

Abstract
Dehaloperoxidase (DHP) from the terebellid polychaete Amphitrite ornata is a bifunctional enzyme that possesses both hemoglobin and peroxidase activities. Of the two DHP isoenzymes identified to date, much of the recent focus has been on DHP A, whereas very little is known pertaining to the activity, substrate specificity, mechanism of function, or spectroscopic properties of DHP B. Herein, we report the recombinant expression and purification of DHP B, as well as the details of our investigations into its catalytic cycle using biochemical assays, stopped-flow UV-visible, resonance Raman and rapid-freeze-quench electron paramagnetic resonance spectroscopies, and spectroelectrochemistry. Our experimental design reveals mechanistic insights and kinetic descriptions of the dehaloperoxidase mechanism which have not been previously reported for isoenzyme A. Namely, we demonstrate a novel reaction pathway in which the products of the oxidative dehalogenation of trihalophenols (dihaloquinones) are themselves capable of inducing formation of oxyferrous DHP B, and an updated catalytic cycle for DHP is proposed. We further demonstrate that unlike the traditional monofunctional peroxidases, the oxyferrous state in DHP is a peroxidase competent starting species, which suggests that the ferric oxidation state may not be an obligatory starting point for the enzyme. The data presented herein provide a link between the peroxidase and oxygen transport activities which furthers our understanding of how this bifunctional enzyme is able to unite its two inherent functions in one system.
Keywords: Dehaloperoxidase, Peroxidase, Hemoglobin, Compound ES, Iron(IV)-oxo, Protein Radical
The ‘metallo-proteome’ contains a number of enzymes which possess more than one inherent catalytic function. Dehaloperoxidase (DHP), the coelomic oxygen-transport hemoglobin from the terebellid polychaete Amphitrite ornata (1), is the first globin identified to possess a biologically relevant peroxidase activity (2). In the benthic ecosystems in which A. ornata is commonly found, DHP functions both as the O2-transport protein and to protect this marine worm against biogenically-produced halometabolites which act as repellents secreted by other organisms. As the monomeric (non-cooperative) intracellular coelomic hemoglobin, DHP binds dioxygen (P50 = 2.8 torr) that is delivered to the coelom by the extracellular, multi-subunit vascular erythrocruorin (P50 = 11 torr) (1, 3). Hemoglobin (Hb) phylogeny reveals a common genetic ancestry across species from bacteria to plants and animals extending back 1.8 billion years (4-6). However, despite DHP being categorized as a globin according to the Structural Classification of Proteins (SCOP) database (7), DHP has little sequence homology to other known Hbs. Moreover, DHP bears little resemblance to the fold of cytochrome c peroxidase (CcP), the prototype for the heme peroxidase family (8). Thus, as a representative of globins found in marine organisms, its relationship to other Hbs and peroxidases may aid in establishing the scientific foundation and new paradigms of protein structure-function relationships specific to bi/multi-functional proteins.
A number of sediment-dwelling marine polychaetes and hemichordates employ haloperoxidases to produce high levels of volatile brominated secondary metabolites as defense mechanisms (9, 10). Examples include Notomastus lobatus (polychaeta) (11-13), which contaminates the sediments with mono-, di-, and tribromophenols and mono- and dibromovinylphenols, and Saccoglossus kowalevskii (hemichordata) (14, 15), which also produces bromopyrroles. Thus, environmental sediments that are contaminated with this diverse array of toxic haloaromatic compounds represent a significant challenge to other infaunal organisms that co-inhabit these coastal mudflats. One such annelid, A. ornata, is able to oxidize a wide variety of mono-, di-, and trisubstituted halophenols that possess bromine, chlorine, or fluorine substituents (2). A. ornata is exposed to these contaminants on two fronts, by contact upon burrowing into the sediments and through ingestion as a deposit feeder consuming contaminated surface deposits, yet survives due to the production of dehalogenating enzymes that allow it to tolerate such environmental haloaromatic toxins. One such enzyme is dehaloperoxidase, a dual-function hemoprotein that, in addition to being the coelomic hemoglobin of A. ornata (1, 16, 17), possesses a broad substrate specificity for the oxidation of the aforementioned trihalophenols (2, 18). The dehaloperoxidase function of this hemoglobin was first determined by fractionation of the A. ornata proteome to determine which component of the organism was capable of degrading bromophenols (2). The high specific activity of the purified protein signified that DHP was solely responsible for the observed oxidative dehalogenation reaction, and recombinant expression of the protein further indicated that such enzymatic activity is intrinsic to DHP (18). Soon thereafter, Ely and co-workers identified and characterized two separate genes (dhpA and dhpB) that encoded for a pair of DHP isoenzymes, termed DHP A and DHP B (19). Both enzymes were found to contain 137 amino acid residues, but DHP B differs from DHP A at five positions: I9L, R32K, Y34N, N81S and S91G (Figure 1). It was hypothesized at that time that the differences between DHP A and B may result in altered co-substrate specificity, but no follow up studies were conducted. Thus, while DHP A has been the focus of many studies for well over a decade, DHP B has received minimal attention.
Figure 1.
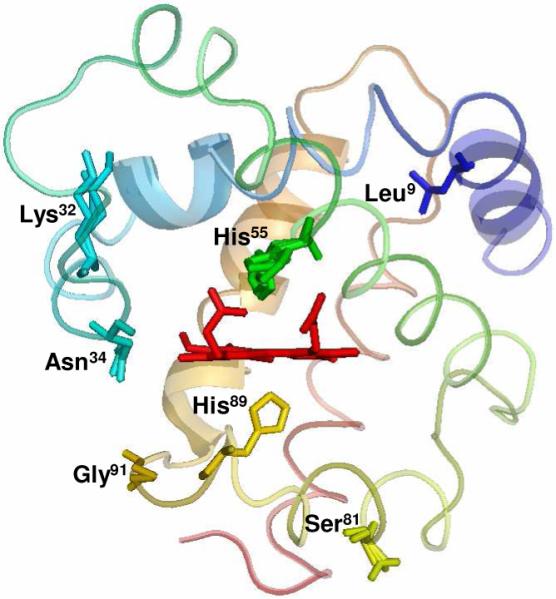
Crystal Structure of DHP B (PDB accession code 3ixf). The location of the five residues (Leu9, Lys32, Asn34, Ser81 and Gly91) relative to the heme active site that differ in DHP A are shown, as well as the proximal (His89) and distal (His55) histidines.
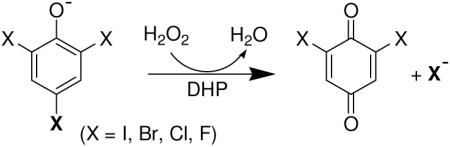
DHP catalyzes the oxidative degradation of 2,4,6-trihalogenated phenols to the corresponding 2,6-dihalo-1,4-benzoquinones in the presence of hydrogen peroxide (see insert). Several recent studies have focused on the characterization of DHP, as well as elucidating the mechanism of this reaction (18, 20-34). Using stopped-flow UV-visible and rapid freeze-quench EPR spectroscopic methods, we have previously demonstrated that ferric DHP reacts with hydrogen peroxide to yield Compound ES, an iron(IV)-oxo heme center with an amino acid radical (20). The catalytic competency of that intermediate in oxidizing the co-substrate 2,4,6-tricholorphenol (TCP) was also shown, and we proposed a peroxidase-like catalytic cycle for DHP at that time. It was also found that in the absence of co-substrate, there is the formation of a new species named as Compound RH, which is unique to dehaloperoxidase and has not found in any other globin. With regards to co-substrate oxidation, Dawson and co-workers have recently reported that the overall two-electron oxidation of TCP by DHP proceeds through discrete one-electron steps (31), which is consistent with the hypothesis that the trihalogenated co-substrate likely binds at an external site similar to other peroxidases (22). However, despite the fact that these and a number of other studies on DHP (20, 26, 31, 35) have helped to elucidate its mechanistic pathways, it is still not understood how this bi-functional protein can act as both a hemoglobin and a peroxidase, and a number of questions still remain.
The primary focus of this report is to provide a detailed picture of the chemistry of DHP B (Scheme 1) with results and experimental details not previously described for either isoenzyme: (a) dichloroquinone (DCQ), the purported product of trihalophenol dehalogenation, further reacts with the putative oxidant Compound ES leading to oxyferrous DHP; (b) DCQ reacts with ferric DHP also leading to oxyferrous DHP formation; (c) oxyferrous DHP catalyzes the oxidative dehalogenation of TCP in the presence of hydrogen peroxide; (d) 4-bromophenol, a presumed co-substrate known to bind in the distal cavity of the heme active site (36), is an inhibitor of 2,4,6-trichlorophenol dehalogenation; (e) spectroelectrochemistry of DHP B reveals an unusually high redox potential for this globin-peroxidase. As investigations of isoenzyme B may also provide important clues and significant advances in understanding the catalytic mechanism of both isoforms of DHP, the secondary focus of this report is to present additional spectroscopic studies of DHP B, including resonance Raman, stopped-flow UV-visible, and rapid-freeze-quench electron paramagnetic resonance spectroscopies, which when coupled with biochemical assays provide further evidence in support of the proposed catalytic cycle (Scheme 1). While the secondary focus parallels our recent study of isoenzyme A (20), our experimental design in this report differs, and reveals new mechanistic insights and kinetic descriptions of the intermediates in DHP B which have not been previously reported for DHP A. When interpreted in light of our recent X-ray crystallographic study of DHP B which demonstrates a mechanistic role for the conformational flexibility of the distal histidine (His55) (37), the results presented herein advance our understanding of how DHP as a bifunctional enzyme is able to concurrently perform its two inherent peroxidase and oxygen transport activities in one system.
Scheme 1.

Proposed Catalytic Cycle for Dehaloperoxidase B.
Materials And Methods
Materials
Buffer salts were purchased from Fisher Scientific. All other reagents and biochemicals, unless otherwise specified, were of the highest grade available from Sigma Aldrich. The QIAprep® Spin Miniprep Kit was from QIAGEN Sciences (Valencia, CA) and the Quikchange® II site-directed mutagenesis kit was purchased from Stratagene (La Jolla, CA). The required oligonucleotides were synthesized by IDT DNA Technologies, Inc. N,N,N’,N’-tetramethyl-p-phenylenediamine (TMPD) was purchased from Sigma, [Ru(en)3][ZnCl4] was synthesized according to previously reported methods (38-40), and the reagents (i.e. RuCl3, ethylenediamine and zinc dust) for its reaction were also purchased from Sigma. EPR tubes were purchased from Norell (Landisville, NJ). Solutions of trihalogenated phenols were freshly prepared prior to each experiment in 100 mM potassium phosphate (KPi) buffer, pH 7 and kept at 4 °C and protected against light. UV-visible spectra were recorded periodically to ensure that the co-substrate had not degraded by monitoring its absorbance: trifluorophenol, 270 nm (1027 M−1cm−1); trichlorophenol, 312 nm (3752 M−1cm−1) (20) ; tribromophenol, 316 nm (5055 M−1cm−1). Hydrogen peroxide stock solutions were also freshly prepared daily prior to each experiment and maintained at 4 °C. The hydrogen peroxide stability was monitored by UV-visible spectroscopic analysis at 240 nm (ε240 = 43.6 M−1 cm−1) (41). The stock H2O2 solution was diluted to the corresponding premixing concentrations for each experiment.
Plasmid Preparation, Protein Expression and Purification
All the mutations were generated with the Quikchange® II site-directed mutagenesis kit. Mutagenesis [melt (95 °C, 50 s), anneal (60 °C, 50 s), and extension (68 °C, 6 min] was performed for 18 cycles. The plasmid encoding wild-type (WT) DHP A (His tag) was used as a template to generate the mutation pDHPA (R32K/Y34N) using the mutagenic primers [5′-G AAT AAG TAT CCG GAC GAG AAA CGC AAC TTC AAA AAC TAT GTC-3′ (sense) and 5′-GAC ATA GTT TTT GAA GTT GCG TTT CTC GTC CGG ATA CTT ATT C-3′ (antisense)]. The resulting plasmid was then subsequently utilized to generate the triple mutant DHPA (R32K/Y34N/S91G) using the primers [5′-G ATG AAA CAG CAT TCC GGC CTG ACG ACT GGA AAC-3′ (sense) and 5′-GTT TCC AGT CGT CAG GCC GGA ATG CTG TTT CAT C-3′ (antisense)]. The R32K/Y34N/S91G plasmid was then employed for the mutation pDHPA(R32K/Y34N/S91G/I9L) using 5′-CAA GAT ATT GCC ACC CTC CGC GGT GAT CTC CGC-3′ (sense) and 5′-GCG GAG ATC ACC GCG GAG GGT GGC AAT ATC TTG-3′ (antisense). Finally, the template R32K/Y34N/S91G/I9L was used to generate the plasmid pDHPA(R32K/Y34N/S91G/I9L/N81S), also referred to as pDHPB, with 5′-CTT GCG TCC GAC GCC AGC ACA CTC GTC CAG ATG-3′ (sense) and 5′-CAT CTG GAC GAG TGT GCT GGC GTC GGA CGC AAG-3′ (antisense). The plasmids were extracted using QIAprep® spin miniprep kit after each round of mutagenesis, and the presence of the desired mutations and lack of secondary mutations were confirmed by sequencing. Wild-type DHP B and DHP A (6x-His-tagged proteins) were expressed and purified as previously described (20, 22) with only minor modification.
Molecular Weight Determination
The molecular weight of DHP B was determined by a 6210 LC-TOF mass spectrometer in positive-ion electrospray ionization (Agilent Technologies, Santa Clara, CA). The protein sample was in 50 mM ammonium acetate buffer, pH 7.0 The mobile phase consisted of HPLC grade solvents: water + 0.1% formic acid (v/v), and water:acetonitrile 5:95 + 0.1 % formic acid (v/v). The injection volume was 5 μL and the flow rate was 300 μL per minute.
Preparation of Ferric DHP
DHP B was treated with an excess of potassium ferricyanide in order to obtain a homogeneous solution of the enzyme in the ferric state. Ferri/ferrocyanide was removed using a PD-10 desalting column prepacked with Sephadex G-25 medium. The protein was concentrated using Amicon Ultra centrifugal filter equipped with a 10 kDa cutoff molecular weight membrane, and the purity of DHP was determined as previously published (20, 22). Only protein samples that exhibited Reinheitzahl values (Rz) greater than 4.0 were utilized in this study. Protoheme content was measured by the pyridine hemochrome assay using Δε557 = 20.7 mM−1cm−1 (reduced - oxidized) for iron protoporphyrin IX (42, 43), leading to a molar absorptivity for DHP B of 117.6 mM−1cm−1 (λmax = 407 nm), in good agreement with that of DHP A (ε406 = 116.4 mM−1cm−1) (18).
Preparation of DHP B Complexes for UV-visible Spectroscopic Studies
Optical spectra were recorded using quartz microcuvettes (1 cm pathlength) on a Cary 50 UV-visible spectrophotometer equipped with thermostatted cell holders at 25 °C. Complexes of DHP B (10 μM final concentration) were prepared in 100 mM KPi buffer (pH 7). The ferric-CN complex was generated upon addition of NaCN (50 mM final concentration) to ferric DHP B. Oxyferrous DHP B was obtained by the aerobic addition of either 2 equivalents of the reducing agent tris(2-carboxyethyl)phosphine (TCEP) or ascorbic acid to a solution of ferric DHP B, followed by application of the enzyme over a PD-10 desalting column. Ferrous DHP B was prepared by mixing the ferric enzyme with sodium dithionite anaerobically using standard Schlenk techniques. Ferrous DHP B prepared thusly was then exposed to CO gas to yield the DHP B ferrous-CO complex.
Mono and trihalophenol complexes of DHP B were obtained upon the addition of stock solutions of the halophenols to a solution of DHP B in 100 mM KPi buffer (pH 7). Final concentrations were as follows: DHP B, 8 μM; 2,4,6-trifluorophenol, 4 mM; 2,4,6-trichlorophenol, 2 mM or 300 μM; 2,4,6-tribromophenol, 255 μM; phenol, 4-fluorophenol, 4-chlorophenol, 4-bromophenol, 8 mM; 4-iodophenol, 850 μM.
Preparation of Oxyferrous DHP B
Oxyferrous DHP B was generated through incubation of the ferric enzyme with 5 equivalents of ascorbic acid in 100 mM potassium phosphate buffer (variable pH). The formation of the oxyferrous species was monitored by its UV-visible spectrum [418 (Soret), 542, 578 nm]. Excess reducing agent was removed by using a PD-10 desalting column prepacked with Sephadex G-25 medium. The protein was concentrated using an Amicon Ultra centrifugal device equipped with a 10 kDa cutoff molecular weight membrane. The oxyferrous form of DHP B was found to be stable for at least two weeks when prepared in the fashion and stored at 4 °C.
UV-visible Spectroscopic Studies and Dehaloperoxidase Activity Assays
Optical spectra were recorded using quartz microcuvettes (1 cm pathlength) on a Cary 50 UV-visible spectrophotometer equipped with thermostatted cell holders at 25 °C. The apparent values of Km and kcat for DHP B for various concentrations of hydrogen peroxide at a fixed saturating concentration of the trihalophenol co-substrate were calculated by triplicate measurements of initial rate at each H2O2 concentration. The experimental data was fitted to the Michaelis-Menten model using the enzyme kinetics software GraFit (Erithacus Software). The enzymatic activity was assayed on the basis of the disappearance of co-substrate (trichlorophenol, 312 nm; tribromophenol, 316 nm) or formation of product (difluoroquinone, 330 nm) monitored for 15 min at 25 °C. The 1-mL reaction mixture contained 0.5 μM of enzyme (ferric or oxyferrous), 150 μM of trihalophenol and varying H2O2 concentrations in 100 mM potassium phosphate buffer at pH 7.
Stopped-flow UV-Visible Spectrophotometric Studies
Experiments were performed on a Bio-Logic SFM-400 Triple Mixing Stopped-Flow instrument equipped with a diode array UV-visible spectrophotometer, and were carried out at 20 °C in 100 mM KPi buffer, pH 7. Constant temperature was maintained using a circulating water bath. Data were collected (900 scans total) over three time-domain regimes (2.5 ms, 25 ms, 250 ms; 300 scans each) using the Bio Kinet32 software package (Bio-Logic). Single-mixing experiments were performed as follows: i) ferric DHP B at a final concentration of 10 μM was reacted with 2.5 – 25 equivalents of H2O2; or ii) 10 μM ferric DHP B was reacted with 5, 25, 70 and 100 μM of DCQ (all final concentrations). Experiments were performed in double-mixing mode using an aging line prior to the second mixing step. The design of the experiments allowed for the mixing of DHP B with either TCP/DCQ or H2O2 for various aging times, followed by the second mix with the remaining (co-) substrate: i) DHP B + TCP/DCQ ➔ Delay ➔ + H2O2; or ii) DHP B + H2O2 ➔ Delay ➔ + TCP/DCQ. Concentrations after mixing were [DHP B]f = 10 μM, [H2O2]f = 100 μM, and [TCP]f = 300 μM, or [DHP B] = 10 μM, [H2O2] = 25 μM, [DCQ] = 70 μM. All data were evaluated using the Specfit Global Analysis System software package (Spectrum Software Associates) and fit with SVD analysis as either one-step, two species or two-step, three species irreversible mechanisms, where applicable. Kinetics data were baseline corrected using the Specfit autozero function.
Preparation of Reaction Intermediates by Freeze-Quench Methods
Rapid freeze-quench experiments were performed with a BioLogic SFM 400 Freeze-Quench apparatus by mixing a 50 μM enzyme solution (final concentration) with a 10-fold excess of H2O2 solution in 100 mM potassium phosphate buffer (pH 7) at 25 °C. Reaction times were as follows: 100, 300, 500, 800 ms, 2, 38.25, 60 s. The dead time of the instrument as configured was 10 ms. A standard 4 mm O.D. quartz EPR tube was connected to a Teflon funnel, and both the tube and the funnel were completely immersed in an isopentane bath at −110 °C. The reaction mixtures were quenched by spraying them into cold isopentane, and the frozen material so obtained was packed at the bottom of the quartz tube using a packing rod equipped with a Teflon plunger. In this manner, a packing factor of 60 ± 2% was consistently achieved. Samples were then transferred to a liquid nitrogen storage dewar until analyzed.
X-band EPR Spectroscopy
EPR spectra were recorded with an X-band (9 GHz) Varian E-9 EPR spectrometer (Varian, El Palo, CA). A standard 4 mm o.d. quartz EPR tube containing sample was placed into a quartz finger dewar insert filled with liquid nitrogen. The temperature of the samples was maintained at 77 K for the duration of the data acquisition, which required periodic refilling of the dewar due to the evaporation of the liquid nitrogen during longer acquisition runs. The typical spectrometer settings were as follows: field sweep 200 G, scan rate 3.33 Gauss/s, modulation frequency 100 KHz, modulation amplitude 4.0 G, and microwave power 2 mW. The exact resonant frequency of each EPR experiment was measured by an EIP-578 (PhaseMatrix, San Jose, CA) in-line microwave frequency counter. Typically, 20 to 200 individual scans were averaged to achieve sufficient signal-to-noise for the spectra obtained at short-quench and long-quench times, respectively.
Spectroelectrochemistry (SEC)
For a given experiment, a solution of ferric DHP B at a concentration of 100 – 200 μM [ε406nm: 117,600 M−1cm−1 (18)] was prepared in a supporting electrolyte of 100 mM KPi, pH 7.0. SEC was carried out in a previously designed anaerobic UV-visible spectroelectrochemical cell made of cast acrylic (Small Parts, Inc.) that utilized an optically-transparent thin-layer electrode (OTTLE) of antimony-tin oxide (ATO, Delta Technologies, Limited) (44). The pathlength for the cell was maintained at 0.1 mm by use of Teflon coated fiberglass tape (Small Parts, Inc.). The ATO electrode was thoroughly cleaned before use by 10 minute successive sonications in 1% (v/v) Contrex™ solution (Decon Labs, Inc.), 95% ethanol and twice in deionized water. The SEC cell was stored in a nitrogen atmosphere drybox (Vacuum Atmospheres Company; H2O < 1 ppm, O2 < 1 ppm) for at least eight hours prior to experimentation. Electronic spectra were recorded with a Hewlett-Packard 8453 spectrophotometer and applied potentials were controlled with a Model 273 Princeton Applied Research potentiostat. The SEC cell made use of an Ag/AgCl (sat. KCl) reference electrode (Microelectrodes, Inc.) and a platinum wire auxiliary electrode (Alfa Aesar). All formal reduction potentials are referenced to the standard hydrogen electrode (SHE). Electron transfer mediators such as TMPD and [Ru(en)3]2+ were used to facilitate electron transfer between DHP B and the working electrode. The formal reduction potentials for TMPD and [Ru(en)3]2+ as determined by cyclic voltammetry were +0.250 V and +0.111 V vs SHE, respectively. The ratio of DHP B-to-mediators was 2 to 1.
Resonance Raman Spectroscopy
Protein samples (~100 μM) were prepared in 100 mM KPi buffer at pH 7. Para-halogenated phenols were introduced to final concentrations of 8 mM for 4-BP, 4-CP, 4-FP, and phenol, and to 1 mM for 4-IP. 2,4,6-Trihalophenols were added to final concentrations of 4 mM for TFP, 3 mM for TCP and 200 μM for TBP. The samples were placed into 5 mm diameter glass NMR tubes and stored on ice until used.
Resonance Raman spectra were obtained by Soret band excitation using a Coherent Mira 900 titanium sapphire (Ti:sapphire) laser. The Ti:sapphire laser was pumped using a Coherent Verdi 10 frequency doubled diode pumped Nd:Vanadate (Nd:VO4) laser generating 10 W at 532 nm. The beam generated from the Ti:sapphire was sent through a Coherent 5-050 doubler to generate a normal working range of 400-430 nm for Soret band excitation of each of the DHP complexes. The beam was collimated and cylindrically focused to a vertical line of ~0.5mm on the sample. Laser power at the sample was 60 mW. Scattered light was collected with a Spex 1877 triple spectrometer equipped with a liquid nitrogen-cooled CCD detector controlled by Spectramax software.
Kinetics of 4-Bromophenol Inhibition of the DHP B Catalyzed Oxidation of TCP
Inhibition assays were conducted in 100 mM KPi buffer at pH 7 using an Agilent 8453 UV-vis spectrometer equipped with temperature control and Hewlett Packard UV-Visible Chemstation Software set to kinetic mode. The concentration of DHP and TCP in each sample was 2.5 μM and 125 μM, respectively. Temperature was equilibrated to 20 °C before initiation. A 100-fold excess of H2O2 (250 μM) was added to the cuvette to initiate the assay. Electronic absorption spectra were taken every 2 seconds for 2 minutes, monitoring the 273 nm peak of the 2,6-dichloro-1,4-dibenzoquinone product (2,4-DCQ). This process was repeated with the addition of 250 μM 4-BP to serve as the inhibitor.
RESULTS
Overexpression, Purification and Characterization of DHP B
The plasmid encoding wild-type DHP A with an N-terminal poly-His tag (pDHPA) was subjected to four successive rounds of PCR amplification using mutagenic primers. DNA sequencing of the entire resulting dhpB gene (pDHPB) confirmed the success of the site-directed mutagenesis and the absence of secondary mutations. Recombinant DHP B was obtained by expression in E. coli and resulted in a protein yield of ~9 mg/L culture. The two-part purification strategy (immobilized metal affinity followed by ion-exchange chromatographies) resulted in purification levels >95% homogeneity, with DHP B being indistinguishable by SDS-PAGE gel from DHP A. As was found for isoenzyme A, DHP B was initially isolated as a mixture of the ferric and oxyferrous forms, but subsequent treatment with an excess of potassium ferricyanide permitted the isolation of the ferric form. The optical purity ratio (Reinheitzahl or Rz, defined as ASoret/A280) for WT DHP B was found to be 4.1, in good agreement with the literature value for DHP A. The monomeric molecular weight of DHP B was determined by electrospray ionization MS to be 16,274.38 g/mol, which agrees with the theoretical expected value (16,274.37 g/mol).
UV-Visible Characterization of DHP B
The electronic absorption spectra of different heme states of DHP B at pH 7 are presented in Figure SD1 and Table 1. Ferric metaquo DHP B at pH 7 exhibits electronic absorption features typical of a high spin ferric heme [UV-visible: 407 (Soret), 508, 633 nm], and similar to those previously observed for DHP A under identical conditions (18, 20). Addition of cyanide, a strong field ligand, led to a red-shift of the Soret band [UV-visible: 423 (Soret), 544 nm], suggestive of the presence of a 6cLS ferric heme. Reduction of ferric DHP B with sodium dithionite yielded an absorption spectrum typical of a 5cHS ferrous heme [UV-visible: 433 (Soret), 554 nm] (27), which blue-shifted in the presence of carbon monoxide [UV-visible: 422 (Soret), 537, 567 nm] and dioxygen [UV-visible: 417 (Soret), 542, 578 nm] to give the ferrous-CO and oxyferrous complexes, respectively, both of which are 6cLS hemes (24, 27). Structural studies of the ferric [1ewa and 1ew6 (45), 2qfk (27), 3ixf (37)], deoxy (21), and oxyferrous (27) analogues of DHP A and B further support these spectroscopic assignments here. Periodic recording of the optical spectrum of the oxyferrous form of DHP B found it to be stable for at least two weeks (when stored at 4 °C).
Table 1.
UV-Visible Spectroscopic Data for DHP B Complexes at pH 7
|
Soret λmax (nm) |
Visible λmax (nm) |
|
|---|---|---|
| Ferric | 407 | 508, 633 |
| Ferric-CN | 423 | 544 |
| Ferrous | 433 | 554 |
| Ferrous-CO | 422 | 537, 567 |
| Oxyferrous | 417 | 542, 578 |
Mono- and Trihalophenol Binding in DHP B
The UV-visible spectra of ferric DHP B and its mono- and trihalophenol complexes at pH 7 are shown in Figure SD2, and relevant spectral features and analysis can be found in Table 2. In general, examination of a hemoprotein electronic absorption spectrum reveals the relative populations of 6c vs. 5c HS heme species present: overall, 5cHS hemes exhibit a slightly blue shifted and lower extinction coefficient Soret band than their 6cHS counterparts, as well as a shoulder at 380 nm (46). Furthermore, the charge transfer band denoted as CT1 in a 5cHS heme is found at ~640 nm (or higher), while that of a 6cHS heme is generally closer to 630 nm. LS heme systems exhibit a red-shifted Soret, the absence of a CT1 feature, and visible features at ~540 and 580 nm. Here, the effect of (tri)halophenol binding on DHP B was demonstrated by following specifically the ASoret/A380 ratio (47, 48). In the presence of phenol, the ASoret/A380 ratio decreases from 1.81 for ferric DHP B to 1.70, indicative of an increase in 5cHS heme upon increasing occupancy of the distal active-site binding pocket (Figure SD2A). Phenols bearing 4-halo substituents led to a progressive decrease in this ratio (1.53, 1.54, 1.39, and 1.35) which correlates well with the periodicity of the halogen (F, Cl, Br, and I, respectively). These findings suggested an increase in the relative population of 5cHS heme as one moves down the periodic table. We interpret these results as an increase in the binding affinity of the monohalophenol based upon the size of the substituent present, which in turn displaces the heme bound water ligand, and leads to a greater population of the ‘open’ conformation (5cHS) of the distal histidine, His55 (21) (Scheme 1, step i-a).
Table 2.
UV-Visible Spectroscopic Data for DHP B Halophenol Complexes at pH 7
| λmax (nm) | ASoret/A380 | |
|---|---|---|
| Ferric DHP B | 407, 508, 540 (sh), 633 | 1.81 |
| + phenol | 406, 503, 538 (sh), 612 | 1.70 |
| + 4-fluorophenol | 404, 502, 538 (sh), 612 | 1.53 |
| + 4-chlorophenol | 404, 503, 538 (sh), 624 | 1.54 |
| + 4-bromophenol | 404, 504, 538 (sh), 640 (sh) | 1.39 |
| + 4-iodophenol | 402, 503, 538 (sh), 640 (sh) | 1.35 |
|
| ||
| + trifluorophenol | 406, 500, 536 (sh), 617 | 1.80 |
| + trichlorophenol a | 406, 503, 536 (sh), 624 | 1.57 |
| + trichlorophenol b | 406, 504, 536 (sh), 619 | 1.81 |
| + tribromophenol | 407, 506, 536 (sh), 623 | 1.74 |
2 mM
300 μM
In contrast to the above results obtained for the monohalophenols complexes, none of the trihalophenol complexes exhibited a significant decrease in the ASoret/A380 ratio (Table 2 and Figure SD2B). As it is unlikely that the exogenously added trihalophenol and distal histidine are able to occupy the internal binding pocket simultaneously, the data suggest that trihalophenols are simply excluded from entering the distal cavity in DHP B due to their bulky size, precluding them from both displacing the heme-bound water ligand and generating a sufficient quantity of the 5cHS species as to affect the ASoret/A380 ratio. In light of the X-ray crystallographic evidence for the binding of monohalogenated phenols at an internal site (36), as well as evidence for a trihalophenol external binding site (22, 25, 49), we suggest that our findings here support the latter supposition that trihalophenols bind externally (predominantly, although we cannot rule out exclusively), and monohalophenols bind internally. Further evidence is provided by our resonance Raman study (vide infra).
Resonance Raman Spectroscopy of DHP B and its (Tri)halophenol Complexes
The high frequency heme vibrational modes were investigated for DHP B at pH 7 using resonance Raman spectroscopy, and the results are presented in Figure 2. These resonance Raman spectroscopic results provide a complementary approach to our electronic absorption study with respect to elucidating the electronics of the heme cofactor. Binding of monohalogenated phenols to DHP B yielded the same trend as that observed in our electronic absorption study above. Specifically, a systematic shift to 5cHS heme resulted from the internal binding of the monohalophenols and followed the halogen series (5cHS heme population: I > Br > Cl > F), concomitant with the distal histidine assuming an open conformation. Internal binding is indicated by the loss of the 6cHS heme population (ν3, 1477 cm−1; ν2, 1563 cm−1) and gain of the 5cHS heme population (ν3, 1494 cm−1; ν2, 1569 cm−1; ν10, 1630 cm−1) (Figure 2B). These findings are in excellent agreement with those observed previously for DHP A under similar conditions (50).
Figure 2.

Resonance Raman spectra of halophenol complexes of DHP B (30-100 μM) at pH 7.0. (A) Trihalophenol complexes of TFP (4 mM), TCP (3 mM), and TBP (200 μM). (B) Monohalophenol complexes of 4-FP (8 mM), 4-CP (8 mM), 4-BP (8 mM), 4-IP (1 mM) and phenol (8 mM).
In contrast, the binding of trihalophenols resulted in an increase of 6cHS heme population (ν3, 1477 cm−1; ν2, 1563 cm−1; ν10, 1607 cm−1) (Figure 2A). While the effect is more limited for TBP and TCP, the binding of TFP to DHP B resulted in nearly complete production of 6cHS heme. We attribute the increase in 6cHS population to the movement of the distal histidine into the closed conformation upon external trihalophenol binding, thereby stabilizing the heme coordinated water molecule (Scheme 1, step ii-a). However, the data differ slightly from those observed for DHP A, which showed that although TBP and TCP were excluded from binding internally, TFP was able to do so, albeit not to the same extent as for the monohalophenols. We surmise that the subtle structural differences between these two isoforms give rise to the greater active-site accessibility in DHP A vs B for TFP, the smallest of the three trihalophenols examined. We rule out a contamination of free fluoride ion in the TFP solution as the cause of these changes in DHP B given that the same solution did not produce an identical spectrum in DHP A (50).
In the above resonance Raman studies, a minor population of 6cLS heme was observed, as denoted by the ferrous heme vibrational mode at 1504 cm−1 (Figure 2). We attribute this to the presence of oxyferrous DHP B, which we presume is formed from the autoreduction of the ferric enzyme (vide infra). The oxyferrous low-spin heme component was also observed, albeit not in all spectra, as a minor feature in the above UV-visible study at ~576 nm. Interestingly, internal binding of monohalogenated phenols did not appear to affect the oxyferrous component of the enzyme (i.e. the signal intensity present in the monohalophenol-free sample remained upon 4XP binding), whereas external binding of trihalophenols led to loss of the 6cLS heme. This suggests that monohalophenols either do not enter the distal cavity of the oxyferrous enzyme, or if they do, they are present in the cavity along with the heme-ligated O2 molecule. Why external binding of the trihalophenol would displace the O2 molecule, while internal binding of monohalophenols does not, remains a question of interest. We surmise that this action might contribute in some way to the trigger mechanism that switches the function of the enzyme from globin to peroxidase. Evidence for this supposition is derived from our UV-visible spectroscopic study which shows that oxyferrous DHP B does not form the catalytically competent Compound ES intermediate in the presence of H2O2 (as ferric DHP does) when trihalophenols are absent, but oxyferrous DHP does exhibit catalytic activity when trihalophenols and hydrogen peroxide are present concurrently. This functional-switch triggering event predicated upon co-substrate binding is discussed in more detail below.
Enzymatic Activity of DHP B
The hydrogen peroxide-dependent oxidative dehalogenation of 2,4,6-trihalogenated phenols (TXP) catalyzed by DHP B at pH 7 was monitored by UV-visible spectroscopy (Figure SD3). Three TXP co-substrates were examined (X = F, Cl, Br), and the reaction mixture yielded the corresponding 2,6-dihalo-1,4-benzoquinone (DXQ) products as expected. The enzymatic reaction was initiated by the addition of H2O2 as the substrate. Both enzyme and TXP co-substrate concentrations were held constant while H2O2 concentration was varied. For the reactions employing TCP or TBP, co-substrate loss was monitored. In the case of TFP, however, the reaction was monitored using product formation [2,6-difluoro-1,4-benzoquinone (DFQ), λmax = 330 nm] due to the lack of a clear absorption maximum for the TFP co-substrate (Figure SD3A). In the absence of DHP (non-enzymatic control), no product was observed under the conditions examined, in agreement with previous reports that showed a requirement for the enzyme (18, 20).
Kinetic parameters (kcat, Km, and catalytic efficiency, kcat/Km) for the dehaloperoxidase activity of DHP B are presented in Table 3, together with those determined for DHP A under the same conditions of fixed trihalophenol co-substrate concentration and varied hydrogen peroxide substrate concentration. Both isoenzymes exhibited saturable dehaloperoxidase activity under the conditions employed, and the data were fit to standard Michaelis-Menten kinetics using the method of initial rates. For the reactions with TBP and TCP, d[S]/dt was computed using the known epsilon values for these two co-substrates, thus allowing for the determination of kcat. For TFP, however, neither −d[S]/dt nor d[P]/dt were determined as the co-substrate absorption maximum underwent a shift during catalytic turnover, and the molar absorptivity of the product is unknown. Thus, while initial rates for the oxidation of TFP could be determined by following product formation (Figure SD3A), the corresponding kcat value could not be determined. Comparison of the enzymatic activity between DHP B and DHP A was therefore based on the ratio of their corresponding kcat (TCP, TBP) or initial rate (TFP) values as listed in Table 3.
Table 3.
Kinetics data for the oxidative catalytic reaction of the different co-substrates: TCP, TBP and TFP with 0.5 μM DHP B and 150 μM of the corresponding co-substrates and varying H2O2 concentrations in 100 mM KPB pH 7
| Co-Substrate | DHP B |
DHP A |
Ratio c kcatB/kcatA |
|||||
|---|---|---|---|---|---|---|---|---|
| KMH2O2 (μM) |
KMTXP (μM) |
kcat (s−1) |
kcat/KMH2O2 (μM−1 s−1) |
KMH2O2 (μM) |
kcat (s−1) |
kcat/KMH2O2 (μM−1 s−1) |
||
| TFP a | 96 ± 18 |
N/A | N/A | N/A | 59 ± 10 |
N/A | N/A | 2.1 |
| TCP a | 22 ± 2 | 210 ± 23 |
1.53 ± 0.03 |
0.070 | 23 ± 1 | 0.61 ± 0.01 |
0.027 | 2.6 |
| TCP b | 35 ± 6 | N/A | 1.17 ± 0.05 |
0.034 | 16 ± 1 | 0.57 ± 0.01 |
0.036 | 2.1 |
| TBP a | 63 ± 15 |
315 ± 11 |
1.3 ± 0.1 | 0.021 | 11 ± 1 | 0.30 ± 0.01 |
0.027 | 4.3 |
N/A = not available
ferric starting oxidation state
oxyferrous starting oxidation state
as ratio of initial rate
When starting from the ferric form, DHP B exhibited a consistently higher catalytic rate than DHP A for the three different co-substrates examined in this study (Scheme 1, step ii-b). We observed the general trend of an increased rate of reaction as the periodicity of the halogen substituent was increased. Specifically, DHP B is able to dehalogenate TFP, TCP and TBP 2.1-, 2.6-, and 4.3-fold faster than DHP A. The fastest rate of conversion was found for the brominated co-substrate, and is not surprising since TBP has been reported to be the natural co-substrate for DHP under physiological conditions (2). Interestingly, the Km values for DHP B were 2-6 fold higher than for DHP A for the substrate H2O2, leading to comparable catalytic efficiencies (kcat/Km) between the two isoenzymes.
The above study was repeated for the co-substrate 2,4,6-TCP using oxyferrous DHP as the starting oxidation state rather than the ferric form of the enzyme (Scheme 1, step viii). Oxyferrous DHP B exhibited a catalytic efficiency that was only 2-fold lower than that observed for ferric DHP, whereas it was found that oxyferrous DHP A had a kcat/Km value 1.3 times greater than that of the ferric enzyme. Overall, these values for kcat and Km between the two different starting oxidation states (ferric and oxyferrous) are virtually the same despite having started in the former from what is the traditional peroxidase resting state, and in the latter from what is normally a catalytically inactive state for peroxidases. Given the similarity of kinetic parameters, it reasons that although the first turnover may be initiated from either of the two oxidations states, that subsequent turnovers proceed through a common pathway (i.e., a traditional peroxidase mechanism).
Inhibition of Trichlorophenol Oxidation by 4-BP
The kinetics of 2,4,6-TCP oxidation in the absence (Figure 3A) and presence (Figure 3B) of 4-bromophenol (4-BP) are shown. The binding affinity of 4-halophenols to DHP A follows the trend I > Br > Cl > F > H, with apparent dissociation constants of 0.536, 1.15, 1.78, 3.72, and 10.0 mM, respectively (51). In agreement with our recent observations for DHP A (51), the presence of 4-monohalophenols led to a significant attenuation of the rate of trihalophenol oxidation as catalyzed by DHP B when compared to the absence of 4-bromophenol. The effect was observed regardless of whether the formation of 2,6-dichloroquinone (DCQ, 276 nm) (Figure 3C) or the loss of TCP (312 nm, data not shown) was followed. The kinetic results are interpreted as internal monohalophenol binding (confirmed above by resonance Raman spectroscopy) which precludes the activation of hydrogen peroxide due to the distal histidine His55 being swung out of the active site (Scheme 1, step i-c). In this ‘open’ conformation, His55 is unable to facilitate the O-O bond cleavage necessary for Compound ES formation. In support of this supposition, the control experiment showed no reaction between ferric DHP B (10 μM) and H2O2 (100 μM) when 30 equivalents of 4-BP were present (data not shown) (Scheme 1, step i-b). Additionally, inhibition was observed regardless as to whether the monohalophenol inhibitor was added to DHP before (Scheme 1, step i-c) or after (Scheme 1, step ii-c) the trihalophenol co-substrate.
Figure 3.

Oxidative dehalogenation of 2,4,6-trichlorophenol (125 μM) as catalyzed by DHP (2.5 μM) and hydrogen peroxide (250 μM) in the absence (A) and presence (B) of 4-bromophenol (250 μM). The formation 2,6-dichloroquinone was monitored at 276 nm (C).
Stopped-Flow UV-Visible Characterization of DHP B Compounds ES and RH
Formation of DHP B transient species were investigated by single-mixing stopped-flow UV-visible spectroscopy at pH 7. Upon rapid mixing (2 ms) of a ferric DHP B solution [UV-visible spectrum: 407 (Soret), 508, 630 nm] with H2O2 (Scheme 1, steps iii and iv), a new species was observed (Figure 4 and Table 4) whose spectral features [UV-visible: 419 (Soret), 545, 585 nm] we attributed to the ferryl-containing DHP B intermediate Compound ES based upon our previous assignment of this reaction intermediate in DHP A (20, 26, 29, 35) as well as the spectroscopic properties of other Fe(IV)-oxo species commonly found in hemoproteins. Specifically, non-globin peroxidases (e.g. HRP (52, 53), CcP (54-56), APX (57, 58) and others) exhibit a red-shifted Soret feature (~417-422 nm) and characteristic visible bands (~530, ~ 560 nm) for Compound ES/II, whereas globins, such as myoglobin (59, 60) and hemoglobin (61, 62), display a similar red-shifted Soret feature but have visible bands that appear at lower energy, generally 540-5 and 580-590 nm. Not surprisingly, DHP exhibits features for Compound ES that are similar to those observed for ferrylglobins. Assignment of this species as a traditional Compound I intermediate was ruled out based on the lack of hypochromicity and absence of a strong red shift of the Soret band that is typical for an iron(IV)-oxo porphyrin π-cation radical (52, 57, 58). Compound 0 (ferric-hydroperoxide) was also ruled out due to time-scale considerations (63, 64) and the lack of an observed hyperporphyrin spectrum (65, 66). As Compound ES and Compound II, both ferryl-containing intermediates, are not distinguishable by UV-visible spectroscopy, our assignment for the intermediate described here as Compound ES is based upon the results of our EPR spectroscopic study (vide infra). The experimental values of kobs for Compound ES formation varied linearly with hydrogen peroxide concentration in the range of 2.5 – 25 molar equivalents (Figure SD4). From this dependence, the bimolecular rate constant was determined to be 1.29 ± 0.11 × 105 M−1s−1, and is approximately 3.6-fold greater in DHP B than in DHP A. At higher peroxide concentrations (> 1 mM), heme bleaching was noted (data not shown).
Figure 4.

(A) Stopped-flow UV-visible spectroscopic monitoring (900 scans, 85 sec) of the reaction between ferric DHP B (10 μM) and a 10-fold excess of H2O2 at pH 7. See Materials and Methods for details. (B) Calculated UV-visible spectra for ferric (black), Compound ES (red), and Compound RH (blue) DHP B are shown; the rapid-scanning data from panel A were compiled and fitted to a two-step, three species sequential irreversible model using the Specfit global analysis program. (C) Relative concentration profile determined from the three component fit used in panel B.
Table 4.
UV-Visible Spectroscopic Data and Kinetic Parameters for the Oxidized Intermediates of DHP B and DHP A at pH 7
| DHP B | DHP A | |||
|---|---|---|---|---|
| λmax (nm) | kobsa | λmax (nm) | kobsa | |
| Ferric | 407, 508, 633 | not applicable | 407, 504, 538, 635 | not applicable |
| Compound ES | 419, 545, 585 | (1.08 ± 0.02) × 105 | 420, 545, 585 | (3.56 ± 0.02) × 104 |
| Compound RH | 411, 554, 599 | (1.9 ± 0.1) × 10−2 | 411, 530, 564 | (1.7 ± 0.03) × 10−2 |
| Oxyferrous | 417, 541, 577 | not determined | 417, 542, 578 | not determined |
M−1s−1 and s−1 for Compounds ES and RH, respectively
In the absence of co-substrate, DHP B Compound ES was found to be unstable and converted to a new species [UV-visible: 411 (Soret), 554, 599 nm; kobs = 0.010 ± 0.001 s−1] (Scheme 1, step vi-a). We have termed this stable endpoint in the DHP B catalytic cycle as Compound RH based upon analogy to a similar, but not identical, species observed in DHP A (20). Both Compound RH species form from Compound ES, but have different UV-visible spectroscopic properties and different chemical reactivity (vide infra).
Reaction of Preformed Compound ES with TCP Co-Substrate
Double-mixing stopped-flow UV-visible spectroscopic methods were used to investigate the reaction of pre-formed DHP B Compound ES with TCP co-substrate. The results of the UV-visible and resonance Raman studies suggest that TCP binds at an external binding site under the conditions employed in these stopped-flow studies reported herein, although such an assumption does not directly affect the interpretation of the results presented. Ferric DHP B was reacted with 10 molar equivalents of H2O2 at pH 7, incubated for 500 ms to allow for the maximum accumulation of Compound ES (see Figure 4C), and subsequently mixed with 30 molar equivalents of TCP, which resulted in the regeneration of ferric DHP B (Figure SD5) after 18.5 s (Scheme 1, step v). The disappearance of Compound ES was concomitant with the formation of the DCQ product. Furthermore, no Compound RH was observed here, and the final spectrum [UV-visible: 409 (Soret), 502, 538, 576 nm] suggested the presence of a mixture of ferric and oxyferrous DHP B (vide infra). Additionally, varying the concentration of TCP co-substrate (5 and 15 equivalents, data not shown) in an attempt to determine the second order rate constant for the reduction of Compound ES by TCP was also investigated. However, while the reaction employing lower equivalents of TCP was qualitatively the same with respect to the formation of ferric DHP B (and subsequently oxyferrous DHP B) as to that observed for 30 equivalents of co-substrate, the reaction kinetics were poorly behaved and did not allow for a quantitative determination of the bimolecular rate constant. We attribute this to the multiple reactions (i.e., steps v, vii, ix of Scheme 1) that may be occurring both sequentially and concurrently that give rise to the overall observed transition of Compound ES to oxyferrous DHP B.
Repetition of the above double-mixing experiment with increasingly longer incubation times (800 ms – 5 min) that allowed for the conversion of Compound ES to RH exhibited progressively less co-substrate loss (312 nm) and less product (275 nm) formation (Figure SD6). Shorter incubation times (100 and 300 ms) that did not allow for complete formation of Compound ES still exhibited the same amount of TCP loss and DCQ formation as was observed for when Compound ES was maximally formed. Thus, the extent of product formation is directly correlated with the amount of Compound ES present or capable of being formed, strongly indicating that this intermediate is an active oxidant in DHP. Interestingly, DHP B still retained a small fraction of activity even after 5 min of pre-incubation (as evidenced by both co-substrate loss and product formation) at which point our component analysis indicates only Compound RH and no Compound ES present (Figure 4C). DHP A Compound RH exhibited no such activity under identical experimental conditions (20).
In situ DHP B Compound ES Formation in the Presence of TCP
Double-mixing stopped-flow UV-visible spectroscopic methods were employed to allow for pre-incubation of ferric DHP B with TCP (30 molar equivalents; 500 ms incubation), followed by the addition of a 10-fold excess of H2O2 (Figure SD7). This set of conditions allowed for the in situ formation of Compound ES in the presence of TCP, as opposed to the above experiments in which Compound ES was pre-formed. DCQ product formation (275 nm) was observed, and exhibited a pseudo-first-order rate constant (kobs) of 0.19 ± 0.03 s−1 at pH 7. However, the amount of product formation was significantly less than when Compound ES was pre-formed and then reacted with TCP (Figure SD6). This observation contrasts with DHP A which showed identical amounts of DCQ generated regardless of whether pre-formed or in situ-formed Compound ES was employed (20). In order to explore this chemistry further, the incubation time for ferric DHP B and TCP was varied from 0.1 – 60 s (Figure SD8). Interestingly, while the data do not support a clear temporal dependence on the amount of product formed or co-substrate reacted, they do show an attenuation of the dehaloperoxidase reaction which is possibly indicative of co-substrate inhibition, but this chemistry was not further explored.
DHP B Compound ES was not observed under these conditions, and is likely indicative of its immediate reduction in the presence of TCP co-substrate. DHP A, however, did yield an observable in situ formed Compound ES at pH 7, but not pH 5, and we attribute this difference to the higher activity of DHP B when compared to DHP A. Of significant note, however, was the observation of an electronic absorption spectrum that did not match that of ferric DHP B at the conclusion of this experiment (Figure SD7; 83 s). The spectral features [UV-visible: 410 (Soret), 541, 577 nm] were more similar to those of oxyferrous DHP B (Table 1), particularly with respect to the visible absorption features at ~541/577 nm, and were also similar to what was observed in the reaction of preformed Compound ES with TCP (Figure SD5). We interpret this spectrum to be a mixture of both oxyferrous and ferric DHP B based primarily on these data and our further investigations on the reaction between DCQ product and ferric DHP (vide infra).
Characterization of Protein Radicals in DHP B Compound ES
In order to confirm the presence of a protein radical in DHP B Compound ES, continuous wave (CW) EPR spectroscopy was employed to probe the intermediate of the reaction of ferric DHP B and a 10-fold excess of H2O2 at pH 7 using rapid-freeze-quench (RFQ) techniques. The X-band RFQ-CW-EPR spectra for DHP B Compound ES with quench times of 100 ms, 300 ms, 500 ms, 800 ms, and 2 s are presented in Figure 5. The position of the signal is characterized by an effective g-value of 2.0057, and the shape of the signal is best described as an anisotropic septet, although both the shape and intensity of the signal change as a function of quench time. At the longest quench time (60 s), when our component analysis (Figure 4C) suggests little to no remaining Compound ES present by UV-visible spectroscopy, the signal intensity dropped significantly, and it was not possible to resolve the hyperfine structure. Previously, we observed that the protein radical in DHP A Compound ES exhibited an anisotropic quintet at pH 7, and an anisotropic septet at pH 5 (both at g = 2.0058) (20). Thus, the DHP B signal collected here at pH 7 more closely resembles that observed for DHP A at pH 5. Furthermore, while their intensities decreased as a function of quench time, neither signal for DHP A exhibited the variation in lineshape observed here for DHP B. We tentatively assign the EPR signal observed here at short quench times (100 – 800 ms) as a tyrosyl radical based on the obtained g value and the partially resolved hyperfine structure with a peak-to-peak line width of approximately 21 G (67, 68).
Figure 5.
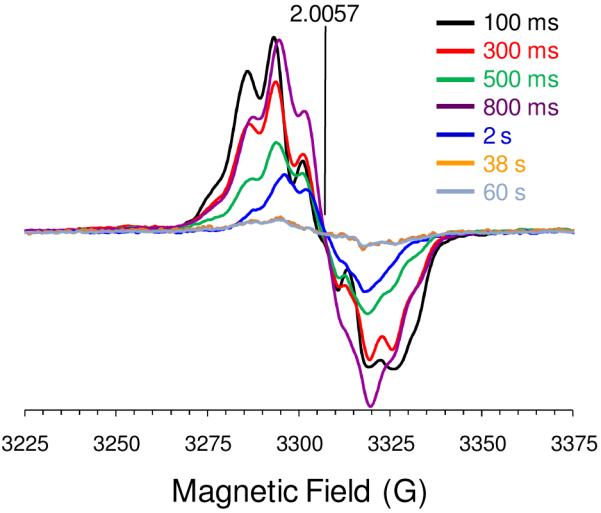
EPR spectra of the radical(s) in DHP B Compound ES at pH 7. Rapid-freeze quench samples were prepared from the reaction of ferric DHP B (50 μM final concentration) with a 10-fold molar excess of H2O2 at 25 °C, and rapidly frozen in an isopentane slurry. Spectra were recorded at 77 K using the spectrometer settings described in the Materials and Methods section. The cavity resonant frequency was 9.28005 GHz.
While we are able to demonstrate its presence, the low g factor spectral resolution of these RFQ-CW X-band experiments here prevents an unequivocal identification of the radical species based solely upon the observed magnetic parameters. However, in a separate report on DHP A (69), the assignment of the Compound ES radical location was performed using the aid of the Tyrosyl Radical Spectra Simulation Algorithm (TRSSA) (68). In this method, two input parameters, the phenoxyl ring rotation angle (θ) and the spin density on atom C1 of the radical (ρC1), were used to calculate twelve different EPR spectral simulation parameters (gx,y,z, Aβ1x,y,z, Aβ2x,y,z, and ΔHx,y,z) using semi-empirical dependences. These TRSSA-derived simulation parameters were combined with another 18 (ϕ and Ax,y,z for C3, C5, C2, and C6; ϕβ1 and ϕβ2) that are set invariant in the algorithm for all tyrosyl radicals, and were then used to simulate the EPR spectra of Compound ES using Simpow6 (70). The derived θ-value that led to an accurate simulation was then compared to tyrosine residues with similar θ angles using the Phenol Ring Rotation Angle Database (71), which when correlated with the known geometries of all the tyrosine residues from the available crystal structure of DHP, led to an assignment of the tyrosine candidate(s) likely to host the radical. Using this approach for DHP A, two different radicals were identified as being both present in Compound ES, a primary tyrosyl radical that is characterized by a phenoxyl ring rotation angle of 45° or 75° and was suggested to correspond to either Tyr34 or Tyr28, and a secondary radical that was suggested to reside on Tyr38. Furthermore, spin quantification revealed that the Tyr34 radical is formed with a very high relative yield (almost 100% of heme), atypical of other globins. By extending this interpretation to the observed heterogeneity of the EPR lineshape in DHP B, which lacks a tyrosine at position 34, we suggest that the radical initially forms on Tyr28 and Tyr38. Although tentative, these assignments are in good agreement with our preliminary mutagenesis studies [R. Dumarieh, J. D’Antonio, D. A. Svistunenko, R. A. Ghiladi, unpublished results] in which the mutation of these residues leads to Compound I formation (see below). However, in light of the limitations of simulating the poorly resolved hyperfine splitting at low field, it will be necessary to utilize high-field EPR spectroscopic measurements in tandem with this site-directed mutagenesis study in order to unambiguously assign the nature of the protein radical in DHP B Compound ES.
EPR spectroscopic characterization of DHP Compound ES at liquid helium temperature has also been reported (69). In that study, the kinetic dependences of the ferric heme state and the free radical concentrations were investigated at low microwave power. Upon addition of hydrogen peroxide to ferric DHP, a nearly quantitative loss of the HS ferric heme signal (upon formation of the EPR silent ferryl state of Compound ES) was observed at pH 7, concomitant with the nearly quantitative formation of the tyrosyl radical signal. Moreover, the recovery of the ferric heme signal was noted to begin after 1 minute, which coincides with the formation of Compound RH from ES. Overall, the reported results strongly support the proposed mechanism in Scheme 1 in which the formation of a tyrosyl radical and ferryl heme occur upon the two-electron oxidation of ferric DHP by hydrogen peroxide.
Reactivity of Compound ES with DCQ
To explore the possibility that DHP B Compound ES could react directly with the product DCQ (Scheme 1, step vii), the reaction of pre-formed Compound ES with 7 equivalents of DCQ was monitored by double-mixing stopped-flow UV-visible spectroscopy. Compound ES was first formed upon rapid mixing of a solution of ferric DHP B with 2.5 equivalents H2O2, allowed to incubate for 800 ms to ensure its full formation, and was subsequently mixed with a solution of DCQ (7-fold excess). As shown in Figure 6, loss of DCQ (275 nm) was observed, concomitant with the conversion of Compound ES [UV-visible spectrum: 420 (Soret), 546, 588 nm] to a new species [UV-visible spectrum: 415 (Soret), 541, 577 nm] whose spectral features closely resemble those observed for oxyferrous DHP B, particularly with respect to the visible absorption features at ~541/577 nm. We interpret this spectrum to be a mixture of both oxyferrous and ferric DHP B. Under these conditions, no Compound RH was observed, strongly suggesting that Compound ES was reduced in the presence of DCQ. Varying concentrations of DCQ (data not shown) were also employed in an attempt to determine the bimolecular rate constant for the reduction of Compound ES by DCQ. Again, although the reactions were qualitatively the same, the kinetics were poorly behaved, likely indicative of several reactions with varying dependencies on DCQ concentration occurring in step vii of Scheme 1, and the quantitative determination of the 2nd order rate constant was not possible.
Figure 6.

(A) Stopped-flow UV-visible spectroscopic monitoring (900 scans, 85 sec) of the double-mixing reaction between preformed DHP B Compound ES (10 μM; 500 ms) and a 7-fold molar excess of DCQ at pH 7. (B) Calculated UV-visible spectra for Compound ES (black) and a ferric/oxyferrous DHP B mixture (grey) are shown; the rapid-scanning data from panel A were compiled and fitted to a one-step, two species sequential irreversible model using the Specfit global analysis program.
To examine the effect of DCQ pre-incubation on in situ formed Compound ES, the above experiment was repeated by first incubating ferric DHP B with 7 equivalents of DCQ for 500 ms, followed by the addition of 2.5 equivalents of H2O2 (Figure SD9). The first spectrum observed exhibited spectral features that matched those of a mixture of oxyferrous and ferric DHP B [UV-visible spectrum: 408 (Soret), 508, 541 (sh), 577 (sh) nm]. This spectrum converted cleanly to DHP B Compound ES [UV-visible spectrum: 419 (Soret), 546, 588 nm], before returning once again to a ferric/oxyferrous mixture [UV-visible spectrum: 413 (Soret), 541, 577 nm]. As was noted above, DCQ loss during this experiment was observed. Furthermore, it was possible to observe formation of the Compound ES intermediate here in situ in the presence of DCQ (as a co-substrate), whereas this was not possible when TCP was employed as co-substrate. Overall, the reaction chemistry with DCQ, regardless of whether Compound ES is preformed or generated in situ, leads to the reduction of this intermediate and the formation of oxyferrous DHP, likely through a transiently formed deoxyferrous species.
Incubation of DCQ with Ferric DHP B
As noted in the previous double-mixing experiment in which ferric DHP B was pre-incubated with DCQ prior to their reaction with H2O2, a mixture of oxyferrous and ferric DHP B was initially observed. As the hydrogen peroxide chemistry described herein has been shown to yield Compound ES, and not oxyferrous DHP, the reaction between ferric DHP B and DCQ in the absence of H2O2 (Scheme 1, step ix) was investigated using single-mixing stopped-flow techniques in order to further elucidate the origin of formation of the oxyferrous species shown in Figures SD5, SD7 and 6. The reaction initiated upon rapid mixing of ferric DHP B with 0.5 – 10 equivalents DCQ was monitored by UV-visible spectroscopy over 85 (Figure SD10) and 800 seconds (Figure 7) at pH 7. The data exhibited saturable-kinetics with a clear formation of oxyferrous DHP B [UV-visible spectrum: 413-5 (Soret), 541, 577 nm; kcat = (8.49 ± 0.26) × 10−2 s−1; Km = 5.4 ± 0.9 μM], with concomitant loss of the DCQ feature (275 nm). In a separate non-enzymatic control experiment (data not shown), DCQ loss due to autooxidation was observed to be less than 1% over 5 minutes at pH 7, indicative of a DCQ degradation pathway under the conditions employed in this study, but one which is not relevant to the timescale of the stopped-flow experiment. Thus, it appears that DCQ itself may be able to reduce ferric DHP B in the presence of dioxygen to generate the observed oxyferrous complex. Although outside of the scope of this current study, it is not clear at this time what the products of DCQ degradation are in the presence and absence of ferric DHP B, and these questions will be addressed separately in future experiments. The reactivity of DCQ demonstrated here complicates the determination of a DXQ binding affinity for ferric DHP B.
Figure 7.

(A) Stopped-flow UV-visible spectroscopic monitoring (900 scans, 800 sec) of the reaction between ferric DHP B (10 μM) and a 7-fold excess of DCQ at pH 7. (B) Calculated UV-visible spectra for ferric (black) and the ferric/oxyferrous DHP B mixture (grey) are shown; the rapid-scanning data from panel A were compiled and fitted to a one-step, two species sequential irreversible model using the Specfit global analysis program. (C) Double-reciprocal plot.
Spectroelectrochemistry of DHP B
The reduction potential of DHP B at pH 7 was determined by spectroelectrochemistry. The UV-visible spectroelectrochemical plot of DHP B at various applied potentials (Eapplied versus SHE) are shown in Figure 8A. In the absence of dioxygen, ferric DHP B (λmax = 407 nm) converted to the ferrous enzyme (λmax = 432 nm) as the applied reduction potential was lowered from 497 mV to −130 mV. The corresponding Nernst plot (Figure 8B) reveals a formal reduction potential of 206 ± 6 mV (vs. SHE), with a slope of 72 ± 5 mV consistent with an n=1 process. The spectroelectrochemical measurement for obtaining the reduction potential was found to be reversible when the electrochemical stepping procedure (titration) was initiated from either the fully oxidized (ferric) or fully reduced (deoxyferrous) state (results not shown). The observed formal reduction potential for DHP B is nearly identical to that of DHP A (202 ± 6 mV) (72), and suggests that the five mutations between these two isoenzymes do not strongly influence their electrochemical properties. The relatively high reduction potential of this oxygen-transport globin may be a contributing factor as to why the reaction of DCQ with ferric DHP B yields the oxyferrous enzyme.
Figure 8.

Spectroelectrochemical determination of the formal reduction potential of the ferric/ferrous couple in DHP B. (A) The UV-visible spectroelectrochemical plot of DHP B at various applied potentials (Eapplied versus SHE) are shown. In the absence of dioxygen, ferric DHP B (λmax = 407 nm) converted to the ferrous enzyme (λmax = 432 nm) as the applied reduction potential was lowered from 497 mV to −130 mV. (B) The corresponding Nernst plot of the data in panel A.
Compound RH Formation and Chemical Reactivity
Previously, we observed that DHP A Compound RH was able to be reduced in the presence of sodium dithionite at pH 7 to subsequently form the oxyferrous complex (20), providing a possible link between the two activities (oxygen carrier and peroxidase) of this bifunctional hemoprotein. To explore whether this chemistry was possible with DHP B Compound RH, this species was formed as described above from the decay of DHP B Compound ES, itself formed from the reaction of the ferric enzyme with 10 equivalents of H2O2 at pH 7. Upon its full formation (as confirmed by UV-visible spectroscopy), catalase was then added to remove any unreacted hydrogen peroxide, yielding Compound RH. Neither excess sodium dithionite nor ascorbate were able to generate the (oxy)ferrous enzyme within 15 minutes. Rather, a new species was observed (Scheme 1, step vi-b) which exhibited a Soret band maximum at 426 nm and no distinct visible bands (Figure SD11). For discussion within this report, we have termed this new species Compound P426.
By contrast, when the above experiment was repeated using Compound RH that was formed from 2.5 equivalents of H2O2 rather than the ten equivalents employed previously, treatment with catalase followed by addition of excess sodium dithionite did reveal the formation of ferrous DHP B (Figure SD12), which then converted to the oxyferrous complex upon consumption of the excess reductant (Scheme 1, step vi-c). Thus, the extent of the formation of the 426 nm species is dependent upon the amount of H2O2 used to generate the precursor Compound RH. Given the conditions needed to form it, there is a high likelihood that Compound P426 is a non-physiologically relevant species, and its chemistry was not explored further.
DISCUSSION
The terebellid polychaete Amphitrite ornata has been shown to possess two genes, dhpA and dhpB (19), which encode for dehaloperoxidase isoenzymes A and B, respectively. While the molecular details of the dehaloperoxidase catalytic cycle have been investigated for some time for isoenzyme A, no such mechanistic studies have been performed for DHP B. The primary focus of this report is to provide the details of dehaloperoxidase chemistry which have yet to be reported for either isoenzyme, and to augment that with a full study of dehaloperoxidase B.
The activity studies presented herein demonstrate that dehaloperoxidase B is able to catalyze the oxidative dehalogenation of 2,4,6-trihalogenated phenols to their corresponding 2,6-dihalo-1,4-benzoquinones in the presence of hydrogen peroxide. As brominated aromatics are the most abundant halometabolites present in the environment of A. ornata (10), it was surprising that 2,4,6-tribromophenol was found to be oxidized with the lowest catalytic efficiency under the conditions examined, a result that differs from that reported previously for DHP A (2). Overall, DHP B exhibits a higher catalytic rate for all co-substrates (TFP, TCP and TBP) examined when compared to DHP A under identical conditions. However, the catalytic efficiencies for the dehalogenation of these co-substrates are similar between the two isoenzymes due to the higher Km for hydrogen peroxide binding in DHP B over that determined for DHP A. Km for trihalophenol co-substrate was determined to be in the high micromolar range. In the absence of data regarding protein expression levels in vivo, it is not possible to speculate whether the differences in activity relate to selective isoenzyme expression under conditions of high oxidative or excessive trihalophenol stresses.
Enzymatic activity was observed regardless of whether the catalytic cycle was initiated from the ferric or oxyferrous states. As DHP is a globin-peroxidase, this is of critical importance given that the two inherent functions of the enzyme traditionally require different iron oxidation states: ferrous for O2-transport, ferric for peroxidase activity. Furthermore, the oxyferrous state is normally a catalytically incompetent species for monofunctional peroxidases (73), which makes this observation even more unique to DHP. Thus, we surmise that dehaloperoxidase may have evolved its peroxidase function to begin from the oxyferrous state, which is the normal oxidation state for this hemoglobin in A. ornata. How the enzyme accomplishes this and functions as both an O2-carrier and a peroxidase may be linked to the highly unusual conformational flexibility of the distal histidine in dehaloperoxidase. Typically, the Fe-His Nε2 distances in globins range between 4.1–4.6 Å, and 5.5–6.0 Å for peroxidases (74-77). By comparison, in dehaloperoxidase the Fe-His Nε2 distance in ferric DHP B is 5.5 Å (37) and between 4.8 and 5.5 Å in DHP A (27, 36, 45) for when the distal histidine is in the closed position. Although not available for DHP B, the Fe-His Nε2 distance in oxyferrous DHP A is 5.1 Å. Thus, the distal histidine-to-heme distance in dehaloperoxidase, being intermediate between those found in globins and peroxidases, is well positioned to function as both a stabilizer of the bound dioxygen ligand in the former, and as the general acid/base that facilitates both the deprotonation and heterolytic O-O bond cleavage of hydrogen peroxide in the latter. Interestingly, using site-directed mutagenesis Watanabe and co-workers repositioned the distal histidine in sperm whale myoglobin from 4.3 Å in the wild-type protein to 5.7 Å in the F43H/H64L Mb mutant, effectively converting Mb from an oxygen transport protein to a peroxidase (74). They concluded that the interaction of the closer distal histidine in wild-type Mb with both oxygen atoms of an iron-bound peroxide would not facilitate the charge separation that is necessary for heterolysis, whereas the repositioned distal histidine in the F43H/H64L mutant, being further from the iron-bound oxygen atom, was suggested to interact exclusively with the terminal oxygen atom, thereby leading to the heterolytic O-O bond cleavage necessary in peroxidases. With His55-O(1) (iron-bound) and His55-O(2) (terminal) distances of 3.2 and 2.8 Å (27), respectively, oxyferrous DHP exhibits similarities to wild-type Mb in that the distal histidine interacts with both of the oxygen atoms of the end-on bound dioxygen ligand, while at the same time ferric DHP has a longer Fe-His Nε2 distance that more resembles those of peroxidases. Taken together, these characteristics that are particular to the highly flexible distal histidine in dehaloperoxidase are likely vital to answering the larger question of how DHP performs its dual functions.
Stopped-flow UV-visible spectroscopic studies were employed to monitor the reaction of ferric DHP with hydrogen peroxide (the putative physiological oxidant) in the absence of co-substrate. Similar to DHP A, the first intermediate observed exhibited spectral features which matched those for a ferryl-containing species that lacked a porphyrin π-cation radical (20), suggestive of the formation of Compound ES or Compound II. We interpret this result as the transient formation of Compound I upon heterolysis of the O-O bond of hydrogen peroxide, followed by rapid endogenous electron transfer yielding the observed Compound ES intermediate. An alternative explanation as to the lack of an identifiable Compound I intermediate under the conditions explored is that the reaction with hydrogen peroxide proceeds homolytically, thereby directly generating the observed ferryl species. However, several lines of evidence argue against this. First, the observation of the protein radical in Compound ES concomitant with the ferryl state favors a two-electron oxidation process (i.e., heterolytic cleavage). Second, Compound I has been identified as a transient species in myoglobin (66, 78), and it is likely that dehaloperoxidase, being a globin itself, proceeds through similar reaction chemistry. Third, a recent report from Dawson and co-workers demonstrated a significant amount of heterolytic O-O bond cleavage of cumene hydroperoxide upon its reaction with DHP A, consistent with the formation of a transient Compound I species (31). Finally, our preliminary characterization of mutants of DHP A and B which lack both Tyr34 and Tyr38 exhibit by optical spectroscopy an intermediate that possesses the spectral features of Compound I [R. Dumarieh, J. D’Antonio, D. A. Svistunenko, R. A. Ghiladi, unpublished results].
In the presence of co-substrate, the disappearance of Compound ES was observed in parallel with the formation of the quinone product, however differences were noted between preformed and in situ-generated Compound ES. Specifically, the amount of product formed when Compound ES was generated in situ was less than one-third of that produced by preformed Compound ES. These results for DHP B differ from the observation reported previously for DHP A, where equivalent amounts of product were generated regardless of how Compound ES was formed (20). One possible interpretation of the results here is that the binding of a small molecule to DHP B interferes with the subsequent activation of H2O2 at the heme center. This may occur if the small molecule binding sterically blocks access of the hydrogen peroxide to the heme iron. Also of note is that both reactions involving either preformed or in situ generated Compound ES yielded oxyferrous DHP B in the presence of co-substrate; these results differ from DHP A in which both methods for forming Compound ES yielded ferric enzyme upon reduction with co-substrate (20). While it is tempting to suggest that these results may represent two different reaction pathways for co-substrate oxidation, i.e. the reactivity of a transiently formed Compound I versus that of pre-formed Compound ES, our findings regarding the reactivity of DCQ with ferric DHP B (vide infra) point to a second reaction which was not known when DHP A was first being investigated.
In the absence of co-substrate, DHP B Compound ES was found to decay to a new, stable species termed Compound RH. Again, differences with DHP A were observed. First, the electronic absorption spectrum of DHP B Compound RH differs from that observed in isoenzyme A (20), possibly indicative of a different electronic structure present. Moreover, whereas the oxyferrous enzyme could be regenerated from DHP A Compound RH upon reduction, providing a link between peroxidase and O2-transport functions, DHP B Compound RH cannot. Rather, a new species unique to DHP B was observed. The formation of ‘Compound P426’, so termed for the absorption maximum of the Soret band, was found to be dependent on the amount of hydrogen peroxide used to first generate the precursors, Compounds ES and RH, with increasing amounts of H2O2 leading to full formation of this species. The significance of Compound P426 in the dehaloperoxidase catalytic cycle, if any, is not known at this time.
Our findings point to DHP B Compound ES as a catalytically competent species in the dehaloperoxidase reaction cycle. Dichloroquinone product formation may result from either a single two-electron oxidation of the trichlorophenol co-substrate by Compound ES, or two sequential one-electron oxidations, both pathways being indistinguishable under the multiple-turnover conditions examined here. However, Dawson and co-workers (31) recently reported evidence supporting a peroxidase cycle for DHP A involving two sequential one-electron oxidations of TCP to yield DCQ when preformed Compound ES was employed. Thus, it is likely that the same reaction pathway with respect to co-substrate oxidation may be occurring here for DHP B. Our data also show significantly attenuated rates of co-substrate loss and product formation when Compound ES is allowed to decay to Compound RH, either partially or fully, prior to the addition of co-substrate. The implication here is that Compound RH is not a catalytically relevant species in the DHP reaction cycle, although it does appear to possess a very low level of activity.
RFQ-CW-EPR spectroscopic studies were employed to confirm the presence of the protein radical in DHP B Compound ES. The absence of an observable Compound I species under the conditions employed here suggests the presence of an endogenous reductant which we have tentatively ascribed to as a tyrosine side chain (20), yielding the EPR-observable tyrosyl radical. Indeed, we have demonstrated the presence of such a carbon-centered signal here for DHP B Compound ES at pH 7 based upon our analysis of the signal g-factor, partially resolved hyperfine structure, and peak-to-peak line width (~21 G). Of the four tyrosine residues in DHP B [Tyr16, Tyr28, Tyr38, and Tyr107], Tyr28 and Tyr38 are the most likely candidates for the initial site of radical formation given their relative proximity of 10.24 Å and 7.57 Å, respectively, to the heme edge, while the remaining two tyrosine residues are >15 Å distant. The time-dependent changes observed in the protein radical signal, which are more pronounced for DHP B than for DHP A, may suggest a migration of the radical out of the active site to other redox active protein side chains (including Trp120, 10.47 Å distant from the heme edge) upon decay of the Compound ES intermediate (20). Thus, we highlight that there may be a difference between the site of initial radical formation and that of the observed radical in Compound ES. With respect to the EPR parameters (g-value, hyperfine splitting), a comparison of the radical signal observed here for DHP B at pH 7 reveals that it is more similar to that signal noted for DHP A at pH 5 rather than at pH 7 (20). Given that the Tyr34 residue present in DHP A is absent in DHP B (replaced with a redox inactive asparagine), we rule out that the location of the radical observed in DHP A at pH 5 given the persistence of the similar signal in DHP B, albeit at the higher pH value. One possible explanation for this pH-dependent observation is that the mutations in DHP B provide an alternative electron-transfer pathway between the heme center and the putative external co-substrate binding site, thus giving rise to EPR signals that originate from different redox active amino acid residues.
The reactivity of dichloroquinone, the presumed end-product of TCP oxidative dehalogenation, with either in situ generated or preformed DHP B Compound ES was also investigated by double-mixing stopped-flow UV-visible spectroscopy. Under the conditions examined, these studies revealed that Compound ES, regardless of how it was generated, reacted with DCQ to ultimately yield oxyferrous DHP B. Thus, in a novel reaction pathway not previously observed for DHP A, the two-electron oxidized Compound ES intermediate was reduced by three electrons in the presence of DCQ, itself already two electrons oxidized compared to the starting co-substrate TCP. Although the mechanistic details of this highly unusual reaction will require a separate full study, one possible explanation is that DCQ is further oxidized, perhaps yielding a polymeric species in solution, and thus presumably able to reduce Compound ES, although one might rather expect a return to the resting ferric state in this case. Even more significant, however, was the finding that ferric DHP B alone was capable of reacting with DCQ to also generate the oxyferrous complex. Here, we hypothesize that the inherent instability of DCQ in solution, as evidenced by its degradation in our non-enzymatic and oxidant-free control reaction, leads to the production of species, possibly radical in nature, that reduce ferric DHP B to ferrous in the presence of dioxygen to yield the observed oxyferrous complex. This would also account for the above formation of oxyferrous DHP B in the reaction of Compound ES with DCQ if one invoked a ferric species in that pathway. The unusually high reduction potential for a peroxidase, as deduced by our spectroelectrochemistry study, likely facilitates reactions that favor reduction of the heme and formation of oxyferrous DHP B. Thus, the dichloroquinone chemistry explored herein may represent a link between the two activities of the bifunctional dehaloperoxidase by allowing for the ferric state to initiate either a peroxidase pathway in the presence of TCP and H2O2, or enabling O2-transport capability by forming the oxyferrous complex in the presence of DCQ (itself generated from the aforementioned peroxidase pathway).
The UV-visible and resonance Raman spectroscopic studies presented here also provide a picture of how small molecule binding affects the electronic structure of the heme active site in DHP B. As inferred from their effect on the relative populations of five and six coordinate high spin heme (50), a clear difference between internal (36) and external (22, 25, 49) small molecule binding was observed. In particular, monohalophenol binding led to a greater population of 5cHS heme, which we interpret as internal binding leading to the displacement of the heme-bound axial water ligand and locking the distal histidine His55 in the ‘open’ conformation (37, 50). Biophysical studies and molecular dynamics simulations on both oxyferrous and carbonmonoxy Mb illustrate the conformational flexibility of the distal histidine in globins (79-81). In particular, the molecular dynamics simulations on the His64 NδH tautomer in Mb showed it to exist in both ‘open’ and ‘closed’ conformations (80), and we envision a similar scenario in dehaloperoxidase B. In the ‘open’ conformation, a channel between the solvent and the heme active site is formed (37), allowing for monohalophenol to enter and sterically block the distal histidine from assuming the ‘closed’ conformation. Trihalophenol binding, however, led to a greater population of 6cHS, which we interpret as external binding where the heme-bounded water molecule is stabilized by this ‘closed’ conformation of the distal histidine (50). The findings presented herein for DHP B are in good agreement with previous studies from which one can surmise internal and external binding sites in DHP A (22, 25, 36, 49). The conformational flexibility of the distal histidine in DHP may be critical in understanding how this hemoprotein functions as both a peroxidase and an oxygen-transport globin (21, 37), as activation of hydrogen peroxide is predicated upon the ability of the distal histidine to facilitate O-O bond cleavage (i.e., the ‘pull’ of the push-pull mechanism). Presumably, monohalophenol binding would preclude the distal histidine from participating in the activation of hydrogen peroxide due to it being locked into the ‘open’ conformation (Scheme 1, step i-a), leading to an attenuation of the oxidative dehalogenation reaction as catalyzed by DHP B. Indeed, in agreement with this supposition are the results of our inhibition study here which strongly suggest that 4-bromophenol is an inhibitor of the oxidation of 2,4,6-trichlorophenol. If correct, then other factors pertaining to the in vivo biological relevance of monohalophenols, which are outside the scope of this in vitro study, need to be elucidated in order to better understand the role of monohalophenols in the environment of A. ornata.
In light of the spectroscopic and biochemical findings presented here, we propose the following updated mechanism for the in vitro peroxide-dependent oxidation of ferric DHP B from A. ornata in the presence and absence of trihalophenol and dichloroquinone (Scheme 1): In the presence of a monohalophenol, ferric DHP forms a 5cHS inhibited complex with the distal histidine (His55) in the open conformation (step i-a). This inhibited form of the enzyme is unreactive towards H2O2 alone (i-b) or in the presence of TXP (i-c). When trihalophenol is present, ferric DHP forms a 6cHS active species with the distal histidine in the closed conformation (ii-a). If H2O2 is added to this active complex, catalytic turnover results (ii-b). However, if a monohalophenol is added prior to the addition of H2O2, then the complex is again inhibited (ii-c).
In the absence of (tri)halophenol, ferric DHP B reacts with one equivalent of H2O2, yielding a putative Compound I intermediate (step iii), which then undergoes rapid endogenous electron transfer from an amino acid side chain to generate the observed Compound ES and a protein radical (iv). From this intermediate, we suggest three possible pathways that are co-substrate and/or product dependent. In the presence of trichlorophenol, Compound ES is reduced by two electrons [likely in two separate one-electron reduction steps as described by Dawson and co-workers (31)], regenerating the ferric state and forming dichloroquinone, chloride, and water as products (v). The DCQ product thus formed, either due to its inherent instability or as an alternative co-substrate for the enzyme, leads to the reduction of the ferric state to the ferrous, generating oxyferrous DHP B in the presence of dioxygen (ix). Alternatively, Compound ES may directly react with DCQ as a co-substrate, leading to the generation of the oxyferrous complex (vii). In the absence of co-substrate, DHP B Compound ES was found to form Compound RH (vi-a), which upon reduction yields either oxyferrous DHP B (vi-c) or the novel Compound P426 state (vi-b), depending on the number of oxidizing equivalents that went into first forming Compound RH. Critically, oxyferrous DHP B is a catalytically competent starting state for the oxidative dehalogenation of TCP in the presence of H2O2 (step viii).
Conclusion
The chemistry reported herein furthers our understanding of the mechanism of dehaloperoxidase. That oxyferrous DHP is a competent starting oxidation state for enzymatic turnover, and that the presumed products of trihalophenol oxidation, namely dihaloquinones, are themselves co-substrates for the ferric enzyme that lead to oxyferrous DHP, are two notable observations which suggest how this globin-peroxidase is able to reconcile its two activities, each with a distinctive oxidation state, in one system. An intriguing interpretation of the results is that the ferric state may not be the only functionally relevant starting oxidation state for the peroxidase activity of DHP in vivo. Additionally, the conformation flexibility of the distal histidine appears to serve as a functional trigger between active and inhibited states of the enzyme, dependent upon the type of halophenol, mono vs. tri, present. While DHP B does exhibit some differences between its reactivity and that observed for DHP A, the most significant findings provide the necessary link between the peroxidase and O2-transport activities present in this bifunctional hemoprotein. The new insights provided by our findings in vitro help rationalize how dehaloperoxidase may maintain both of these roles in vivo, although additional questions still need to be addressed. For example, it is still not known why A. ornata possesses two dehaloperoxidase isoenzymes. Given the parallels between hemoglobin and DHP, one intriguing possibility is that DHP A and B form a cooperative dimer or tetramer, similar to the α2β2 structure of Hb, and future studies employing mixtures of DHP A/B are planned. Additionally, the spectroscopic findings presented here, when viewed in context of our recent structural investigations of DHP, provide a clearer picture of the structure-function relationship in dehaloperoxidase, and may have potential implications for globin protein engineering or directed evolution of DHP for bioremediation.
Supplementary Material
Abbreviations
- DHP
dehaloperoxidase
- Hb
hemoglobin
- Mb
myoglobin
- DCP
2,4-dichlorophenol
- TBP
2,4,6-tribromophenol
- TCP
2,4,6-trichlorophenol
- TFP
2,4,6-trifluorophenol
- TXP
trihalophenol
- DXQ
dihalophenol
- RFQ-CW-EPR
rapid-freeze-quench continuous wave electron paramagnetic resonance
- Compound I
two-electron oxidized heme center when compared to the ferric form, commonly as an FeIV=O porphyrin π-cation radical
- Compound II
one-electron oxidized heme center when compared to the ferric form, commonly as an FeIV=O or FeIV–OH
- Compound III
oxyferrous [FeII-O2 or FeIII-(O2−)] state of the enzyme
- Compound ES
two-electron-oxidized state containing both a ferryl center [FeIV=O] and an amino acid (tryptophanyl or tyrosyl) radical, analogous to Compound ES in cytochrome c peroxidase
- Compound RH
‘Reversible Heme’ state of dehaloperoxidase, formed from the decay of Compound ES in the absence of co-substrate
- Compound P426
state of DHP B formed upon reduction of Compound ES with Na2S2O4
- 5cHS
five-coordinate high spin heme
- 6cHS
six-coordinate high spin heme
- 6cLS
six-coordinate low spin heme
Footnotes
This project was supported by the North Carolina State University Molecular Biotechnology Training Program through an NIH T32 Biotechnology Traineeship grant (J. D.), the Army Research Office Grant 52278-LS (S. F.) and Grant 51432-CH-SR (E. B.), and the North Carolina State University Department of Chemistry (R. G.).
Supporting Information Available – UV-visible spectra of different heme states of DHP B (10 μM) at pH 7.0 (Figure SD1); UV-visible spectra of the (tri)halophenol complexes of DHP B (Figure SD2); UV-visible spectroscopic monitoring of the oxidative dehalogenation of trihalophenols as catalyzed by DHP B in the presence of hydrogen peroxide (Figure SD3); dependence of kobs for the reaction between ferric DHP B with hydrogen peroxide (2.5 – 25 equivalents) at pH 7 yielding Compound ES (Figure SD4); stopped-flow UV-visible spectroscopic monitoring of (Figure SD5), and DCQ product formation and TCP co-substrate loss for (Figure SD6), the double-mixing reaction between preformed DHP B Compound ES and TCP at pH 7; stopped-flow UV-visible spectroscopic monitoring of (Figure SD7), and DCQ product formation and TCP co-substrate loss for (Figure SD8), the double-mixing reaction between ferric DHP B pre-incubated with TCP for 500 ms prior to its reaction with a 10-fold excess of H2O2 (in situ generated Compound ES) at pH 7; stopped-flow UV-visible spectroscopic monitoring of the double-mixing reaction between ferric DHP B pre-incubated with a 7-fold molar excess of DCQ for 500 ms prior to its reaction with a 2.5-fold excess of H2O2 (in situ generated Compound ES) (Figure SD9); stopped-flow UV-visible spectroscopic monitoring of the reaction between ferric DHP B and a 7-fold excess of DCQ at pH 7 (Figure SD10); reduction of Compound RH yielding Compound P426 (Figure SD11), reduction of Compound RH yielding oxyferrous DHP B (Figure SD12). This material is available free of charge via the Internet at http://pubs.acs.org
Department of Chemistry, North Carolina State University, Raleigh, North Carolina, 27695-8204
References
- (1).Weber RE, Mangum C, Steinman H, Bonaventura C, Sullivan B, Bonaventura J. Hemoglobins of two terebellid polychaetes: Enoplobranchus sanguineus and Amphitrite ornata. Comp Biochem Physiol A Comp Physiol. 1977;56:179–187. doi: 10.1016/0300-9629(77)90182-7. [DOI] [PubMed] [Google Scholar]
- (2).Chen YP, Woodin SA, Lincoln DE, Lovell CR. An unusual dehalogenating peroxidase from the marine terebellid polychaete Amphitrite ornata. J Biol Chem. 1996;271:4609–4612. [PubMed] [Google Scholar]
- (3).Weber RE, Vinogradov SN. Nonvertebrate hemoglobins: functions and molecular adaptations. Physiol Rev. 2001;81:569–628. doi: 10.1152/physrev.2001.81.2.569. [DOI] [PubMed] [Google Scholar]
- (4).Hardison R. Hemoglobins from bacteria to man: evolution of different patterns of gene expression. J Exp Biol. 1998;201:1099–1117. doi: 10.1242/jeb.201.8.1099. [DOI] [PubMed] [Google Scholar]
- (5).Bailly X, Chabasse C, Hourdez S, Dewilde S, Martial S, Moens L, Zal F. Globin gene family evolution and functional diversification in annelids. Febs J. 2007;274:2641–2652. doi: 10.1111/j.1742-4658.2007.05799.x. [DOI] [PubMed] [Google Scholar]
- (6).Chabasse C, Bailly X, Sanchez S, Rousselot M, Zal F. Gene structure and molecular phylogeny of the linker chains from the giant annelid hexagonal bilayer hemoglobins. J Mol Evol. 2006;63:365–374. doi: 10.1007/s00239-005-0198-9. [DOI] [PubMed] [Google Scholar]
- (7).Murzin AG, Brenner SE, Hubbard T, Chothia C. SCOP: a structural classification of proteins database for the investigation of sequences and structures. J Mol Biol. 1995;247:536–540. doi: 10.1006/jmbi.1995.0159. [DOI] [PubMed] [Google Scholar]
- (8).Poulos TL, Kraut J. A hypothetical model of the cytochrome c peroxidase. cytochrome c electron transfer complex. J Biol Chem. 1980;255:10322–10330. [PubMed] [Google Scholar]
- (9).Woodin SA. Recruitment of Infauna - Positive or Negative Cues. Am Zool. 1991;31:797–807. [Google Scholar]
- (10).Woodin SA, Walla MD, Lincoln DE. Occurrence of Brominated Compounds in Soft-Bottom Benthic Organisms. J Exp Mar Biol Ecol. 1987;107:209–217. [Google Scholar]
- (11).Chen YP, Lincoln DE, Woodin SA, Lovell CR. Purification and Properties of a Unique Flavin-Containing Chloroperoxidase from the Capitellid Polychaete Notomastus-Lobatus. J. Biol. Chem. 1991;266:23909–23915. [PubMed] [Google Scholar]
- (12).Roach MP, Chen YP, Woodin SA, Lincoln DE, Lovell CR, Dawson JH. Notomastus lobatus chloroperoxidase and Amphitrite ornata dehaloperoxidase both contain histidine as their proximal heme iron ligand. Biochem. 1997;36:2197–2202. doi: 10.1021/bi9621371. [DOI] [PubMed] [Google Scholar]
- (13).Lincoln DE, Fielman KT, Marinelli RL, Woodin SA. Bromophenol accumulation and sediment contamination by the marine annelids Notomastus lobatus and Thelepus crispus. Biochem Syst Ecol. 2005;33:559–570. [Google Scholar]
- (14).King GM. Inhibition of Microbial Activity in Marine-Sediments by a Bromophenol from a Hemichordate. Nature. 1986;323:257–259. [Google Scholar]
- (15).Fielman KT, Targett NM. Variation of 2,3,4-Tribromopyrrole and Its Sodium Sulfamate Salt in the Hemichordate Saccoglossus-Kowalevskii. Mar Ecol-Prog Ser. 1995;116:125–136. [Google Scholar]
- (16).Chiancone E, Ferruzzi G, Bonaventura C, Bonaventura J. Amphitrite-Ornata Erythrocruorin.2. Molecular Controls of Function. Biochim. Biophys. Acta. 1981;670:84–92. doi: 10.1016/0005-2795(81)90052-0. [DOI] [PubMed] [Google Scholar]
- (17).Chiancone E, Brenowitz M, Ascoli F, Bonaventura C, Bonaventura J. Amphitrite-Ornata Erythrocruorin.1. Structural-Properties and Characterization of Subunit Interactions. Biochim. Biophys. Acta. 1980;623:146–162. doi: 10.1016/0005-2795(80)90017-3. [DOI] [PubMed] [Google Scholar]
- (18).Osborne RL, Taylor LO, Han KP, Ely B, Dawson JH. Amphitrite ornata dehaloperoxidase: enhanced activity for the catalytically active globin using MCPBA. Biochem Biophys Res Commun. 2004;324:1194–1198. doi: 10.1016/j.bbrc.2004.09.174. [DOI] [PubMed] [Google Scholar]
- (19).Han K, Woodin SA, Lincoln DE, Fielman KT, Ely B. Amphitrite ornata, a marine worm, contains two dehaloperoxidase genes. Mar Biotechnol (NY) 2001;3:287–292. doi: 10.1007/s10126-001-0003-8. [DOI] [PubMed] [Google Scholar]
- (20).Feducia J, Dumarieh R, Gilvey LB, Smirnova T, Franzen S, Ghiladi RA. Characterization of dehaloperoxidase compound ES and its reactivity with trihalophenols. Biochem. 2009;48:995–1005. doi: 10.1021/bi801916j. [DOI] [PubMed] [Google Scholar]
- (21).Chen Z, de Serrano V, Betts L, Franzen S. Distal histidine conformational flexibility in dehaloperoxidase from Amphitrite ornata. Acta Crystallogr D Biol Crystallogr. 2009;65:34–40. doi: 10.1107/S0907444908036548. [DOI] [PubMed] [Google Scholar]
- (22).Davis MF, Gracz H, Vendeix FA, de Serrano V, Somasundaram A, Decatur SM, Franzen S. Different modes of binding of mono-, di-, and trihalogenated phenols to the hemoglobin dehaloperoxidase from Amphitrite ornata. Biochem. 2009;48:2164–2172. doi: 10.1021/bi801568s. [DOI] [PubMed] [Google Scholar]
- (23).Smirnova TI, Weber RT, Davis MF, Franzen S. Substrate binding triggers a switch in the iron coordination in dehaloperoxidase from Amphitrite ornata: HYSCORE experiments. J Am Chem Soc. 2008;130:2128–2129. doi: 10.1021/ja0772952. [DOI] [PubMed] [Google Scholar]
- (24).Miksovska J, Horsa S, Davis MF, Franzen S. Conformational dynamics associated with photodissociation of CO from dehaloperoxidase studied using photoacoustic calorimetry. Biochem. 2008;47:11510–11517. doi: 10.1021/bi8012033. [DOI] [PubMed] [Google Scholar]
- (25).Nienhaus K, Nickel E, Davis MF, Franzen S, Nienhaus GU. Determinants of substrate internalization in the distal pocket of dehaloperoxidase hemoglobin of Amphitrite ornata. Biochem. 2008;47:12985–12994. doi: 10.1021/bi801564r. [DOI] [PubMed] [Google Scholar]
- (26).Franzen S, Gilvey LB, Belyea JL. The pH dependence of the activity of dehaloperoxidase from Amphitrite ornata. Biochim. Biophys. Acta. 2007;1774:121–130. doi: 10.1016/j.bbapap.2006.09.019. [DOI] [PubMed] [Google Scholar]
- (27).de Serrano V, Chen Z, Davis MF, Franzen S. X-ray crystal structural analysis of the binding site in the ferric and oxyferrous forms of the recombinant heme dehaloperoxidase cloned from Amphitrite ornata. Acta Crystallogr D Biol Crystallogr. 2007;63:1094–1101. doi: 10.1107/S0907444907043417. [DOI] [PubMed] [Google Scholar]
- (28).Franzen S, Jasaitis A, Belyea J, Brewer SH, Casey R, MacFarlane A. W. t., Stanley RJ, Vos MH, Martin JL. Hydrophobic distal pocket affects NO-heme geminate recombination dynamics in dehaloperoxidase and H64V myoglobin. J Phys Chem B. 2006;110:14483–14493. doi: 10.1021/jp056790m. [DOI] [PubMed] [Google Scholar]
- (29).Franzen S, Belyea J, Gilvey LB, Davis MF, Chaudhary CE, Sit TL, Lommel SA. Proximal cavity, distal histidine, and substrate hydrogen-bonding mutations modulate the activity of Amphitrite ornata dehaloperoxidase. Biochem. 2006;45:9085–9094. doi: 10.1021/bi060020z. [DOI] [PubMed] [Google Scholar]
- (30).Belyea J, Belyea CM, Lappi S, Franzen S. Resonance Raman study of ferric heme adducts of dehaloperoxidase from Amphitrite ornata. Biochem. 2006;45:14275–14284. doi: 10.1021/bi0609218. [DOI] [PubMed] [Google Scholar]
- (31).Osborne RL, Coggins MK, Raner GM, Walla M, Dawson JH. The mechanism of oxidative halophenol dehalogenation by Amphitrite ornata dehaloperoxidase is initiated by H2O2 binding and involves two consecutive one-electron steps: role of ferryl intermediates. Biochem. 2009;48:4231–4238. doi: 10.1021/bi900367e. [DOI] [PubMed] [Google Scholar]
- (32).Osborne RL, Coggins MK, Walla M, Dawson JH. Horse heart myoglobin catalyzes the H2O2-dependent oxidative dehalogenation of chlorophenols to DNA-binding radicals and quinones. Biochem. 2007;46:9823–9829. doi: 10.1021/bi700684u. [DOI] [PubMed] [Google Scholar]
- (33).Osborne RL, Raner GM, Hager LP, Dawson JH. C. fumago chloroperoxidase is also a dehaloperoxidase: oxidative dehalogenation of halophenols. J Am Chem Soc. 2006;128:1036–1037. doi: 10.1021/ja056213b. [DOI] [PubMed] [Google Scholar]
- (34).Osborne RL, Sumithran S, Coggins MK, Chen YP, Lincoln DE, Dawson JH. Spectroscopic characterization of the ferric states of Amphitrite ornata dehaloperoxidase and Notomastus lobatus chloroperoxidase: His-ligated peroxidases with globin-like proximal and distal properties. J Inorg Biochem. 2006;100:1100–1108. doi: 10.1016/j.jinorgbio.2006.02.008. [DOI] [PubMed] [Google Scholar]
- (35).Belyea J, Gilvey LB, Davis MF, Godek M, Sit TL, Lommel SA, Franzen S. Enzyme function of the globin dehaloperoxidase from Amphitrite ornata is activated by substrate binding. Biochem. 2005;44:15637–15644. doi: 10.1021/bi051731k. [DOI] [PubMed] [Google Scholar]
- (36).Lebioda L, LaCount MW, Zhang E, Chen YP, Han K, Whitton MM, Lincoln DE, Woodin SA. An enzymatic globin from a marine worm. Nature. 1999;401:445. doi: 10.1038/46728. [DOI] [PubMed] [Google Scholar]
- (37).de Serrano V, D’Antonio J, Franzen S, Ghiladi RA. Structure of dehaloperoxidase B at 1.58 A resolution and structural characterization of the AB dimer from Amphitrite ornata. Acta Crystallogr D Biol Crystallogr. 2010;66:529–538. doi: 10.1107/S0907444910004580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Meyer TJ. Stanford University; 1967. [Google Scholar]
- (39).Smolenaers PJ, Beattie JK. Inorg. Syn. 1979;19:117–121. [Google Scholar]
- (40).Smolenaers PJ, Beattie JK, Hutchinson ND. Crystal and molecular structure of racemic tris(ethylenediamine)ruthenium(II) tetrachlorozincate(II) Inorg. Chem. 1981;20:2202–2206. [Google Scholar]
- (41).Beers RF, Jr., Sizer IW. A spectrophotometric method for measuring the breakdown of hydrogen peroxide by catalase. J Biol Chem. 1952;195:133–140. [PubMed] [Google Scholar]
- (42).Falk JE. Porphyrins and Metalloporphyrins: Their General, Physical, and Coordination Chemistry and Laboratory Methods. Elsevier Publishing; New York: 1964. Haems. I. Determination as pyridine hemochromes; pp. 181–188. [Google Scholar]
- (43).Fuhrhop JH, Smith KM. Laboratory Methods. In: Smith KM, editor. Porphyrins and Metalloporphyrins. Elsevier Publishing; New York: 1975. pp. 804–807. [Google Scholar]
- (44).Bowden EF, Cohen DJ, Hawkridge FM. Anaerobic Thin-Layer Electrochemical-Cell for Planar Optically Transparent Electrodes. Anal. Chem. 1982;54:1005–1008. [Google Scholar]
- (45).LaCount MW, Zhang E, Chen YP, Han K, Whitton MM, Lincoln DE, Woodin SA, Lebioda L. The crystal structure and amino acid sequence of dehaloperoxidase from Amphitrite ornata indicate common ancestry with globins. J Biol Chem. 2000;275:18712–18716. doi: 10.1074/jbc.M001194200. [DOI] [PubMed] [Google Scholar]
- (46).Gouterman M. Optical spectra and electronic structure of porphyrins and related rings. In: Dolphin D, editor. The Porphyrins. Academic Press; New York: 1978. pp. 1–165. [Google Scholar]
- (47).Chouchane S, Girotto S, Kapetanaki S, Schelvis JP, Yu S, Magliozzo RS. Analysis of heme structural heterogeneity in Mycobacterium tuberculosis catalase-peroxidase (KatG) J Biol Chem. 2003;278:8154–8162. doi: 10.1074/jbc.M208256200. [DOI] [PubMed] [Google Scholar]
- (48).Ghiladi RA, Knudsen GM, Medzihradszky KF, Ortiz de Montellano PR. The Met-Tyr-Trp cross-link in Mycobacterium tuberculosis Catalase-Peroxidase (KatG): Autocatalytic Formation and Effect on Enzyme Catalysis and Spectroscopic Properties. J. Biol. Chem. 2005;280:22651–22663. doi: 10.1074/jbc.M502486200. [DOI] [PubMed] [Google Scholar]
- (49).Nienhaus K, Deng P, Belyea J, Franzen S, Nienhaus GU. Spectroscopic study of substrate binding to the carbonmonoxy form of dehaloperoxidase from Amphitrite ornata. J Phys Chem B. 2006;110:13264–13276. doi: 10.1021/jp060278z. [DOI] [PubMed] [Google Scholar]
- (50).Nicoletti FP, Thompson MK, Howes BD, Franzen S, Smulevich G. New insights into the role of distal histidine flexibility in ligand stabilization of dehaloperoxidase-hemoglobin from Amphitrite ornata. Biochem. 2010;49:1903–1912. doi: 10.1021/bi9020567. [DOI] [PubMed] [Google Scholar]
- (51).Thompson MK, Davis MF, De Serrano V, Nicoletti FP, Howes BD, Smulevich G, Franzen S. Internal binding of halogenated phenols in dehaloperoxidase-hemoglobin inhibits peroxidase function. Biophys J. 2010 doi: 10.1016/j.bpj.2010.05.041. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Hewson WD, Hager LP. Oxidation of horseradish peroxidase compound II to compound I. J Biol Chem. 1979;254:3182–3186. [PubMed] [Google Scholar]
- (53).Blumberg WE, Peisach J, Wittenberg BA, Wittenberg JB. The electronic structure of protoheme proteins. I. An electron paramagnetic resonance and optical study of horseradish peroxidase and its derivatives. J Biol Chem. 1968;243:1854–1862. [PubMed] [Google Scholar]
- (54).Wittenberg BA, Kampa L, Wittenberg JB, Blumberg WE, Peisach J. The electronic structure of protoheme proteins. II. An electron paramagnetic resonance and optical study of cytochrome c peroxidase and its derivatives. J Biol Chem. 1968;243:1863–1870. [PubMed] [Google Scholar]
- (55).Balny C, Anni H, Yonetani T. A stopped-flow study of the reaction of cytochrome c peroxidase with hydroperoxides. FEBS Lett. 1987;221:349–354. doi: 10.1016/0014-5793(87)80954-7. [DOI] [PubMed] [Google Scholar]
- (56).Coulson AF, Erman JE, Yonetani T. Studies on cytochrome c peroxidase. XVII. Stoichiometry and mechanism of the reaction of compound ES with donors. J Biol Chem. 1971;246:917–924. [PubMed] [Google Scholar]
- (57).Marquez LA, Quitoriano M, Zilinskas BA, Dunford HB. Kinetic and spectral properties of pea cytosolic ascorbate peroxidase. FEBS Lett. 1996;389:153–156. doi: 10.1016/0014-5793(96)00562-5. [DOI] [PubMed] [Google Scholar]
- (58).Yadav RK, Dolai S, Pal S, Adak S. Role of tryptophan-208 residue in cytochrome c oxidation by ascorbate peroxidase from Leishmania major-kinetic studies on Trp208Phe mutant and wild type enzyme. Biochim. Biophys. Acta. 2008;1784:863–871. doi: 10.1016/j.bbapap.2008.02.006. [DOI] [PubMed] [Google Scholar]
- (59).Lardinois OM, Ortiz de Montellano PR. Autoreduction of ferryl myoglobin: discrimination among the three tyrosine and two tryptophan residues as electron donors. Biochem. 2004;43:4601–4610. doi: 10.1021/bi036241b. [DOI] [PubMed] [Google Scholar]
- (60).Giulivi C, Cadenas E. Ferrylmyoglobin: formation and chemical reactivity toward electron-donating compounds. Methods Enzymol. 1994;233:189–202. doi: 10.1016/s0076-6879(94)33022-0. [DOI] [PubMed] [Google Scholar]
- (61).Herold S, Rehmann FJ. Kinetics of the reactions of nitrogen monoxide and nitrite with ferryl hemoglobin. Free Radic Biol Med. 2003;34:531–545. doi: 10.1016/s0891-5849(02)01355-2. [DOI] [PubMed] [Google Scholar]
- (62).Giulivi C, Davies KJ. Hydrogen peroxide-mediated ferrylhemoglobin generation in vitro and in red blood cells. Methods Enzymol. 1994;231:490–496. doi: 10.1016/0076-6879(94)31032-7. [DOI] [PubMed] [Google Scholar]
- (63).Belevich I, Borisov VB, Verkhovsky MI. Discovery of the true peroxy intermediate in the catalytic cycle of terminal oxidases by real-time measurement. J Biol Chem. 2007;282:28514–28519. doi: 10.1074/jbc.M705562200. [DOI] [PubMed] [Google Scholar]
- (64).Shintaku M, Matsuura K, Yoshioka S, Takahashi S, Ishimori K, Morishima I. Absence of a detectable intermediate in the compound I formation of horseradish peroxidase at ambient temperature. J Biol Chem. 2005;280:40934–40938. doi: 10.1074/jbc.M503472200. [DOI] [PubMed] [Google Scholar]
- (65).Baek HK, Van Wart HE. Elementary steps in the formation of horseradish peroxidase compound I: direct observation of compound 0, a new intermediate with a hyperporphyrin spectrum. Biochem. 1989;28:5714–5719. doi: 10.1021/bi00440a003. [DOI] [PubMed] [Google Scholar]
- (66).Egawa T, Yoshioka S, Takahashi S, Hori H, Nagano S, Shimada H, Ishimori K, Morishima I, Suematsu M, Ishimura Y. Kinetic and spectroscopic characterization of a hydroperoxy compound in the reaction of native myoglobin with hydrogen peroxide. J Biol Chem. 2003;278:41597–41606. doi: 10.1074/jbc.M210383200. [DOI] [PubMed] [Google Scholar]
- (67).Svistunenko DA. Reaction of haem containing proteins and enzymes with hydroperoxides: the radical view. Biochim. Biophys. Acta. 2005;1707:127–155. doi: 10.1016/j.bbabio.2005.01.004. [DOI] [PubMed] [Google Scholar]
- (68).Svistunenko DA, Cooper CE. A new method of identifying the site of tyrosyl radicals in proteins. Biophys J. 2004;87:582–595. doi: 10.1529/biophysj.104.041046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Thompson MK, Franzen S, Ghiladi RA, Reeder BJ, Svistunenko DA. Free radical mechanism of hydrogen peroxide induced activation of dehaloperoxidase-hemoglobin from Amphitrite ornata. 2010 submitted for publication. [Google Scholar]
- (70).Nilges M. SimPow6. Illinois EPR Research Center; http://ierc.scs.uiuc.edu/~nilges/software.html. [Google Scholar]
- (71).Svistunenko DA. Tyrosine residues in different proteins: Phenol ring rotation angle database. 2004 http://privatewww.essex.ac.uk/~svist/lev1/tyrdb/home.shtml.
- (72).D’Antonio EL, Franzen S, Bowden EF. Redox Thermodynamics of the Fe(III)/Fe(II) Couple of Dehaloperoxidase-hemoglobin from Amphitrite ornata. 2010 submitted. [Google Scholar]
- (73).Dunford HB. Heme Peroxidases. Wiley-VCH; New York: 1999. [Google Scholar]
- (74).Matsui T, Ozaki S, Liong E, Phillips GN, Jr., Watanabe Y. Effects of the location of distal histidine in the reaction of myoglobin with hydrogen peroxide. J Biol Chem. 1999;274:2838–2844. doi: 10.1074/jbc.274.5.2838. [DOI] [PubMed] [Google Scholar]
- (75).Finzel BC, Poulos TL, Kraut J. Crystal structure of yeast cytochrome c peroxidase refined at 1.7-A resolution. J Biol Chem. 1984;259:13027–13036. [PubMed] [Google Scholar]
- (76).Phillips GN, Jr., Arduini RM, Springer BA, Sligar SG. Crystal structure of myoglobin from a synthetic gene. Proteins. 1990;7:358–365. doi: 10.1002/prot.340070407. [DOI] [PubMed] [Google Scholar]
- (77).Gajhede M, Schuller DJ, Henriksen A, Smith AT, Poulos TL. Crystal structure of horseradish peroxidase C at 2.15 A resolution. Nat Struct Biol. 1997;4:1032–1038. doi: 10.1038/nsb1297-1032. [DOI] [PubMed] [Google Scholar]
- (78).Egawa T, Shimada H, Ishimura Y. Formation of compound I in the reaction of native myoglobins with hydrogen peroxide. J Biol Chem. 2000;275:34858–34866. doi: 10.1074/jbc.M004026200. [DOI] [PubMed] [Google Scholar]
- (79).Olson JS, Phillips GN., Jr. Kinetic pathways and barriers for ligand binding to myoglobin. J Biol Chem. 1996;271:17593–17596. doi: 10.1074/jbc.271.30.17593. [DOI] [PubMed] [Google Scholar]
- (80).Jewsbury P, Kitagawa T. The distal residue-CO interaction in carbonmonoxy myoglobins: a molecular dynamics study of two distal histidine tautomers. Biophys J. 1994;67:2236–2250. doi: 10.1016/S0006-3495(94)80708-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Perutz MF. Myoglobin and haemoglobin: role of distal residues in reactions with haem ligands. Trends Biochem Sci. 1989;14:42–44. doi: 10.1016/0968-0004(89)90039-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.