

Abstract
Methods for Ni-catalyzed hydroalumination of terminal alkynes, leading to the formation of α-vinylaluminum isomers efficiently (>98% conv in 2–12 h) and with high selectivity (95% to >98% α), are described. Catalytic α-selective hydroalumination reactions proceed in the presence of a reagent (diisobutylaluminum hydride; dibal–H) and 3.0 mol % metal complex (Ni(dppp)Cl2) that are commercially available and inexpensive. Under the same conditions, but with Ni(PPh3)2Cl2, hydroalumination becomes highly β-selective, and, unlike uncatalyzed transformations with dibal–H, generates little or no alkynylaluminum byproducts. All hydrometallation reactions are reliable, operationally simple and practical, and afford an assortment of vinylaluminums that are otherwise not easily accessible. The derived α-vinyl halides and boronates can be synthesized through direct treatment with the appropriate electrophiles [e.g., Br2 and methoxy(pinacolato)boron, respectively]. Ni-catalyzed hydroaluminations can be performed with as little as 0.1 mol % catalyst and on gram scale with equally high efficiency and selectivity.

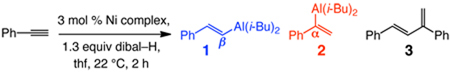
Herein, we disclose that commercially available and inexpensive Ni-based catalysts and diisobutylaluminum hydride (dibal–H) convert alkyl- or aryl-substituted alkynes to internal (or α-) vinylaluminums efficiently and with exceptional selectivity (95% to >98% α isomer). The catalytic protocol offers a one-pot, high yielding and selective method for conversion of terminal alkynes to α-vinyl halides1 and boronates2 – valuable entities otherwise prepared by less efficient routes that are intolerant of several functional groups.3 Moreover, aryl-substituted terminal β-vinylaluminums, formed along with substantial amounts of byproducts in uncatalyzed hydroaluminations, can be accessed in high yield by the catalytic process.
Selective and practical methods for alkyne hydrometallations are of substantial value.4 The resulting vinylmetals might be utilized to access vinyl halides and boronates, intermediates critical to catalytic cross-coupling, one of the most important classes of transformations in chemical synthesis.5,6 Vinylaluminums are mildly nucleophilic reagents that can be used in catalytic C–C bond forming processes,7 including those that are enantioselective.8 Alkyl-substituted vinylaluminums can be prepared9 by reaction of alkynes with dibal–H;10 the corresponding processes with aryl alkynes, however, suffer from competitive alkyne deprotonation, affording significant amounts of alkynylaluminums.11,12 Regardless of varying efficiencies, all existing terminal alkyne hydroaluminations favor β-substituted isomers strongly or, often, exclusively. In contrast, a procedure that directly delivers internal or α-substituted vinylaluminums (eq 1) reliably and selectively does not exist. We reasoned that the above shortcomings might be resolved by rendering alkyne hydroalumination catalytic (eq 1), and that α selectivity might be achievable through manipulation of a catalyst’s structure and the energetics of the catalytic cycle.13 An efficient catalytic hydroalumination might also facilitate metal hydride addition to such a degree that adventitious alkyne deprotonation is avoided.
 |
(1) |
To initiate our search for an effective alkyne hydroalumination catalyst, we focused on Ni complexes, partly based on a report by Eisch regarding reactions of a small number of disubstituted alkynes promoted by Ni(acac)2.14 The outcome of our screening studies, with phenylacetylene serving as the substrate, is summarized in Table 1. Reaction with Ni(acac)2 and 1.3 equiv of dibal–H in tetrahydrofuran (thf) at 22 °C for two hours leads to efficient hydrometallation (entry 1) but with a modest preference for the terminal vinylaluminum (68% 1 and 19% 2) along with 13% of 1,3-diene 3. Use of NiCl2•6H2O (entry 2) or Ni(0) complexes (entries 3–4) lead to similarly low selectivity and significant amounts of 3. With 3 mol % Ni(PPh3)2Cl2 (entry 5), hydroalumination proceeds with a strong preference for the terminal vinylaluminum (1:2 = 93:7) and with <2% of the diene contaminant (1H NMR analysis). More importantly, as depicted in entries 6–9 of Table 1, when Ni catalysts bear a bidentate phosphine ligand, hydrometallation occurs with total reversal of site selectivity. Vinylaluminum 2 is formed efficiently (>98% conv) and with >98% α selectivity when Ni(dppp)Cl2 is used (entry 7).
Table 1.
Ni-Catalyzed Hydroalumination of Phenylacetylenea
 | |||
|---|---|---|---|
| entry | Ni complex | conv (%)b | 1:2:3 (%)b |
| 1 | Ni(acac)2 | 97 | 68:19:13 |
| 2 | NiCl2•6H2O | 95 | 45:35:20 |
| 3 | Ni(cod)2 | >98 | 69:14:17 |
| 4 | Ni(PPh3)4 | >98 | 55:32:13 |
| 5 | Ni(PPh3)2Cl2 | >98 | 93:7:<2 |
| 6 | Ni(dppe)Cl2 | 92 | 3:97:<2 |
| 7 | Ni(dppp)Cl2 | >98 | <2:>98:<2 |
| 8 | Ni(dppb)Cl2 | 76 | 8:92:<2 |
| 9 | Ni(dppf)Cl2 | 75 | 5:95:<2 |
Reactions performed under N2 atm; <2% alkynyl aluminum product observed in all cases.
By analysis of 400 MHz 1H NMR spectra of unpurified mixtures after quench with D2O (0 °C, 30 min); see the Supporting Information for details.
It must be noted that, in the absence of a catalyst, dibal–H reacts with phenylacetylene at 50 °C (hexanes or toluene)15 to afford only ~50% of the β-vinylaluminum (1) along with 25% styrene and 25% alkynylaluminum (due to deprotonation of alkyne by the vinylaluminum). In sharp contrast, alkyne deprotonation is not observed in any of the Ni-catalyzed reactions (<2%). Thus, in addition to allowing effective control of α or β selectivity, Ni complexes enhance hydrometallation rates to the extent that adventitious deprotonation is entirely obviated.
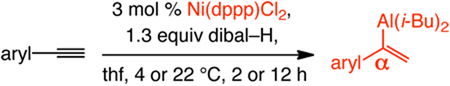
Catalytic hydroaluminations can be performed with a considerable range of aryl alkynes (Table 2), affording α-vinylmetals efficiently and selectively. Transformations are performed at 4–22 °C and require ≤12 hours. Alkynes bearing electron-donating (entries 1–3) or electron-withdrawing units (entries 4–8) or sterically congested aryl acetylenes (entries 7–9) are suitable substrates. N- or S-containing heterocyclic substrates readily undergo hydroalumination with high selectivity (entries 10–11). Two additional points are noteworthy: (1) None of the transformations with Ni(dppp)Cl2 generate the alkynylaluminum side product, which is obtained in significantly larger amounts in the absence of the catalyst (see above). (2) Reactions can be performed with commercial grade thf and Ni salts that are used as received (see below for example).
Table 2.
α-Selective Ni-Catalyzed Hydroalumination of Arylacetylenesa
 | |||||
|---|---|---|---|---|---|
| entry | aryl | temp (°C) | time (h)b | conv (%)c | α:βc |
| 1 | o-OMeC6H4 | 4 | 12 | >98 | 98:2 |
| 2 | m-OMeC6H4 | 22 | 2 | >98 | >98:2 |
| 3 | p-OMeC6H4 | 4 | 12 | >98 | >98:2 |
| 4 | m-CF3C6H4 | 4 | 12 | >98 | 95:5 |
| 5 | p-CF3C6H4 | 22 | 2 | >98 | 97:3 |
| 6 | p-FC6H4 | 22 | 2 | >98 | >98:2 |
| 7 | o-ClC6H4 | 22 | 2 | >98 | >98:2 |
| 8 | o-BrC6H4 | 22 | 2 | >98 | >98:2 |
| 9 | o-MeC6H4 | 4 | 12 | >98 | >98:2 |
| 10 | 3-pyridyl | 22 | 2 | >98 | >98:2 |
| 11 | 3-pyridyl | 22 | 2 | >98 | >98:2 |
Reactions under N2 atm.
Reaction times correspond to hydroalumination portion of the process (not including D2O quench).
By analysis of 400 MHz 1H NMR spectra of unpurified mixtures (after D2O); <2% alkynylaluminum observed.
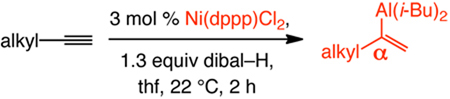
α-Selective catalytic hydroaluminations of alkyl-substituted alkynes proceed readily and with high selectivity as well (Table 3);16 only in one case (entry 6), ~5% alkyne deprotonation is observed (<2% otherwise). Alkynes bearing a linear (entries 1–4), α- (entries 5–6) or β-branched (entry 7) alkyl unit can be used.
Table 3.
α-Selective Ni-Catalyzed Hydroalumination of Alkylacetylenesa
 | |||
|---|---|---|---|
| entry | product | conv (%)b | α:βc |
| 1 | >98 | >98:2 | |
| 2 |  |
>98 | 97:3 |
| 3 | >98 | >98:2 | |
| 4 | 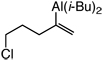 |
>98 | >98:2 |
| 5 | >98 | >98:2 | |
| 6 | 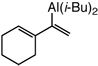 |
>98d | 98:2 |
| 7 | >98 | 96.5:3.5 | |
See Table 2; 2.3 equiv dibal–H in entry 2; 3 h for entry 4.
Performed at 4 °C for 12 h; ~5% alkyne deprotonation observed.
The same range of substrates shown in Table 2 undergo hydroalumination readily in the presence of Ni(PPh3)2Cl2, proceeding with high β-selectivity (85.5% to >98%) and with minimal alkyne deprotonation; complete data is available in the Supporting Information. Only alkyl-substituted β-vinylaluminums, which can be accessed by (uncatalyzed) reaction of dibal–H with a terminal alkyne, have so far been utilized in metal-catalyzed enantioselective C–C bond formation.8 Efficient access to a wider range of α- and β-vinylaluminums would substantially expand the utility of such reagents. Cu-catalyzed enantioselective alkylations in Scheme 1 demonstrate this point. An aryl-substituted β-vinylaluminum, obtained through the above Ni-catalyzed process, can be used to synthesize 1,4-diene 4 with high efficiency and in 96:4 er. In contrast, when the vinylmetal prepared by uncatalyzed hydroalumination is utilized, 26% of the undesired alkyne (vs vinyl) addition product is generated. Furthermore, synthesis of enantiomerically enriched 1,4-dienes 5 and 6 is rendered feasible because the requisite α-vinylaluminums can now be easily prepared.
Scheme 1.

Applications of Ni-Catalyzed Hydroaluminationsa
a Synthesis of 5 and 6 at –15 °C. Ar = 2,4,6-(i-Pr)3-C6H2.
Ni-catalyzed vinylaluminum synthesis is more efficient than other alternatives, which involve initial synthesis of a vinyl halide, followed by metal-halogen exchange (n-BuLi) and treatment with an aluminum chloride. In addition to being a lengthier route, the latter two-vessel protocol requires α-vinyl halides prepared from terminal alkynes by hydroiodation (HI)1c–d or halo-boration (BBr3)/proto-deboration with 15–20 equiv HOAc),17 which constitute strongly acidic conditions (specific examples below).
Ni-catalyzed hydroalumination does more than obviate the need for intermediacy of vinyl halides and the attendant harsh conditions leading to vinylaluminums; it provides an efficient route for the synthesis of this valuable class of halogenated building blocks. Treatment of the α-vinylaluminums with N-bromo- or N-iodosuccinimide furnishes vinyl bromides or iodides in >98% α-selectivity and 79–88% yield after chromatography (Scheme 2). Synthesis of α-vinylbromide 7 or iodide 11 would not be feasible through the BBr3/HOAc,1a hydroiodation1c–d,18 (concomitant removal of methyl or silyl ether). Vinyl halide synthesis through Ni-catalyzed hydroalumination is a practical process. Iodide 8 is obtained in 83% yield and >98% α selectivity with commercial grade thf and Ni catalyst (not purified); similarly, the alkyl-substituted iodide formed by catalytic hydroalumination of 1-octyne and treatment with N-iodosuccinimide is obtained in 89% yield and 96% α selectivity.
Scheme 2.

Conversion of Terminal Alkynes to α-Vinyl Halides
a Reaction with N-iodosuccinimide under otherwise identical conditions. See the Supporting Information for experimental details.
Ni-catalyzed hydroalumination and vinyl halide synthesis is amenable to scale up and requires low catalyst loadings. As an example, with only 0.1 mol % Ni(dppp)Cl2 and with inexpensive Br2 as the halide source, α-vinyl bromide 12 is obtained in 69% yield after purification (eq 2; <2% β).19
 |
(2) |
α Vinylboronates can be readily accessed through Ni-catalyzed alkyne hydroalumination (Scheme 3). Direct subjection of vinylaluminums with commercially available and inexpensive methoxy(pinacolato)borane delivers the desired products in >98% α selectivity and 68–94% yield after purification.20
Scheme 3.

One Pot Synthesis of α-Vinyl Boronatesa
a Yields refer to purified α-vinylboronates; see the Supporting Information for experimental details. B(pin) = pinacolatoboron.
Several issues that underline the unique attributes of the α-vinyl boronate synthesis method described herein merit specific mention: (1) Hydroborations of terminal alkynes with pinacolborane, which require rigorously dried CH2Cl2,21 or Rh-, Ir-22 and Zr-catalyzed23 variants, furnish β-vinyl boronates exclusively or as the major isomer (<10% α).24 (2) Alkyl-substituted α-vinyl boronates can be accessed through hydroborations that require stoichiometric amounts of a Cu complex (1.1 equiv CuCl, KOAc, LiCl or a phosphine), and only up to 91% selectivity (typically 9–71%).25 (3) α-Vinylboronates can be synthesized from α-vinylbromides by metal/halogen exchange (n-BuLi) and subjection to i-propoxypinacolborane.17b Cross-coupling of vinyl halides with B2(pin)2, promoted by the more costly Pd salts, also afford α-vinylboronates.2 In addition to being two-vessel protocols (vs one-pot in Scheme 3), the latter processes require vinyl halides prepared by severely acidic procedures, rendering several vinyl halides inaccessible (see above). The present approach furnishes the vinylmetal directly. Finally, a vinylboronate that bears an aryl halide (e.g., 15, Scheme 3) cannot be selectively prepared by the aforementioned metal/halogen exchange/boronate trap or Pd-catalyzed cross-coupling of the corresponding vinyl halide.
Investigations regarding the basis of selectivity reversal in Ni-catalyzed hydroaluminations26 and applications to other classes of alkynes as well as hydride sources are in progress.
Supplementary Material
Acknowledgment
Financial support was provided by the NIH (GM-47480). Mass spectrometry facilities at Boston College are supported by the NSF (DBI-0619576).
Footnotes
Supporting Information Available: Experimental procedures and spectral, analytical data for all products (PDF). This material is available on the web: http://www.pubs.acs.org
References
- 1.For acyclic α-vinyl halide synthesis, see: (a) Bromoboration/protodeboration of alkyl-substituted alkynes: Hara S, Dojo H, Takinami S, Suzuki A. Tetrahedron Lett. 1983;24:731. (b) Cr-mediated conversion of aldehydes to iodides: Takai K, Nitta K, Utimoto K. J. Am. Chem. Soc. 1986;108:7408. doi: 10.1021/ja00279a068. (c) Terminal alkynes to iodides by hydroiodation (HI formed in situ from TMSI): Kamiya N, Chikami Y, Ishii Y. Synlett. 1990:675. (d) Ketones and aldehydes to iodides with phosphitohalides: Spaggiari A, Vaccari D, Davoli P, Torre G, Prati F. J. Org. Chem. 2007;72:2216. doi: 10.1021/jo061346g. (e) Terminal alkynes (no branched alkyl-substituted cases) to iodides by HI addition (generated from iodine/hydrophosphine): Kawaguchi S-i, Ogawa A. Org. Lett. 2010;12:1893. doi: 10.1021/ol1005246.
- 2.For synthesis of various cyclic and acyclic vinylboronates through Pd-catalyzed cross-coupling reactions involving the corresponding vinyl bromides and triflates, see: Takagi J, Takahashi K, Ishiyama T, Miyaura N. J. Am. Chem. Soc. 2002;124:8001. doi: 10.1021/ja0202255.
- 3.For α-selective Ru-catalyzed hydrosilylation of alkynes, see: Trost BM, Ball ZT. J. Am. Chem. Soc. 2005;127:17644. doi: 10.1021/ja0528580.
- 4.For a review on hydroaluminations of alkynes and alkenes, see: Eisch JJ. In: Comprehensive Organic Synthesis. Trost BM, Fleming I, Schreiber SL, editors. Vol. 8. Oxford: Pergamon; 1991. p. 733.
- 5.For representative transformations where vinyl iodides are used in complex molecule synthesis, see: Liu X, Henderson JA, Sasaki T, Kishi Y. J. Am. Chem. Soc. 2009;131:16678. doi: 10.1021/ja9079308. and references cited therein.
- 6.For applications of vinylboronates in C–C bond formation, see: Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. Kotha S, Lahiri K, Kashinath D. Tetrahedron. 2002;58:9633.
- 7.For example, see: Baba S, Negishi E-i. J. Am. Chem. Soc. 1976;98:6729. Negishi E-i, Okukado N, King AO, Van Horn DE, Spiegel BI. J. Am. Chem. Soc. 1978;100:2254. Lipshutz BH, Bülow G, Lowe RF, Stevens KL. Tetrahedron. 1996;52:7265. Langille NF, Jamison TF. Org. Lett. 2006;8:3761. doi: 10.1021/ol0613721.
- 8.Allylic alkylations: Lee Y, Akiyama K, Gillingham DG, Brown MK, Hoveyda AH. J. Am. Chem. Soc. 2008;130:446. doi: 10.1021/ja0782192. Akiyama K, Gao F, Hoveyda AH. Angew. Chem., Int. Ed. 2010;49:419. doi: 10.1002/anie.200905223. Carbonyl additions: Biradar DB, Gau H-M. Org. Lett. 2009;11:499. doi: 10.1021/ol801999u.
- 9.Vinylmetals (including vinylaluminums) can be accessed through carbometallation of alkynes. For example, see: Wipf P, Lim S. Angew. Chem., Int. Ed. Engl. 1993;32:1068. Fallis AG, Forgione P. Tetrahedron. 2001;57:5899. Lipshutz BH, Butler T, Lower A, Servesko J. Org. Lett. 2007;9:3737. doi: 10.1021/ol701469e.
- 10. Zweifel G, Whitney CC. J. Am. Chem. Soc. 1967;89:2753. For use of alkyl-substituted vinylaluminums in Cu-catalyzed allylic alkylations, see: (b) Ref 8a.
- 11.(a) Zweifel G, Snow JT, Whitney CC. J. Am. Chem. Soc. 1968;90:7139. [Google Scholar]; (b) Uhl W, Er E, Hepp A, Kösters J, Grunenberg J. Organometallics. 2008;27:3346. [Google Scholar]; (c) Uhl W, Er E, Hepp A, Kösters J, Layh M, Rohling M, Vinogradov A, Würthwein E-U, Ghavtadze N. Eur. J. Inorg. Chem. 2009:3307. [Google Scholar]
- 12.Hydroalumination (dibal–H) of Si-substituted aryl alkynes, which proceeds efficiently and site-selectively, has been introduced to address this shortcoming. The C–Si bond is removed by protodesilylation. See ref 8b.
- 13.For selectivity reversal in Ni-catalyzed H–P and H–S addition to terminal alkynes, see: Han L-B, Zhang C, Yazawa H, Shimada S. J. Am. Chem. Soc. 2004;126:5080. doi: 10.1021/ja0494297.
- 14.Eisch JJ, Foxton MW. J. Organomet. Chem. 1968;12:33. [Google Scholar]
- 15.There is <2% reaction when phenylacetylene is treated with dibal–H in thf (at 22 °C, in 2 h).
- 16.In the absence of a catalyst, alkyl-substituted substrates react relatively efficiently (in contrast to aryl alkynes) with dibal–H to afford β-vinylaluminums (>98% selectivity). Such uncatalyzed reactions proceed to ~80% conversion after 6 h at 22 °C and must be heated to 55 °C (2 h) to achieve ~90% conv. The Ni-catalyzed processes (Ni(PPh3)2Cl2) are complete in 2 h at 22 °C. For data regarding this class of catalytic hydroaluminations, see the Supporting Information.
- (a) Ref 1a. Moran WJ, Morken JP. Org. Lett. 2006;8:2413. doi: 10.1021/ol060735u.
- 18.For example, see: Morrill C, Funk TW, Grubbs RH. Tetrahedron Lett. 2004;45:7733.
- 19.In a similar manner, the β-vinyl bromide derived from vinylaluminum 1 is obtained in 75% yield from a reaction performed on 10 mmol of phenylacetylene with 0.5 mol % Ni(PPh3)2Cl2.
- 20.Synthesis of α-vinylboronates by an NHC–Cu-catalyzed hydroboration of a propargyl ether and a propargyl amide was recently reported to proceed with ~90% α selectivity: Lee Y, Jang H, Hoveyda AH. J. Am. Chem. Soc. 2009;131:18234. doi: 10.1021/ja9089928.
- 21.Tucker CE, Davidson J, Knochel P. J. Org. Chem. 1992;57:3482. [Google Scholar]
- 22.(a) Pereira S, Srebnik M. Tetrahedron Lett. 1996;37:3283. [Google Scholar]; (b) Ohmura T, Yamamoto Y, Miyaura N. J. Am. Chem. Soc. 2000;122:4990. [Google Scholar]
- 23.Wang YD, Kimball G, Prashad AS, Wang Y. Tetrahedron Lett. 2005;46:8777. [Google Scholar]
- 24.For a Cu-catalyzed hydroboration of phenylacetylene that affords the terminal vinylboronate, see: Lee J-E, Kwon J, Yun J. Chem. Commun. 2008;733 doi: 10.1039/b716697d. This procedure is ineffective with alkyl-substituted alkynes.
- 25.Takahashi K, Ishiyama T, Miyaura N. J. Organomet. Chem. 2001;625:47. [Google Scholar]
- 26.Initial studies indicate that Ni(0) complexes are involved (vs Ni(II)). Subjection of phenylacetylene to the conditions shown in Table 1, but with 3 mol % Ni(dppp)Cl2 and 6 mol % MeMgI (to generate Ni(0)(dppp) complex in situ), affords 2 with 97% α selectivity (>98% conv). It is plausible that reactions proceed via an Al–Ni hydride [(i-Bu)2Al–Ni–H]; addition of Ni–H across the alkyne and subsequent alkenyl–Al reductive elimination regenerates the Ni(0) complex. See: Eisch JJ, Sexsmith SR, Fichter KC. J. Organomet. Chem. 1990;382:273. Eisch JJ, Ma X, Singh M, Wilke G. J. Organomet. Chem. 1997;527:301.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.