

Abstract
Protein and mRNA copy numbers vary from cell to cell in isogenic bacterial populations. However, these molecules often exist in low copy numbers, and are difficult to detect in single cells. Here we carried out quantitative system-wide analyses of protein and mRNA expression in individual cells with single-molecule sensitivity using a newly constructed yellow fluorescent protein fusion library for Escherichia coli. We found that almost all protein number distributions can be described by the gamma distribution with two fitting parameters which, at low expression levels, have clear physical interpretations as the transcription rate and protein burst size. At high expression levels, the distributions are dominated by extrinsic noise. Strikingly, we found that a single cell's protein and mRNA copy numbers for any given gene are uncorrelated.
Gene expression is often stochastic because gene regulation takes place at a single DNA locus within a cell. Such stochasticity is manifested in fluctuations of mRNA and protein copy numbers within a cell lineage over time, and in variations of mRNA and protein copy numbers among a population of genetically identical cells at a particular time (1, 2, 3, 4). Because both manifestations of stochasticity are connected, measurement of the latter allows the deduction of the gene expression dynamics in a cell (5). We aim to characterize such mRNA and protein distributions in single bacteria cells at a system-wide level.
While single cell mRNA profiling has been carried out with cDNA microarray (6) and mRNA-seq (7), these studies did not have single molecule sensitivity and are not suitable for bacteria, which express mRNA at low copy numbers (8). A fluorescent protein reporter library of Saccharomyces cerevisiae (9) has proven to be extremely useful in protein profiling (10, 11). However, the lack of sensitivity in existing flow cytometry or fluorescence microscopy techniques prevented the quantification of one third of the labeled proteins because of their low copy numbers. In recent years, single-molecule fluorescence microscopy has been used to count mRNA (12-16) or protein (8, 17) molecules in individual cells, especially in bacteria. However, these methods have only been applied to limited number of specific genes.
Here we report single cell global profiling of both mRNA and proteins with single molecule sensitivity using a yellow fluorescent protein (YFP) fusion library for the model organism Escherichia coli.
Single-molecule imaging of a YFP reporter library
We created a chromosomal YFP fusion library (Fig. 1A), in which each strain has a particular gene tagged with the YFP coding sequence. YFP can be detected with single molecule sensitivity in live bacterial cells (8, 18). We converted the C-terminus tags of an existing chromosomally affinity-tagged E. coli library (19, 20) to yfp translational fusions using λ-RED recombination (21). Out of the 1,400 strains attempted, 1,018 strains were confirmed by sequencing and showed no significant growth defects. The list of strains is given in Table S1 (18).
Figure 1.
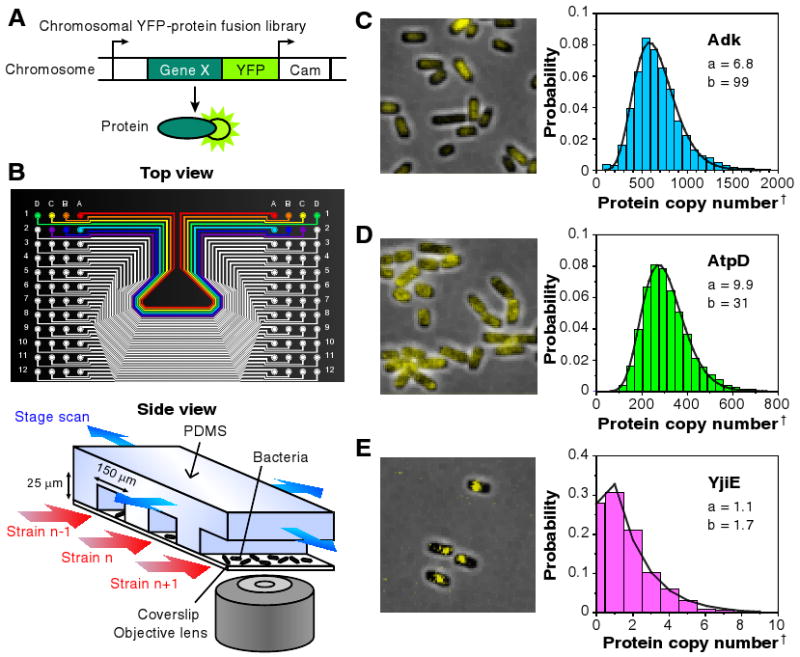
Quantitative imaging of a YFP-fusion library. (A) Each library strain has a yellow fluorescent protein (YFP) translationally fused to the C-terminus of a protein in its native chromosomal position. (B) A poly-dimethylsiloxane (PDMS) microfluidic chip is used for imaging 96 library strains. E. coli cells of each strain are injected into separate lanes and immobilized on a polylysine-coated coverslip for automated fluorescence imaging with single molecule sensitivity. (C-E) Representative fluorescence images overlaid on phase contrast images of three library strains, with respective single cell protein level histograms that are fit to gamma distributions with parameters a and b. Protein levels are determined by deconvolution (18). †The protein copy number per average cell volume, or the concentration, was determined as described in the main text and the SOM. (C) Cytoplasmic protein, Adk, uniformly distributed intracellularly. (D) Membrane protein, AtpD, distributed on the cell periphery. (E) Predicted DNA-binding protein, YjiE, with clear intercellular localization. Single YFPs can be visualized via detection by localization. Note unlike (C) and (D), the gamma distribution asymmetrically peaks near zero if a is close to or less than unity.
To facilitate high-throughput analyses of the YFP library strains, we implemented an automated imaging platform based on a microfluidic device (Fig. 1B) (22) that holds 96 independent library strains attached to poly-lysine coated cover glass. Each device was imaged with a single-molecule fluorescence microscope at a rate of ∼4,000 cells in 25 s per strain (Fig. 1C). Single molecule sensitivity was confirmed by abrupt photobleaching of membrane-bound YFPs expressed at low level (Fig. S14) (8, 18, 23). Automated image analysis was performed to determine the distribution of single cell protein abundance normalized by cell size (Fig. 1D) (18). Normalization by cell size is necessary to account for cell size and gene copy number variation due to the cell cycle. We removed the contribution of cellular autofluorescence by deconvolution (Fig. S14) (18). The absolute protein level was obtained by calibration with single-molecule fluorescence intensities (Fig. S1) (18) to determine the protein concentration (copy numbers per average cell volume). An independent reporter assay confirmed that the resulting fluorescence accurately reports on native protein abundance (Fig. S4).
The fluorescence images show the intracellular localization of protein (Fig. 1C-E). Most cytoplasmic proteins localized to the inner regions of the cell (Fig. 1C), whereas many membrane-bound or periplasmic proteins showed localization along the outer contours of the cell (Fig. 1D, see Table S3). Other proteins, including some DNA-bound proteins and low copy membrane proteins, showed punctate localization (Fig.1E).
Average protein abundances span five orders of magnitude, ranging from 10-1 to 104 molecules per cell (Fig 2A). The average protein abundances of essential genes are higher than those for all genes. Of the 121 essential proteins in the library (24), 108 express at ten or more molecules per cell (Fig 2A), whereas about half of all the measured proteins are present at fewer than ten molecules per cell (18). Of the low expression genes, 60% have been annotated to date (18), and at least 25% were found to have a genetic interaction in a recent double knockout study (25). The prevalence of proteins with very low copy number suggests that single-molecule experiments are necessary for bacteriology.
Figure 2.
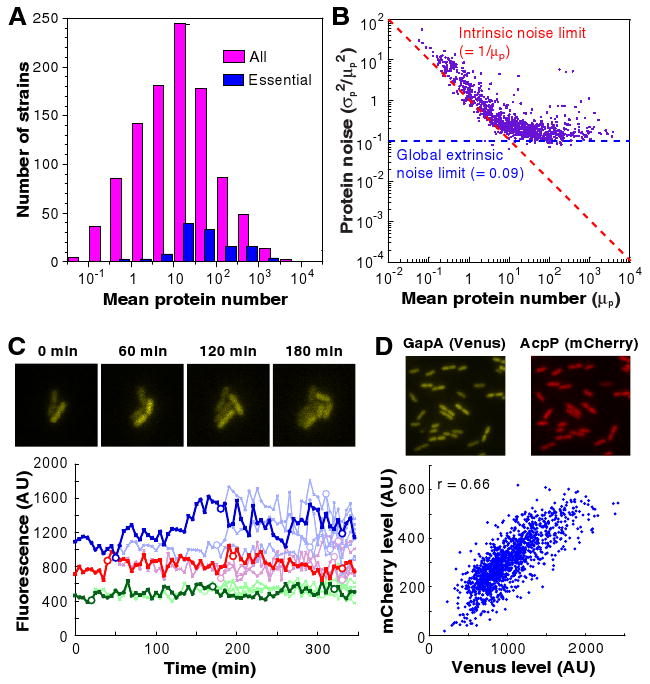
Profiling of protein abundance and noise. (A) Histogram of average copy numbers of essential proteins (blue) and all proteins (pink) for 1,018 library strains. Almost all essential proteins are expressed at above an average of 10 copies / cell, while some non-essential proteins have lower abundances. (B) Protein expression noise (σp2/μp2) versus the mean copy number per cell (μp). When μp <10, protein expression noise is inversely proportional to the mean, with as lower noise limit (red dashed line), as expected for intrinsic noise. When μp >10, noise becomes independent of the mean and is always above ∼0.1 (noise limit indicated by blue dashed lines), because of extrinsic noise. (C) Real time observation of slow fluctuation of protein levels at a time scale longer than a cell cycle, originating from extrinsic noise. Each time trace of fluorescence is normalized by cell size, and represents a cell lineage of strain expressing AcpP-YFP. The dark line follows a single lineage; the rest of the descendents have a lighter colored line. Each circle represents a cell division event. The variation among different cells arises primarily from slowly-varying extrinsic noise because the fluctuations within one cell over a cell cycle are comparatively small. (D) Two-color measurements of correlations of two different proteins in the same cell. Two highly-expressed proteins, GapA and AcpP, are respectively labeled with Venus (YFP) and mCherry (RFP) in the same E. coli strain. The protein levels are correlated, with a correlation coefficient of 0.66, supporting the hypothesis that the dependence on global extrinsic factors like ribosomes, rather than gene-specific factors, dominates the extrinsic noise at high expression levels (see (18)).
Analysis of protein distributions
To obtain intrinsic properties of gene expression dynamics, we analyzed the protein expression distributions of different genes. We consider the kinetic scheme
| (1) |
Here k1 and k2 are the transcription and translation rates, respectively. γ1 is the mRNA degradation rate, and γ2 is the protein degradation rate. For stable proteins, including fluorescent protein fusions, γ2 is dominated by the rate of dilution due to cell division, and is insensitive to protein lifetime, which could be different for the fusion and native protein. The number of mRNA produced per cell cycle is given by a = k1/γ2, and the protein molecules produced per mRNA is given by b = k2/γ1. It was shown theoretically (5, 26) that, under the steady-state condition of Poissonian production of mRNA and an exponentially distributed protein burst size, as previously observed (8, 17), Eq. 1 results in a gamma distribution of protein copy numbers, x, which is normalized by the average cell volume.
| (2) |
Here Γ is a gamma function. The gamma distribution has the property that a is equal to the inverse of noise (σp2/μp2) and b is equal to the Fano factor (σp2/μp), where σp2 and μp are the variance and mean of the protein number distributions, respectively. Specific cases have provided experimental support for gamma distribution, but it has not been verified on a system-wide manner (17).
The distributions for 1,009 out of the 1,018 strains can be well fit by the gamma distribution, Eq. 2 (Fig. S20) (18). Consistent with the gamma distribution, the observed distributions are skewed with the peak at zero for low abundance proteins, and have non-zero peaks for high abundance proteins (Fig. 1C-E). We note that the bimodal distribution of lac permease was observed in E. coli under certain inducer concentrations (23, 27). The fact that we did not observe clear bimodal distributions among the 1,018 strains under our growth conditions indicates that bimodal distributions are generally rare.
We note that alternative mathematical solution to Eq. 1 gives a negative binomial distribution of protein copy numbers (26). However, the gamma distribution offers a more robust fit of experimental data at low expression levels because the negative binomial fits are very sensitive to measurement error (18). The two distributions have similar fitting at high expression levels. Other functions such as log-normal distributions have been used phenomenologically to fit unimodal distributions (10, 18). However, the gamma distribution fits better than the log-normal distribution for proteins with low expression levels (Fig. S20) (18) and fits similarly well for proteins with high expression levels. Most importantly, the gamma distribution allows extraction of dynamic information from easy measurements of the steady-state distribution at low expression levels. The a-b values and the goodness-of-fits for the 1,018 strains are given in Table S6.
Global scaling of intrinsic and extrinsic protein noise
The protein noise (ηp2 ≡ σp2/μp2) exhibits two distinct scaling properties (Fig. 2B). Below ten molecules per cell, ηp2 is inversely proportional to protein abundance, indicative of intrinsic noise. In contrast, at higher expression levels (>10 molecules per cell), the noise reaches a plateau of ∼0.1 and does not decrease further, suggesting that each protein has at least 30% variation in its expression level.
For lowly expressed proteins, simple Poisson production and degradation of mRNA and protein, commonly termed intrinsic noise, are sufficient to account for the observed scaling of σp2/μp2∝ 1/μp(Fig. 2B) (10, 11, 28-30). This scaling property has also been observed for highly expressed yeast proteins (10, 11). We verified Poisson kinetics by monitoring real-time protein production in single cells for several genes whose expression levels were low (Table S4) (18), which agrees with previous work on the repressed lac operon (8, 17). The observed noise is always greater or equal to 1/μp, suggesting that specific regulatory methods do not decrease noise significantly below this limit.
For abundant proteins, the 1/μp scaling no longer applies, and a large noise floor overwhelms the intrinsic noise contribution (Fig 2B). This means that the interpretation of the two parameters a = μp2/σp2 and b = σp2/μp as the burst frequency (k1/γ2) and burst size (k2/γ1) applies well only at low expression levels, while the protein distributions at high expression levels are dominated by other factors extrinsic to the above model. We found that the noise floor does not result from cell size effects, nor did it arise from measurement noise (18).
We attribute the additional noise to extrinsic noise (3), that is the slow variation of the values of a and b, which we confirm with real time observation of protein levels for four randomly selected high copy library strains. The high expression noise fluctuates more slowly than the cell cycle (Fig. 2C) (18), so that the rate constants in Eq. 1 can be considered to be heterogeneous among cells.
Assuming that there exist static or slowly varying heterogeneities of a and b, with distributions f (a) and g(b), respectively, the protein distribution is
| (3) |
Even if the normalized variances of f (a) and g(b), ηa2 and ηb2, Eq. 3 can still be approximated as a gamma distribution, which explains the generality of the gamma distribution fit of the data (18).
The noise plateau in Fig 2B can be explained by calculating the expected noise from Eq. 3 (18, 26, 31)
| (4) |
The extrinsic noise in the last three terms in Eq. 4 might originate from fluctuations in cellular components such as metabolites, ribosomes, and polymerases (30, 32), and dominates the noise of high copy proteins (μp≫1, Eq. 4.).
We further demonstrate that the extrinsic noise is global to all high expression genes by analyzing the correlations between expression levels of 13 pairs of randomly selected genes. Using YFP and red fluorescent protein (RFP) fusions as a pair of reporters (Fig. 2D), we observed statistically significant correlations between the expression levels of all gene pairs, confirming the existence of a global noise factor. The observed correlation is quantitatively predicted by the observed noise floor (18).
Single-molecule RNA counting
To examine single cell mRNA expression, we performed fluorescence in situ hybridization (FISH) with single molecule sensitivity (33) (Fig. 3A) using a single universal Atto594-labeled 20-mer oligonucleotide probe targeting the yfp mRNA in our library. Because the same probe is used for all strains, the optimized hybridization efficiency is unbiased for every measured gene (18). We confirmed the validity of our transcript measurements with RNA-seq (Table S6) (18).
Figure 3.
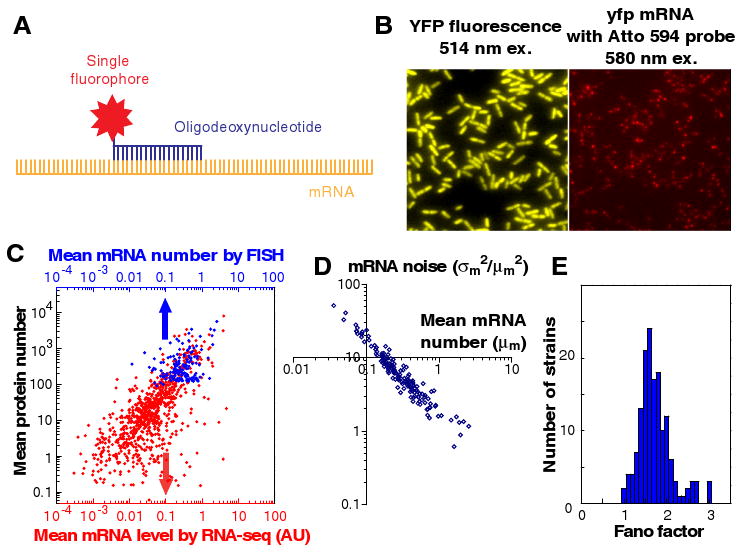
mRNA profiling of the YFP-fusion library with single molecule sensitivity in single cells. (A) The mRNA of a tagged gene can be detected by fluorescence in situ hybridization (FISH) against the yfp mRNA sequence using a DNA oligo probe that is labeled with a single Atto594 fluorophore. (B) Protein (left) and mRNA (right) of the same gene are detected simultaneously in the same fixed cells. (C) Mean mRNA number, measured by RNA-seq (red) and by FISH (blue), and mean protein number are correlated. The respective Pearson correlation coefficients (r) are 0.54 and 0.77. Each dot is average of a gene. The FISH data were taken for genes that express >100 copies of proteins per cell, whereas the RNA-seq data includes all expressed mRNAs, which are not fused to the yfp tag. (D) mRNA noise (σm2/μm2) scales inversely with mRNA mean number (μm), and is higher than expected for Poisson distributions. (E) Histogram of mRNA Fano factors for 137 highly-expressed genes. The mRNA Fano factor (σm2/μm) of the measured strains have similar values centered around 1.6, indicating non-Poissonian mRNA production or degradation.
We show that the YFP (yellow) and the mRNA (red) of the same gene can be simultaneously detected, and spectrally resolved, within a single fixed cell (Fig. 3B). Due to their low copy numbers, mRNA molecules are sparsely distributed within a cell, independent of YFP locations. By measuring the intensity of each fluorescent spot and counting the number of spots per cell, mRNA copy numbers were determined for individual cells. We used this single molecule FISH method to quantify mRNA abundance and noise for 137 library strains with high protein expression levels (>100 proteins/cell).
At the ensemble level, the mean mRNA abundances among these 137 genes range from 0.05 to 5 per cell, and are moderately correlated with the corresponding mean protein expression level at the gene-by-gene basis (correlation coefficient r = 0.77) (Fig. 3C). The lack of complete correlation, as reported previously in other organisms, is often attributed to differences in post-transcriptional regulation. Here, with the ability to determine the absolute number of molecules per cell, we determined the ratio between the mean protein abundance and the mean mRNA abundance to range from 102 to 104.
At the single cell level, the mRNA copy number distributions were broader than the Poisson distributions expected by the random generation and degradation of transcripts with constant rates (18). The mRNA noise scales in inverse proportion to the mean mRNA abundance (Fig. 3D), but mRNA Fano factor values (σm2/μm), are close to ∼1.6 (Fig. 3E), rather than unity as expected for the Poissonian case. We excluded gene dosage effects by gating with the cell size to select the cells that have not yet gone through chromosome replication (18). The non-Poisson mRNA distributions indicate that the rate constant for mRNA generation or degradation fluctuates on a timescale similar or longer than the typical mRNA degradation time, which has an average of ∼5-10 minutes for our growth condition (18).
Simultaneous RNA and protein measurements in single cells
We now examine the extent to which the mRNA copy numbers and the protein levels are correlated in the same cells. We quantified single cell mRNA and protein levels simultaneously (Fig. 3B). Figure 4A shows a 2D scatter plot, in which each cell is plotted as a dot with its mRNA and protein levels on the X and Y axes, respectively, for the translation elongation factor EF-Tu in the TufA-YFP strain. mRNA and protein copy numbers in a single cell are not correlated (r = 0.01 ± 0.03, SEM, N = 5,447). In fact, among many different highly expressed strains surveyed, the correlation coefficients are all centered on zero (Fig. 4B), indicating a general lack of mRNA-protein correlation of the same gene within a single cell.
Figure 4.

No correlation between mRNA and protein levels in a single cell at a particular time. (A) Histograms of mRNA (top) and protein (right) levels. Protein vs. mRNA copy number plot for the TufA-YFP strain, in which TufA is tagged with YFP. Each point represents a single cell of the strain. Strikingly, the correlation coefficient is r = 0.01 ± 0.03 (mean ± SD, N = 5,447). (B) Histogram of correlation coefficients from 129 strains with highly expressed labeled genes whose sampling error for the correlation coefficient is <0.1. The histogram indicates that the lack of correlation between mRNA and protein levels in a single cell is a general phenomenon.
The lack of mRNA-protein correlation can be explained by the difference in mRNA and protein lifetime. In E. coli, mRNA is typically degraded within minutes (Table S6) (18), whereas most proteins, including fluorescent proteins, have a lifetime longer than the cell cycle (34). As a result, the mRNA copy number at any instant only reflects the recent history of transcription activity (∼ a few min), whereas the protein level at the same instant represents the long history of accumulated expression (time scale of a cell cycle). However, extrinsic translational noise or regulatory networks must also be present to account for the near-zero mRNA-protein correlation we observe (18). We note that the observed lack of correlation arises because the experiment only measured the copy numbers of protein and mRNA present at the moment of fixation of a single cell. This is not contradictory to the central dogma, which suggests that mRNA molecules produced in a long period of time should correlate with the protein molecules produced in the same period, as reflected in Fig 3C among different genes (11). However, our result offers a cautionary note in interpreting single-cell transcriptome analysis and argues for the necessity for single cell proteome analysis.
Correlation of expression properties with biological factors
The correlation between the expression parameters and selected gene characteristics is shown in Figure 5. Small a values correspond to a narrow range of b values, and large a values correspond to a wide range of b values (Fig. 5A). Highly expressed proteins (mean > 10) had high b values while low expression proteins had b values of about 1 (Fig. 5B). The protein expression levels had a weak correlation with codon adaptation index (CAI, r = 0.42), but had little correlation with GC content (r = -0.06) and the mRNA lifetime (r = 0.08). The a and b values showed moderate dependence on the chromosome position (Fig. 5F). The correlation coefficients and Z scores between these two and additional parameters are summarized in Table S2.
Figure 5.

Correlation between expression and gene characteristics. (A) Correlation plots of a and b (r = 0.01) and (B) mean protein expression versus b (r = 0.72). a and b values are calculated as a = μp2/σp2 and b = σp2/μp, respectively, using the mean, μp, and standard deviation, σp, of the protein number histograms. (C) Correlation plots of mean protein expression versus codon adaptation index (CAI) (r = 0.40), (D) GC content (r = -0.06) and (E) mRNA lifetime (r = 0.08). (F) Chromosomal dependence of a and b values. Z scores of more than 3 (indicated by red) represent a significantly larger value compared with the whole genome distribution with >99.9% confidence, while Z scores less than -3 (indicated by blue) represent a significantly smaller value.
In addition, we characterized the statistical bias of the expression and localization parameters for functional gene categories, as measured by a Z-score in Table 1. Some functional categories are strongly correlated with parameters. For example, essential proteins have a strong correlation with high a (Z = 7.5) and high b (Z = 5.3). As expected, membrane transporters showed high edge/inside ratio (Z = 7.3), and transcriptional repressors indicated high punctate localization (Z = 4.1). Proteins with no known protein-protein interactions have significantly reduced expression (Z = -4.7). We also found that shorter ORFs may have higher protein expression levels (Z = 4.1). RNA expression tends to be higher for genes transcribed from the leading strand parallel to the movement of the replication folk (Z = 4.0). Thus expression and localization properties can be significantly correlated with functional properties.
Table 1.
Trends in expression levels and protein localization. Table of Z scores of subsets of gene classes characterized by protein and RNA mean, RNA lifetime, a, b, ratio of fluorescence detected on the edge compared to inside of the cell (E/I), and the degree of punctuate protein localization (DP). Leading strand corresponds to transcription in the same direction as the replication fork. PPI indicates protein-protein interactions. Z scores of more than 3 (indicated by red) represent a significantly larger value compared with the whole genome distribution with >99.9% confidence, while Z scores less than -3 (indicated by blue) represent a significantly smaller value.
| Category | n | Protein | RNA | |||||
|---|---|---|---|---|---|---|---|---|
| Mean | a | b | E/I | DP | Mean | Lifetime | ||
| All | 1018 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Essential gene | 121 | 7.54 | 8.70 | 5.34 | -2.19 | -3.16 | 5.61 | 1.38 |
| Nonessential gene | 894 | -2.74 | -3.16 | -2.02 | 0.74 | 1.30 | -1.99 | -0.32 |
| Enzyme | 410 | 3.98 | 4.28 | 1.85 | -5.45 | -3.04 | 3.30 | 3.15 |
| Translation | 17 | 4.04 | 3.36 | 3.30 | -0.86 | -3.52 | 3.30 | -2.44 |
| Turnover, degradation | 13 | 3.05 | 3.60 | 2.08 | -0.99 | -1.76 | 2.37 | 2.14 |
| Transcription factor | 98 | 0.61 | 1.27 | 0.71 | -2.95 | 4.29 | -0.53 | -2.53 |
| Transporter | 88 | -0.84 | 0.45 | -1.44 | 7.78 | 0.96 | 2.28 | 3.29 |
| Lagging strand | 425 | -1.42 | -1.95 | -0.82 | 0.74 | -0.07 | -4.42 | -3.63 |
| Leading strand | 593 | 1.12 | 1.62 | 0.61 | -0.64 | 0.02 | 3.96 | 3.17 |
| Gene length < 500 bp | 193 | 4.06 | 1.09 | 3.34 | -1.06 | -2.98 | 2.67 | -1.65 |
| Gene length >= 500 bp | 825 | -1.92 | -0.55 | -1.72 | 0.57 | 1.45 | -1.26 | 0.73 |
| Known PPI | 603 | 3.73 | 3.75 | 1.37 | -3.69 | -3.77 | 1.90 | 0.58 |
| No known PPI | 415 | -4.68 | -4.65 | -1.73 | 4.28 | 4.43 | -2.25 | -0.61 |
Comparison between E. coli and yeast
Protein abundance and noise has been investigated in yeast for >2,500 high-abundance proteins using flow cytometry (10, 11). The single molecule sensitivity in single bacterial cells allowed us to characterize the full range of protein copy numbers in E. coli, which has not been realized in yeast. We found that E. coli proteins generally had larger noise and Fano factors than yeast proteins, even for those present at similar copy numbers (Fig. S6) (18). A noise plateau due to extrinsic factors is present for both, but the extrinsic noise is larger in E. coli.
Conclusion
We have provided quantitative analyses of both abundance and noise in the proteome and transcriptome on a single-cell level for gram negative bacteria E. coli. Given that some proteins and most mRNAs of functional genes are present at low copy numbers in a bacterial cell, the single molecule sensitivity afforded by our measurements is necessary for the understanding of stochastic gene expression and regulation. We discovered large fluctuations in low abundance proteins as well as a common extrinsic noise in high abundance proteins. Furthermore, we found that in a single cell mRNA and protein levels for the same gene are completely uncorrelated. This striking result highlights the disconnect between proteome and transcriptome analyses of a single cell, as well as the need for single cell proteome analysis. Taken together, a quantitative and integral account of single cell gene expression profile is emerging.
Supplementary Material
Acknowledgments
We thank L. Xun, N. K. Lee, D. Court, C. Zong, R. Roy and J. Agresti for experimental assistance; and E. Rubin, L. Cai, and J. Elf for helpful discussions. This work was supported by the Gates Foundation and the NIH Pioneer Director's Award. Y.T. acknowledges additional support from the Japan Society for the Promotion of Science, the Uehara Memorial Foundation, and the Marubun Research Promotion Foundation; and P.J.C. from the John and Fannie Hertz Foundation.
Footnotes
Publisher's Disclaimer: This manuscript has been accepted for publication in Science. This version has not undergone final editing. Please refer to the complete version of record at http://www.sciencemag.org/. The manuscript may not be reproduced or used in any manner that does not fall within the fair use provisions of the Copyright Act without the prior, written permission of AAAS.”
References
- 1.Rao CV, Wolf DM, Arkin AP. Nature. 2002 Nov 14;420:231. doi: 10.1038/nature01258. [DOI] [PubMed] [Google Scholar]
- 2.Raser JM, O'Shea EK. Science. 2004 Jun 18;304:1811. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Science. 2005 Mar 25;307:1962. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- 4.Pedraza JM, van Oudenaarden A. Science. 2005 Mar 25;307:1965. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- 5.Friedman N, Cai L, Xie XS. Physical review letters. 2006 Oct 20;97:168302. doi: 10.1103/PhysRevLett.97.168302. [DOI] [PubMed] [Google Scholar]
- 6.Tietjen I, et al. Neuron. 2003 Apr 24;38:161. doi: 10.1016/s0896-6273(03)00229-0. [DOI] [PubMed] [Google Scholar]
- 7.Tang F, et al. Nature methods. 2009 May;6:377. doi: 10.1038/nmeth.1315. [DOI] [PubMed] [Google Scholar]
- 8.Yu J, Xiao J, Ren X, Lao K, Xie XS. Science. 2006 Mar 17;311:1600. doi: 10.1126/science.1119623. [DOI] [PubMed] [Google Scholar]
- 9.Huh WK, et al. Nature. 2003 Oct 16;425:686. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 10.Bar-Even A, et al. Nature genetics. 2006 Jun;38:636. doi: 10.1038/ng1807. [DOI] [PubMed] [Google Scholar]
- 11.Newman JR, et al. Nature. 2006 Jun 15;441:840. doi: 10.1038/nature04785. [DOI] [PubMed] [Google Scholar]
- 12.Levsky JM, Shenoy SM, Pezo RC, Singer RH. Science. 2002 Aug 2;297:836. doi: 10.1126/science.1072241. [DOI] [PubMed] [Google Scholar]
- 13.Raj A, Peskin CS, Tranchina D, Vargas DY, Tyagi S. PLoS Biol. 2006 Oct;4:e309. doi: 10.1371/journal.pbio.0040309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golding I, Paulsson J, Zawilski SM, Cox EC. Cell. 2005 Dec 16;123:1025. doi: 10.1016/j.cell.2005.09.031. [DOI] [PubMed] [Google Scholar]
- 15.Maamar H, Raj A, Dubnau D. Science. 2007 Jul 27;317:526. doi: 10.1126/science.1140818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zenklusen D, Larson DR, Singer RH. Nat Struct Mol Biol. 2008 Dec;15:1263. doi: 10.1038/nsmb.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cai L, Friedman N, Xie XS. Nature. 2006 Mar 16;440:358. doi: 10.1038/nature04599. [DOI] [PubMed] [Google Scholar]
- 18.Methods, and discussion are available as supporting material on Science Online.
- 19.Butland G, et al. Nature. 2005 Feb 3;433:531. doi: 10.1038/nature03239. [DOI] [PubMed] [Google Scholar]
- 20.Hu P, et al. PLoS Biol. 2009 Apr 28;7:e96. doi: 10.1371/journal.pbio.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu D, et al. Proceedings of the National Academy of Sciences of the United States of America. 2000 May 23;97:5978. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald JC, et al. Electrophoresis. 2000 Jan;21:27. doi: 10.1002/(SICI)1522-2683(20000101)21:1<27::AID-ELPS27>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 23.Choi PJ, Cai L, Frieda K, Xie XS. Science. 2008 Oct 17;322:442. doi: 10.1126/science.1161427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baba T, et al. Mol Syst Biol. 2006;2:2006 0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Butland G, et al. Nature methods. 2008 Sep;5:789. doi: 10.1038/nmeth.1239. [DOI] [PubMed] [Google Scholar]
- 26.Paulsson J, Ehrenberg M. Physical review letters. 2000 Jun 5;84:5447. doi: 10.1103/PhysRevLett.84.5447. [DOI] [PubMed] [Google Scholar]
- 27.Novick A, Weiner M. Proceedings of the National Academy of Sciences of the United States of America. 1957 Jul 15;43:553. doi: 10.1073/pnas.43.7.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulsson J. Nature. 2004 Jan 29;427:415. doi: 10.1038/nature02257. [DOI] [PubMed] [Google Scholar]
- 29.Thattai M, van Oudenaarden A. Proceedings of the National Academy of Sciences of the United States of America. 2001 Jul 17;98:8614. doi: 10.1073/pnas.151588598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Science. 2002 Aug 16;297:1183. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- 31.Shahrezaei V, Swain PS. Proceedings of the National Academy of Sciences of the United States of America. 2008 Nov 11;105:17256. doi: 10.1073/pnas.0803850105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Swain PS, Elowitz MB, Siggia ED. Proceedings of the National Academy of Sciences of the United States of America. 2002 Oct 1;99:12795. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Femino AM, Fay FS, Fogarty K, Singer RH. Science. 1998 Apr 24;280:585. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- 34.Koch L, Levy HR. J Biol Chem. 1955 May 12;217:947. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.