

Abstract
Pharmacophore models for nicotinic agonists have been proposed for four decades. Central to these models is the presence of a cationic nitrogen and a hydrogen bond acceptor. It is now well-established that the cationic center makes an important cation-π interaction to a conserved tryptophan, but the donor to the proposed hydrogen bond acceptor has been more challenging to identify. A structure of nicotine bound to the acetylcholine binding protein predicted that the binding partner of the pharmacophore’s second component was a water molecule, which also hydrogen bonds to the backbone of the complementary subunit of the receptors. Here we use unnatural amino acid mutagenesis coupled with agonist analogs to examine whether such a hydrogen bond is functionally significant in the α4β2 neuronal nAChR, the receptor most associated with nicotine addiction. We find evidence for the hydrogen bond with the agonists nicotine, acetylcholine, carbamylcholine, and epibatidine. These data represent a completed nicotinic pharmacophore and offer insight into the design of new therapeutic agents that selectively target these receptors.
The nicotinic acetylcholine receptor (nAChR) is a pentameric, ligand-gated ion channel activated by the neurotransmitter acetylcholine (ACh), and also by nicotine and structurally related agonists (1–3). Nicotinic receptors mediate fast synaptic transmission at the neuromuscular junction of the peripheral nervous system. In addition, a family of paralogous nAChRs termed the neuronal receptors function in the central nervous system and certain autonomic ganglia, and the addictive and cognitive properties of nicotine are associated with these neuronal receptors (4, 5). Neuronal receptors comprised of α4 and β2 subunits are most strongly associated with nicotine addiction (6–9). They are upregulated during chronic nicotine exposure and are implicated in various disorders, including Alzheimer’s disease and schizophrenia, and in protection against Parkinson disease. Interest in the development of molecules that selectively target α4β2 receptors has been growing, highlighted by the development of the smoking cessation drug, varenicline (6).
Many have undertaken the task of dissecting nicotinic agonists into a core pharmacophore, since the first publication on the topic in 1970 (10). While the details are debated, two aspects are clear. Nicotinic agonists contain a cationic nitrogen and a hydrogen bond acceptor (Fig. 1A) (11, 12). In 1990, we proposed that binding of the cationic nitrogen of acetylcholine would be mediated through a cation-π interaction with an aromatic residue of the nAChRs (13). We subsequently validated this model with the identification of a cation-π interaction to a conserved tryptophan residue for both acetylcholine and nicotine (14, 15). In fact, the cation-π interaction has been shown to be a general contributor to agonist affinity across the entire family of Cys-loop (pentameric) neurotransmitter-gated ion channels (16).
Fig. 1.

Key structures considered in the present work. (A) Structures of agonists used. Hydrogen bond acceptor moieties are red and cationic nitrogens are blue. (B) Backbone amide to ester mutation strategy for perturbing a hydrogen bond.
In the nAChRs, ligand binding occurs at the interface between adjacent principal (α4 in α4β2) and complementary (β2) subunits. Three segments from the α4 subunit (historically referred to as the A, B, and C “loops”) form the principal face of the ligand-binding domain, which contains the cation-π binding site, and three segments from the β2 subunit (D, E, and F) form the complementary face. A major advance in the study of nAChRs was the discovery of the water-soluble acetylcholine binding proteins (AChBP) (17–22). AChBP serves as a structural template for the extracellular, N-terminal, ligand-binding domain of the nAChRs, sharing 20–24% sequence identity with the ligand-binding domain of the much larger ion channel proteins. Several AChBP structures with ligands bound have been published, including structures of AChBP in complex with the ACh analog carbamylcholine (CCh) and with nicotine (18) and the nicotine analog epibatidine (21). Drugs that target the nAChR, such as nicotine and epibatidine, typically contain a protonatable amine rather than the quaternary ammonium seen in ACh. Along with the cation-π interaction, the crystallography indicated a hydrogen bond between the N+H and the backbone carbonyl of the tryptophan that also forms the cation-π interaction, and functional studies on intact receptors confirmed the hydrogen bonding interaction (14, 23).
Concerning the second component of the pharmacophore, the hydrogen bond acceptor, the AChBP structure produced intriguing results. With nicotine bound, the pyridine nitrogen makes a hydrogen bond to a water molecule that is positioned by hydrogen bonds to the main chains of two residues, the CO of N107 and the NH of L119, both in the complementary subunit (α4β2 numbering; residues are in the β2 sequence FYSNAVVSYDGSIFWLPPA) (Fig. 2) (18). In other structures, including those with CCh or epibatidine bound, the overall binding site structure is preserved, although the key water molecule is not always evident, especially in lower-resolution structures.
Fig. 2.

Key interactions seen in the crystal structure of nicotine bound to AChBP (PDB ID code 1UW6). Residue numbering is for the α4β2 receptor, with AChBP homologs in brackets.
A key question, then, is the extent to which predictions based on the AChBPs, which evolved to bind a target molecule, relate to the nAChRs, which evolved to undergo a global structural change (to gate) on binding ACh. Here we describe an approach to probe with high precision a specific structural interaction in a complex receptor protein. Using unnatural amino acid mutagenesis and agonist analogs, we find that nicotine, acetylcholine, epibatidine, and carbamylcholine make the same hydrogen bond involving the backbone NH of β2L119 of the α4β2 receptor, supporting a common pharmacophore for acetylcholine and nicotine.
Results
Strategy.
A well-established strategy for probing potential backbone hydrogen bonds is to replace the residue that contributes the hydrogen bond donor with its α-hydroxy analog (Fig. 1B) (24–28). This mutation converts a backbone amide to a backbone ester, a subtle change that impacts backbone hydrogen bonding in two ways. The backbone NH that can donate a hydrogen bond is removed, and the carbonyl oxygen, by virtue of being part of an ester rather than an amide, is a weaker hydrogen bond acceptor.
In the present context, simply seeing a change in receptor function in response to appropriate backbone ester substitutions would not prove the presence of the proposed interaction. Backbone mutation is certainly subtle, but when installed in an important region of the receptor it could affect function in a number of ways. As such, we sought a way to provide a direct connection between any consequences of backbone mutation and the proposed hydrogen bond. To do this, we considered the molecule S-N-methyl-2-phenylpyrrolidine (S-MPP, Fig. 1A). In this structure a phenyl ring replaces the pyridyl group of nicotine, obliterating the possibility of forming the proposed hydrogen bond. This would allow a “double mutant cycle” analysis that links the backbone NH to the pyridine N. If the mutant cycle analysis shows that the effects of the two changes—the backbone mutation and the modification of the drug—are substantially nonadditive, this would provide compelling evidence for the proposed interaction.
The metric used to evaluate receptors is EC50, the effective concentration of agonist required to achieve half-maximal response. This is a functional measure that can be influenced by changes to drug binding and/or efficacy of activation of the receptor. Previously we have shown that subtle mutations to TrpB of the binding site primarily, if not exclusively, affect agonist binding (14), but we cannot assume the same for Leu119. Because the goal here is to map the pharmacophore for a collection of agonists, we are interested in factors that influence receptor activation. We consider EC50 to be an appropriately useful guide for understanding agonism and designing new agonists, but more detailed studies of the mutations considered here would be valuable.
Optimization of Nonsense Suppression Experiments.
The α4β2 receptor is a pentamer with two possible stoichiometries, (α4)2(β2)3 and (α4)3(β2)2 termed A2B3 and A3B2, respectively. Our studies have focused on the A2B3 receptor, which shows the higher sensitivity to nicotine and is thought to be upregulated during chronic nicotine exposure. Subunit stoichiometry can be managed by controlling mRNA injection ratios. Exclusive expression of A2B3 can be verified by monitoring I-V relationships of agonist-induced currents, as described previously (14).
This study represents the first report of unnatural amino acid mutagenesis in the β2 subunit of α4β2. Since nonsense suppression often produces low protein yields of the subunit where the suppression occurs, it was critical to ensure that a receptor with excess β2 subunit, i.e., the A2B3 stoichiometry, was exclusively produced in nonsense suppression experiments. To that end, mRNA ratios substantially favoring the β2 subunit were explored. We found that an injected mRNA ratio of 1∶20 of α4∶β2 (with β2 containing the nonsense suppression site) gave I-V relationships indicative of A2B3 (14), while still providing enough current to conduct meaningful dose-response experiments. The α4 subunit also contained a known mutation in the M2 transmembrane helix (L9’A), which improves receptor expression and lowers whole-cell EC50 values, but does not influence the binding trends of the receptor (29).
One challenge in incorporating a hydroxy acid at β2L119 was to limit the amount of current observed from oocytes injected with full length tRNA that was not synthetically appended to an amino or α-hydroxy acid. Such current would indicate that the suppressor tRNA was aminoacylated by an endogenous aminoacyl-tRNA synthetase and delivered a natural amino acid at the mutation site. We observed significant background currents attributable to such infidelity when using the suppressor tRNA THG73, which has been the workhorse of our unnatural amino acid mutagenesis experiments (30). Employing the recently developed opal suppressor tRNA TQOpS’ (31, 32) significantly reduced this background current at β2L119. Aminoacylation from TQOpS’ was assessed for each agonist by injection of unacylated TQOpS’, and full dose-response relations were generated for agonists displaying > 20 nA of current. Suppression experiments typically produced ≥1 μA of current and yielded Hill and EC50 values that were markedly different from unacylated TQOpS’ control experiments, and so the small background currents are not expected to distort the reported EC50 values. With these conditions, characterization of mutant receptors was straightforward (Fig. 3).
Fig. 3.

Representative current waveforms and dose-response relations for S-nicotine and S-MPP. Agonist-induced current waveforms for (A) S-Nic on wild-type α4β2. (B) S-Nic on α4β2L119Lah. (C) S-MPP on wild-type α4β2. (D) S-MPP on α4β2L119Lah. Concentrations are in μM. (E) Dose-response relations for S-Nic and S-MPP on wild-type α4β2 or α4β2L119Lah.
Amide to Ester Backbone Mutation at β2L119 Impacts Receptor Function.
To probe the hydrogen bond suggested by the AChBP structures, β2L119 was replaced with its α-hydroxy analog (leucine, α-hydroxy; Lah). Meaningful increases in EC50 for the backbone amide to ester mutation were seen for the conventional agonists nicotine, ACh, CCh, and epibatidine, suggesting a significant functional role for the backbone NH (Table 1 and Fig. 3). In contrast, no shift was seen for the very weak agonist choline.
Table 1.
EC50 values, Hill coefficients, and relative efficacies*
| Agonist | Mutation | EC50, nM | nH |
| S-Nic | WT | 120 ± 5 | 1.3 ± 0.05 |
| Leu | 120 ± 3 | 1.5 ± 0.05 | |
| Lah | 800 ± 30 | 1.3 ± 0.04 | |
| S-MPP | WT | 11,000 ± 400 | 1.7 ± 0.08 |
| Leu | 14,000 ± 900 | 1.5 ± 0.11 | |
| Lah | 1,100 ± 40 | 1.5 ± 0.05 | |
| ACh | WT | 360 ± 20 | 1.3 ± 0.07 |
| Leu | 440 ± 20 | 1.3 ± 0.08 | |
| Lah | 3,000 ± 100 | 1.2 ± 0.04 | |
| CCh | WT | 7,200 ± 80 | 1.3 ± 0.02 |
| Leu | 7,900 ± 200 | 1.2 ± 0.03 | |
| Lah | 29,000 ± 800 | 1.2 ± 0.04 | |
| Ch | WT | 140,000 ± 4,000 | 1.6 ± 0.06 |
| Leu | 140,000 ± 20,000 | 1.2 ± 0.09 | |
| Lah | 150,000 ± 5,000 | 1.4 ± 0.05 | |
| Epi | WT | 0.79 ± 0.04 | 1.4 ± 0.07 |
| Leu | 0.58 ± 0.05 | 1.5 ± 0.15 | |
| Lah | 2.9 ± 0.06 | 1.3 ± 0.03 |
*All studies showed current values at +70 mV, normalized to -110 mV, ≤ 0.08, confirming the A2B3 stoichiometry. Errors are standard error of the mean. Epi is epibatidine. Mutations identified as “Leu” represent recovery of the wild-type receptor by nonsense suppression.
As noted above, we considered S-MPP as a potentially informative structure for probing the pyridine hydrogen bond. As such, we adapted existing synthetic protocols (33) to prepare N-methyl-2-phenylpyrroline (MPP). Recrystallization of the dibenzoyl tartrate salt (at the phenylpyrrolidine stage) gave the S enantiomer.
As expected, S-MPP is a much poorer agonist than nicotine, showing a ∼120-fold higher EC50 with the wild-type receptor. For nicotine, the S enantiomer is the higher affinity enantiomer and the one traditionally used in studies of nicotinic receptors. We find that S-MPP has a twofold lower EC50 than racemic MPP, indicating that the higher affinity enantiomer is being used.
Incorporation of a backbone ester at β2L119 leads to a remarkable change in relative agonist potencies. Instead of the increase in EC50 seen with nicotine, S-MPP actually shows a decrease in EC50; S-MPP is a more potent agonist when the backbone ester is present than when the natural backbone amide is present. In fact, when the backbone ester is present, nicotine and S-MPP display comparable potency.
The AChBP structure also predicts that a second residue in the complementary subunit positions the water molecule in proximity to the pyridine N of nicotine. The backbone carbonyl of β2N107 is expected to make a hydrogen bond to the water molecule in conjunction with the first hydrogen bond made by β2L119 (Fig. 2). As noted above, an established strategy for attenuating the hydrogen bonding ability of a backbone carbonyl is to mutate the (i + 1) residue to its α-hydroxy acid (Fig. 1B). However, nonsense suppression experiments at the β2A108 site gave inconsistent results that suggested we could not reliably control the stoichiometry of the mutant receptor. As such, we have been unable to probe this interaction.
Mutant Cycle Analyses Indicate Strong Receptor-Agonist Interactions at β2L119.
As noted above, a mutant cycle analysis (Fig. 4) is the standard way to determine whether pairs of mutations are independent or are coupled. EC50-based mutant cycle analyses have been performed by our lab and others to investigate multiple interactions in Cys-loop receptors and related structures (28, 34–36). For several different agonist pairs, coupling coefficients (Ω) and coupling energies (ΔΔG) were calculated (Table 2).
Fig. 4.

Double mutant cycle analysis for S-Nic and S-MPP on wild-type α4β2 and α4β2L119Lah.
Table 2.
Coupling parameters (Ω) and ΔΔG values for mutant cycle analyses
| Agonist | Ω | ΔΔG, kcal mol-1 |
| S-Nic/S-MPP | 0.012 | 2.6 |
| ACh/Ch | 0.16 | 1.1 |
| CCh/Ch | 0.29 | 0.73 |
Mutant cycle analysis for the S-nicotine/S-MPP pair and the β2L119/β2L119Lah pair predicts a substantial coupling energy of 2.6 kcal/mol. This is a relatively large energy for a putative hydrogen bond, and it provides strong evidence for a hydrogen bonding interaction between the pyridine N of nicotine and the backbone NH of β2L119.
We also considered double mutant cycle analyses for the agonists ACh and CCh using choline as the reference compound, as it lacks the key hydrogen bond acceptor. This is a much less subtle probe than the S-nicotine/S-MPP pair, but it still could produce relevant results. Indeed, we find that for both the ACh/Ch and CCh/Ch pairs, smaller, but still meaningful, coupling energies are seen (Table 2).
Discussion
The nicotinic receptor has produced one of the longest-known, best-studied pharmacophores. The original study of Beers and Reich (10) proposed that two points, a cationic nitrogen and a hydrogen bond acceptor, were required for successful interaction with biological receptors. Later discussion debated the optimal distance between the two points (deemed the internitrogen distance), and more recent models have alluded to pharmacophore binding partners within the biological receptors. Despite 40 years of interest in the nicotinic pharmacophore, the binding partners of the essential two point pharmacophore have only recently been identified. Pioneering mutagenesis and affinity labeling studies of the receptor from Torpedo rays identified a number of aromatic amino acids near the binding site (1, 3). Early unnatural amino acid mutagenesis studies showed that one of these aromatics, now termed TrpB, makes a cation-π interaction with ACh in the muscle-type nAChR (15), and more recent studies established a comparable interaction to both ACh and nicotine in the α4β2 receptor (14).
The search for the presumed hydrogen bond donor to the acetyl group of ACh and the pyridine N of nicotine was much more challenging. A breakthrough came with the discovery of the AChBPs, and in 2004 a structure of nicotine bound to AChBP was reported (18). As shown in Fig. 2, that AChBP structure confirmed the cation-π interaction to TrpB. It also implicated a hydrogen bond between the pyrrolidine N+H and the backbone carbonyl of TrpB, an interaction that was subsequently confirmed by unnatural amino acid mutagenesis (14, 23).
Importantly, the AChBP structure also suggested the binding partner for the second element of the pharmacophore. In AChBP, the pyridine N of nicotine makes a water-mediated hydrogen bond to a backbone NH and to a backbone carbonyl (Fig. 2). This elegant arrangement emphasizes the interfacial nature of the agonist binding site, as the pyridine N interacts with residues that are on the complementary subunit, while TrpB, which makes the cation-π interaction and the hydrogen bond to the pyrrolidine N+H, lies in the principal subunit. The value of AChBP in guiding nAChR research is undeniably large, especially in the present context. It would have been very challenging to guess the hydrogen bond partner(s) to agonists such as ACh and nicotine before the structure of AChBP with nicotine bound. Nevertheless, AChBP is not a nAChR. AChBP evolved to bind ligands, not to gate an ion channel in response to ACh binding. As such, tests of predictions from AChBP structures in real receptors are always essential.
Here we employ a unique strategy to test the water-mediated hydrogen bonding model of Fig. 2 in the neuronal, α4β2 nAChR. The α4β2 receptor shows high affinity for nicotine, and it is generally accepted to be the dominant receptor subtype that contributes to nicotine addiction. Our studies of α4β2 are made possible by recent advances (14) that allow us to express significant quantities of α4β2 in Xenopus oocytes, to control subunit stoichiometry, and to efficiently incorporate unnatural amino acids into the receptor. Recently, we have shown that the cation-π interaction and the hydrogen bond to TrpB are strong in the (α4)2(β2)3 receptor (14).
To probe the second hydrogen bond suggested by AChBP, we mutated β2L119 to its α-hydroxy analog. This removes the critical NH, and, indeed, the agonists nicotine, ACh, CCh, and epibatidine, all show 5- to 7-fold increases in EC50 in response to the mutation. While consistent with the hydrogen bonding model, these observations certainly do not prove it. It could be that the backbone mutation is simply generically disruptive to receptor function.
To make an explicit connection between the pyridine N of nicotine and the backbone NH of β2L119, we combined backbone mutagenesis with a modification of the agonist, removing the pyridine N to create S-MPP. Of course, S-MPP would never be the target of a medicinal chemistry study; it can be anticipated to be a terrible drug at the nAChR. Here it is used as a chemical probe, to evaluate a key binding interaction of the potent drug nicotine.
Studies with S-MPP produced remarkable results. As expected, it is a very poor agonist at the wild-type receptor. However, completely opposite to what is seen with nicotine, ACh, CCh, or epibatidine, introduction of the backbone ester at β2L119 lowers EC50 for S-MPP. In fact, S-MPP and nicotine are comparably potent at the mutant receptor. Clearly the backbone mutation has had dramatically different effects on the two agonists. The effect can be quantified by a mutant cycle analysis, which reveals a coupling energy of 2.6 kcal/mol between the backbone mutation and the agonist “mutation.” This is a quite substantial energy, especially when one considers that these chemical changes—both in the protein and in the ligand—are more structurally subtle than those typically employed in mutant cycle analysis studies using conventional mutagenesis.
The results with S-MPP provide strong support for the nicotine binding model based on the AChBP structure. As noted above, however, AChBP structures with CCh or epibatidine bound do not include the key water molecule, although other components of the hydrogen bonding network are comparably positioned. We find that ACh, CCh, and epibatidine all respond to the backbone ester mutation in a way that is comparable to that seen for nicotine. In addition, choline, a weak agonist that lacks the hydrogen bond acceptor of ACh and CCh, is not influenced by the backbone mutation. We thus conclude that all the drugs studied here make a hydrogen bonding interaction with the backbone NH of β2L119; the nicotinic pharmacophore has thus been completed by interactions with the complementary subunit. Note that these studies do not establish that the interaction between the hydrogen bond acceptor component of the agonists and the backbone NH of β2L119 is mediated by a water molecule; a direct interaction would be just as compatible with our data. At present, we feel the water-mediated interaction is the most reasonable interpretation, but further experiments to address this point would be valuable.
We have now used chemical-scale investigations of functional receptors to establish a three-point interaction between nicotine and the α4β2 neuronal nAChR, the receptor most strongly associated with nicotine addiction. A cation-π interaction to TrpB has been established by progressive fluorination of the key tryptophan. Backbone mutagenesis has been used to establish two key hydrogen bonds: the pyrrolidine N+H hydrogen bonds to the backbone carbonyl of TrpB and the pyridine N of nicotine hydrogen bonds to the backbone NH of β2L119. Studies of these two hydrogen bonds were inspired by the AChBP structures, emphasizing the substantial impact of AChBP on nAChR research.
At the same time, AChBP is not a neurotransmitter-gated ion channel; it evolved to serve a different function than a nAChR. As such, we should anticipate some differences between the two structures. Indeed, two features of the nicotine-AChBP structure have been shown to be not functionally significant in studies of nAChRs. The AChBP structure clearly shows a cation-π interaction between the CH3 of nicotine and a tyrosine at the agonist binding site termed TyrC2 (Fig. S1) (18). This methyl group—which carries a charge comparable to a CH3 attached to the N+ of ACh—points directly at the center of the aromatic ring of TyrC2 and essentially makes van der Waals contact with the ring, unquestionably a cation-π interaction. However, we find no experimental support for this cation-π interaction in either the muscle-type or the α4β2 nAChR. In each system, inserting 4-CN-Phe at TyrC2 gives essentially wild-type receptor function (14, 15). A CN group is very strongly deactivating in a cation-π interaction, and so this result is in conflict with the AChBP structure. Note that in a different Cys-loop receptor, the residue at position C2 does make a functionally significant cation-π interaction to the natural agonist serotonin (37).
In addition, all AChBP structures—the nicotine, CCh, and epibatidine bound structures considered here as well as the “apo” structure—contain a strong hydrogen bond between the indole NH of TrpB and the backbone carbonyl of the residue that corresponds to β2L119 (Fig. S1). N•••O distances range from 2.7 to 3.0 Å. However, earlier studies of the muscle-type receptor found no evidence for an important interaction of this kind. In particular, TrpB can be substituted by unnatural amino acids in which the indole ring is replaced by a naphthalene or an N-methylindole with very little impact on EC50 (15). All of these analogs lack the critical hydrogen bond-donating NH of the Trp indole ring.
In summary, we have used a combination of unnatural amino acid mutagenesis and chemical synthesis to provide strong evidence for a functionally important hydrogen bond between the pyridine N of nicotine and the backbone NH of β2L119 in the nicotine-sensitive α4β2 receptor. A similar interaction contributes to the binding of ACh, CCh, and epibatidine. We have now used unnatural amino acid mutagenesis to establish three strong contact points between this critical receptor and nicotine: the cation-π interaction to the side chain of TrpB, the hydrogen bond between the pyrrolidine N+H and the backbone carbonyl of TrpB, and the hydrogen bond between the pyridine N and the backbone NH of β2L119. There is much interest in the pharmaceutical industry in developing subtype-selective agonists of neuronal nAChRs, and it seems likely that the complementary subunit will play the dominant role in discriminating among subtypes. As such, these studies of a key binding interaction involving the complementary binding site suggest a general strategy for developing insights that could lead to subtype-specific pharmaceuticals.
Materials and Methods
Molecular Biology Protocols.
Rat α4 and β2 cDNA in the pAMV vector was linearized with the restriction enzyme Not 1. mRNA was prepared by in vitro transcription using the mMessage Machine T7 kit (Ambion). Unnatural mutations were introduced by the standard Stratagene QuickChange protocol, using a TGA mutation at the site of interest. The α4 subunit contained a known mutation in the M2 transmembrane helix (L9’A) that improves receptor expression and lowers whole-cell EC50 values, but does not influence the ligand-binding trends of the receptor (29). Stage V–VI Xenopus laevis oocytes were injected with mRNA in a 1∶1 or 1∶20 ratio of α4L9'A∶β2 for wild-type experiments or suppression with α-hydroxy acids, respectively. Hydroxy or amino acids were appended to the dinucleotide dCA and enzymatically ligated to the truncated 74-nucleotide TQOpS’ tRNA as previously described (30). Each cell was injected with 75 nL of a 1∶1 mixture of mRNA (20–25 ng of total mRNA): tRNA (20–30 ng), with oocytes injected with Leu ligated to TQOpS’ receiving an additional 75 nL after 24 h of incubation at 18 °C. Wild-type recovery experiments (injection of tRNA appended to the natural amino acid) were performed to evaluate the fidelity of the unnatural suppression experiments. Additional controls, mRNA only and 74-mer TQOpS’ ligated to dCA (TQOpS’-dCA), were also examined. While small currents (typically less than 200 nA) were seen for TQOpS’-dCA control experiments, EC50 and Hill values were substantially different from suppression values.
Electrophysiology Protocols.
Electrophysiology experiments were performed 24–48 h after injection using the OpusXpress 6000A instrument (Axon Instruments) in two-electrode voltage clamp mode at a holding potential of -60 mV. The running buffer was Ca2+-free ND96 solution (96 mM NaCl, 2 mM KCl, 1 mM MgCl2, and 5 mM Hepes, pH 7.5). During typical recordings, agonists were applied for 15 s followed by a 116-s wash with the running buffer. For recordings with epibatidine, the first eight drug concentrations were applied for 90 s with a 116-s wash with running buffer, while the remaining concentrations were applied for 15 s with a 116-s wash. Dose-response data were obtained for ≥8 agonist concentrations on ≥6 cells. All EC50 and Hill coefficient values were obtained by fitting dose-response relations to the Hill equation and are reported as averages ± standard error of the fit. A detailed error analysis of nonsense suppression experiments reveals data are reproducible to ± 50% in EC50 (38, 39). Voltage jump experiments were conducted to verify stoichiometry as described previously (14).
Double mutant cycle analyses were performed with EC50 values to calculate coupling coefficients (Ω) using the equation 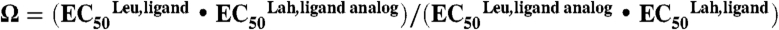 , where Leu, ligand and Leu, ligand analog represent the EC50 of the wild-type receptor with either ligand and Lah, ligand; and Lah, ligand analog represent the EC50 of the ester mutation with either ligand. Coupling energies ΔΔGint were calculated from the equation ΔΔGint = -RTlnΩ.
, where Leu, ligand and Leu, ligand analog represent the EC50 of the wild-type receptor with either ligand and Lah, ligand; and Lah, ligand analog represent the EC50 of the ester mutation with either ligand. Coupling energies ΔΔGint were calculated from the equation ΔΔGint = -RTlnΩ.
Synthesis of N-Methyl-2-Phenylpyrrolidine Hydrochloride.
Racemic 2-phenylpyrrolidine (5.0 g, 34 mmol), prepared according to a published protocol (33), was mixed with dibenzoyl-L-taratric acid (6.1 g, 17 mmol) in a 100-mL round-bottom flask equipped with a reflux condenser. To this was added 35% ethanol in ethylacetate (30 mL). The solution was heated to boiling for 10 min and then cooled to room temperature overnight. The white crystals were collected, rinsed with cold ethylacetate, and then submitted to five sequential recrystallizations. The yield was (10%, 2.2 g). Spectral data are 1H NMR (CDCl3, 300 MHz) δ 8.20 (4H, m), 7.61–7.32 (16H, m), 5.92 (2H, s), 5.03 (4H, b), 4.54 (2H, dd, J = 9.1, 6.7 Hz), 3.38 (4H, m), 2.27–2.00 (8H, m); 13C NMR (CDCl3, 75 MHz) δ 172.58, 166.45, 134.91, 132.73, 130.54, 129.70, 128.83, 128.76, 128.03, 127.37, 75.60, 62.74, 44.80, 30.42, 23.37. High resolution mass spectrometry (HRMS) (FAB+) m/z calculated for C10H14N [M+]: 148.1126, found 148.1081. To obtain enantioenriched 2-phenylpyrrolidine, the product was vigorously stirred in a 1∶1 mixture of 2 M NaOH: CH2Cl2. The organic layer was then extracted with additional CH2Cl2 (3×), washed with brine, dried over Na2SO4, and concentrated to yield enantioenriched 2-phenylpyrrolidine as a yellow oil (yield: 95%). NMR spectra are consistent with previously reported data. HRMS (FAB+) m/z calculated for C10H14N [M + H]: 148.1126, found 148.1134. To establish enantiomeric excess, the product was converted to ethyl 2-phenylpyrrolidine-1-carboxylate via a previously described procedure (40), and this material was evaluated by analytical chiral HPLC analysis using a Chiralcel OD-H column (4.6 mm × 25 cm) from Daicel Chemical Industries, Ltd., with 2% isopropyl alcohol in hexanes, giving an enantiomeric excess of 96%. 1H NMR of ethyl 2-phenylpyrrolidine-1-carboxylate gave (CH3OD, 300 MHz) δ; 7.32–7.15 (5H, m), 4.92 (1H, m), 4.08 (IH, m), 3.92 (1H, m), 3.59 (2H, q, J = 7.7 Hz), 2.34 (1H, m), 1.95–1.86 (4H, m), 1.26 (1H, t, J = 7.0 Hz), 0.94 (1H, t, J = 7 Hz); 13C NMR of ethyl 2-phenylpyrrolidine-1-carboxylate (CDCl3, 75 MHz) δ 155.40, 144.32, 128.22, 126.59, 125.44, 60.85, 47.34, 47.03, 35.71, 23.58, 14.79. HRMS of ethyl 2-phenylpyrrolidine-1-carboxylate (FAB+) m/z calculated for C13H18O2N [M + H]: 220.1338, found 220.1336.
Enantioenriched 2-phenylpyrrolidine from above, (0.13 g, 0.86 mmol) was added to a two-neck, 25-mL round-bottom flask equipped with a reflux condenser. To this was added 4 mL of formic acid and 2 mL of 37 wt% formaldehyde (in H2O). The mixture was stirred and heated to reflux at 80 °C for 3 h. The solution was cooled to room temperature and made basic (pH 12) by the addition of 2 M NaOH. The organics were extracted with CH2Cl2, washed with brine, dried over Na2SO4, and concentrated. The resulting yellow oil was placed into a 25-mL round-bottom flask and dissolved in 5 mL of cold ether. HCl (g) was generated and passed into the solution by slow addition of HCl (aq, 12 M) into H2SO4 (aq). The resulting white crystals were collected by filtration and dried. The yield was 83%, 140 mg, and the spectral data were 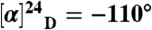 (c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 7.66 (2H, m), 7.32 (3H, m), 4.14 (1H, m), 3.95 (1H, b), 3.05 (2H, m), 2.60 (3H, d, J = 4.7 Hz), 2.29 (4H, m); 13C NMR (CDCl3, 75 MHz) δ 131.99, 129.89, 129.31, 128.76, 73.05, 44.43, 37.67, 31.94, 20.95; HRMS (FAB+) m/z calculated for C11H16N [M+]: 162.1283, found 162.1325.
(c = 1, CHCl3); 1H NMR (CDCl3, 300 MHz) δ 7.66 (2H, m), 7.32 (3H, m), 4.14 (1H, m), 3.95 (1H, b), 3.05 (2H, m), 2.60 (3H, d, J = 4.7 Hz), 2.29 (4H, m); 13C NMR (CDCl3, 75 MHz) δ 131.99, 129.89, 129.31, 128.76, 73.05, 44.43, 37.67, 31.94, 20.95; HRMS (FAB+) m/z calculated for C11H16N [M+]: 162.1283, found 162.1325.
Supplementary Material
Acknowledgments.
We thank Ariele P. Hanek and Sean M. A. Kedrowski for helpful discussions. This work was supported by the National Institutes of Health (NS 34407; NS 11756) and the California Tobacco-Related Disease Research Program of the University of California, Grant 16RT-0160.
Footnotes
This contribution is part of the special series of Inaugural Articles by members of the National Academy of Sciences elected in 2009.
The authors declare no conflict of interest.
See Commentary on page 13195.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1007140107/-/DCSupplemental.
References
- 1.Corringer PJ, Le Novere N, Changeux JP. Nicotinic receptors at the amino acid level. Annu Rev Pharmacol Toxicol. 2000;40:431–458. doi: 10.1146/annurev.pharmtox.40.1.431. [DOI] [PubMed] [Google Scholar]
- 2.Grutter T, Changeux JP. Nicotinic receptors in wonderland. Trends Biochem Sci. 2001;26:459–463. doi: 10.1016/s0968-0004(01)01921-1. [DOI] [PubMed] [Google Scholar]
- 3.Karlin A. Emerging structure of the nicotinic acetylcholine receptors. Nat Rev Neurosci. 2002;3:102–114. doi: 10.1038/nrn731. [DOI] [PubMed] [Google Scholar]
- 4.Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol Sci. 2006;27:482–491. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 5.Romanelli MN, et al. Central nicotinic receptors: Structure, function, ligands, and therapeutic potential. ChemMedChem. 2007;2:746–767. doi: 10.1002/cmdc.200600207. [DOI] [PubMed] [Google Scholar]
- 6.Coe JW, et al. Varenicline: An alpha 4 beta 2 nicotinic receptor partial agonist for smoking cessation. J Med Chem. 2005;48:3474–3477. doi: 10.1021/jm050069n. [DOI] [PubMed] [Google Scholar]
- 7.Mansvelder HD, Keath JR, McGehee DS. Synaptic mechanisms underlie nicotine-induced excitability of brain reward areas. Neuron. 2002;33:905. doi: 10.1016/s0896-6273(02)00625-6. [DOI] [PubMed] [Google Scholar]
- 8.Nashmi R, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: Basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tapper AR, et al. Nicotine activation of alpha4* receptors: Sufficient for reward, tolerance, and sensitization. Science. 2004;306:1029–1032. doi: 10.1126/science.1099420. [DOI] [PubMed] [Google Scholar]
- 10.Beers WH, Reich E. Structure and activity of acetylcholine. Nature. 1970;228:917–922. doi: 10.1038/228917a0. [DOI] [PubMed] [Google Scholar]
- 11.Glennon RA, Dukat M. Central nicotinic receptor ligands and pharmacophores. Pharm Acta Helv. 2000;74:103–114. doi: 10.1016/s0031-6865(99)00022-9. [DOI] [PubMed] [Google Scholar]
- 12.Glennon RA, Dukat M, Liao L. Musings on alpha4beta2 nicotinic acetylcholine (nACh) receptor pharmacophore models. Curr Top Med Chem. 2004;4:631–644. doi: 10.2174/1568026043451122. [DOI] [PubMed] [Google Scholar]
- 13.Dougherty DA, Stauffer DA. Acetylcholine binding by a synthetic receptor. Implications for biological recognition. Science. 1990;250:1558–1560. doi: 10.1126/science.2274786. [DOI] [PubMed] [Google Scholar]
- 14.Xiu X, Puskar NL, Shanata JAP, Lester HA, Dougherty DA. Nicotine binding to brain receptors requires a strong cation-π interaction. Nature. 2009;458:534–537. doi: 10.1038/nature07768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong W, et al. From ab initio quantum mechanics to molecular neurobiology: A cation-π binding site in the nicotinic receptor. Proc Natl Acad Sci USA. 1998;95:12088–12093. doi: 10.1073/pnas.95.21.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dougherty DA. Cys-loop neuroreceptors: Structure to the rescue? Chem Rev. 2008;108:1642–1653. doi: 10.1021/cr078207z. [DOI] [PubMed] [Google Scholar]
- 17.Brejc K, et al. Crystal structure of an ACh-binding protein reveals the ligand-binding domain of nicotinic receptors. Nature. 2001;411:269–276. doi: 10.1038/35077011. [DOI] [PubMed] [Google Scholar]
- 18.Celie PH, et al. Nicotine and carbamylcholine binding to nicotinic acetylcholine receptors as studied in AChBP crystal structures. Neuron. 2004;41:907–914. doi: 10.1016/s0896-6273(04)00115-1. [DOI] [PubMed] [Google Scholar]
- 19.Rucktooa P, Smit AB, Sixma TK. Insight in nAChR subtype selectivity from AChBP crystal structures. Biochem Pharmacol. 2009;78:777–787. doi: 10.1016/j.bcp.2009.06.098. [DOI] [PubMed] [Google Scholar]
- 20.Hansen SB, et al. Structural characterization of agonist and antagonist-bound acetylcholine-binding protein from Aplysia californica. J Mol Neurosci. 2006;30:101–102. doi: 10.1385/JMN:30:1:101. [DOI] [PubMed] [Google Scholar]
- 21.Hansen SB, et al. Structures of Aplysia AChBP complexes with nicotinic agonists and antagonists reveal distinctive binding interfaces and conformations. EMBO J. 2005;24:3635–3646. doi: 10.1038/sj.emboj.7600828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor P, et al. Structure-guided drug design: Conferring selectivity among neuronal nicotinic receptor and acetylcholine-binding protein subtypes. Biochem Pharmacol. 2007;74:1164–1171. doi: 10.1016/j.bcp.2007.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cashin AL, Petersson EJ, Lester HA, Dougherty DA. Using physical chemistry to differentiate nicotinic from cholinergic agonists at the nicotinic acetylcholine receptor. J Am Chem Soc. 2005;127:350–356. doi: 10.1021/ja0461771. [DOI] [PubMed] [Google Scholar]
- 24.Koh JT, Cornish VW, Schultz PG. An experimental approach to evaluating the role of backbone interactions in proteins using unnatural amino acid mutagenesis. Biochemistry. 1997;36:11314–11322. doi: 10.1021/bi9707685. [DOI] [PubMed] [Google Scholar]
- 25.England PM, Zhang Y, Dougherty DA, Lester HA. Backbone mutations in transmembrane domains of a ligand-gated ion channel: Implications for the mechanism of gating. Cell. 1999;96:89–98. doi: 10.1016/s0092-8674(00)80962-9. [DOI] [PubMed] [Google Scholar]
- 26.Deechongkit S, et al. Context-dependent contributions of backbone hydrogen bonding to beta-sheet folding energetics. Nature. 2004;430:101–105. doi: 10.1038/nature02611. [DOI] [PubMed] [Google Scholar]
- 27.Deechongkit S, Dawson PE, Kelly JW. Toward assessing the position-dependent contributions of backbone hydrogen bonding to beta-sheet folding thermodynamics employing amide-to-ester perturbations. J Am Chem Soc. 2004;126:16762–16771. doi: 10.1021/ja045934s. [DOI] [PubMed] [Google Scholar]
- 28.Gleitsman KR, Kedrowski SMA, Lester HA, Dougherty DA. An intersubunit hydrogen bond in the nicotinic acetylcholine receptor that contributes to channel gating. J Biol Chem. 2008;283:35638–35643. doi: 10.1074/jbc.M807226200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fonck C, et al. Novel seizure phenotype and sleep disruptions in knock-in mice with hypersensitive alpha 4* nicotinic receptors. J Neurosci. 2005 Dec 7;25:11396–11411. doi: 10.1523/JNEUROSCI.3597-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nowak MW, et al. In vivo incorporation of unnatural amino acids into ion channels in a Xenopus oocyte expression system. Methods Enzymol. 1998;293:504–529. doi: 10.1016/s0076-6879(98)93031-2. [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez EA, Lester HA, Dougherty DA. Improved amber and opal suppressor tRNAs for incorporation of unnatural amino acids in vivo. Part 1: Minimizing misacylation. RNA. 2007;13:1703–1714. doi: 10.1261/rna.666807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodriguez EA, Lester HA, Dougherty DA. Improved amber and opal suppressor tRNAs for incorporation of unnatural amino acids in vivo. Part 2: Evaluating suppression efficiency. RNA. 2007;13:1715–1722. doi: 10.1261/rna.667607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dunsmore CJ, Carr R, Fleming T, Turner NJ. A chemo-enzymatic route to enantiomerically pure cyclic tertiary amines. J Am Chem Soc. 2006;128:2224–2225. doi: 10.1021/ja058536d. [DOI] [PubMed] [Google Scholar]
- 34.Kash TL, Jenkins A, Kelley JC, Trudell JR, Harrison NL. Coupling of agonist binding to channel gating in the GABA(A) receptor. Nature. 2003;421:272–275. doi: 10.1038/nature01280. [DOI] [PubMed] [Google Scholar]
- 35.Price KL, Millen KS, Lummis SC. Transducing agonist binding to channel gating involves different interactions in 5-HT3 and GABAC receptors. J Biol Chem. 2007;282:25623–25630. doi: 10.1074/jbc.M702524200. [DOI] [PubMed] [Google Scholar]
- 36.Venkatachalan SP, Czajkowski C. A conserved salt bridge critical for GABAA receptor function and loop C dynamics. Proc Natl Acad Sci USA. 2008;105:13604–13609. doi: 10.1073/pnas.0801854105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mu TW, Lester HA, Dougherty DA. Different binding orientations for the same agonist at homologous receptors: A lock and key or a simple wedge? J Am Chem Soc. 2003;125:6850–6851. doi: 10.1021/ja0348086. [DOI] [PubMed] [Google Scholar]
- 38.Torrice MM. Chemical-Scale Studies of the Nicotinic and Muscarinic Acetylcholine Receptors. Pasadena, CA: California Institute of Technology; 2009. [Google Scholar]
- 39.Torrice MM, Bower KS, Lester HA, Dougherty DA. Probing the role of the cation-pi interaction in the binding sites of GPCRs using unnatural amino acids. Proc Natl Acad Sci USA. 2009;106:11919–11924. doi: 10.1073/pnas.0903260106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felpin F-X, et al. Efficient enantiomeric synthesis of pyrrolidine and piperidine alkaloids from tobacco. J Org Chem. 2001;66:6305–6312. doi: 10.1021/jo010386b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.