, Wirth B. In vivo activation of SMN in spinal muscular atrophy carriers and patients treated with valproate. Ann Neurol. 2006;59(6):970–5. doi: 10.1002/ana.20836. doi:10.1002/ana.20836. [DOI] [PubMed] [Google Scholar]
- 84.Liang WC, Yuo CY, Chang JG, Chen YC, Chang YF, Wang HY, Ju YH, Chiou SS, Jong YJ. The effect of hydroxyurea in spinal muscular atrophy cells and patients. J Neurol Sci. 2008;268(1-2):87–94. doi: 10.1016/j.jns.2007.11.012. doi:S0022-510X(07)00736-8 [pii] 10.1016/j.jns.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 85.Pane M, Staccioli S, Messina S, D'Amico A, Pelliccioni M, Mazzone ES, Cuttini M, Alfieri P, Battini R, Main M, Muntoni F, Bertini E, Villanova M, Mercuri E. Daily salbutamol in young patients with SMA type II. Neuromuscul Disord. 2008;18(7):536–40. doi: 10.1016/j.nmd.2008.05.004. doi:S0960-8966(08)00134-X [pii] 10.1016/j.nmd.2008.05.004. [DOI] [PubMed] [Google Scholar]