

Abstract
Specific interactions between post-translational modifications (PTMs) and chromatin-binding proteins are central to the idea of a ‘histone code’. Here, a 5000-member, PTM-randomized, combinatorial peptide library based on the N-terminus of histone H3 was utilized to interrogate multi-site specificity of six chromatin-binding modules, which read the methylation status of K4. We found that T3 phosphorylation, R2 methylation, and T6 phosphorylation are critical additional PTMs that modulate the ability to recognize and bind histone H3. Notably, phosphorylation of T6 yielded the most varied effect on protein binding, suggesting an important regulatory mechanism for readers of the H3 tail. Mass spectrometry and antibody-based evidence indicate that this previously uncharacterized modification exists on native H3, and NMR analysis of ING2 revealed the structural basis for discrimination. These investigations reveal a continuum of binding affinities in which multi-site PTM recognition involves both switch- and rheostat-like properties, yielding graded effects that depend on the inherent ‘reader’ specificity.
Histone proteins package eukaryotic DNA into chromatin and orchestrate virtually all DNA-templated processes (transcription, replication, repair, recombination, etc.). The basic building block of chromatin, the nucleosome, is derived from an octamer of histone proteins: one H3/H4 tetramer and two H2A/H2B dimers1. Histones are composed of α-helical globular domains tethered to unstructured N-terminal tails, which protrude from the DNA gyres. Histone proteins are the targets of a dazzling array of posttranslational modifications (PTMs) that include acetylation, methylation, phosphorylation, and deimination2. The vast majority of histone modifications occur on the unstructured N-terminal tails3. These dynamic modifications regulate the structure and function of chromatin through mechanisms that remain to be completely elucidated. The histone code hypothesis asserts that combinatorial histone modification patterns potentiate biological outcomes via recruitment of chromatin remodeling enzymes and protein complexes4. The language of such a code is interpreted by specialized histone-binding modules such as bromodomains, chromodomains and PHD (plant homeodomain) fingers, which recognize histones in a modification-dependent manner2,3.
Plant Homeo Domain (PHD) fingers are a versatile class of nuclear protein-interaction domains comprised of approximately 60 amino acids. The archetypical PHD finger contains a pair of zinc fingers and features a Cys4-His-Cys3 motif, which is responsible for coordinating two Zn2+ ions. The human genome consists of ~150 PHD fingers5, many of which have not been characterized. Recent findings link loss-of-function mutations in PHD fingers to cancer, immunodeficiency syndromes, and neurological disorders6. For example, a W453R mutation of the Recombination Activating Gene 2 (RAG2)- PHD finger (found in patients with Omenn syndrome) impairs recognition of trimethylated H3K4 and subsequent V(D)J recombination, a critical component of antigen-receptor gene assembly7. Several other PHD fingers are linked to “reading” and interpreting the modification state at H3K4. The autoimmune regulator (AIRE)-PHD finger 1 binds unmethylated H3K4 to activate gene expression8. In contrast, binding of the BRAF-HDAC Complex 80 (BHC80)-PHD finger to unmethylated H3K4 is linked to LSD1-mediated gene repression9. Association with H3K4me3 —a mark commonly associated with transcriptional initiation10 —stimulates chromatin remodeling, gene expression, and gene repression in the cases of the PHD fingers of bromodomain PHD finger transcription factor (BPTF)11, TBP- associated factor 3 (TAF3)12 and inhibitor of growth 2 (ING2)13, respectively.
How can PHD fingers that interact at H3K4 facilitate such disparate outcomes? Recent evidence suggests the coexistence of modifications at nearby sites modulates the binding affinity of PHD fingers. In the case of PHD fingers that interact with H3K4me3, a conserved tryptophan frequently imposes a barrier between H3K4me3 and H3R2, which results in an adjacent binding groove. In several instances, this groove does not accommodate methylation of H3R214. With the RAG2-PHD finger, H3 peptides bind in an orientation in which H3R2 extends away from this binding pocket7,15. The combined H3K4me3 and H3R2me2 marks result in a modest increase in affinity towards the RAG2- PHD finger, while disfavoring interaction with other PHD fingers, such as ING2 and BPTF. The potential for multi-site PTM specificity (i.e. a ‘histone code’) of binding is not limited to PHD fingers. In 2005, it was found that binding of the HP1 chromodomain to H3K9me3 is abolished by phosphorylation of H3S1016. Furthermore, the double chromodomains of CHD1 (recognize H3K4me3) and the WD40-repeats of WDR5 (recognize the N-terminus of H3) are sensitive to modification at and R2 and T314,17–20.
These published examples of the interplay between different PTM sites are only a small sampling of the potential complexity encountered by chromatin binding proteins. The unbiased dissection of the combinatorial PTM patterns recognized by PHD fingers and other chromatin binding proteins requires a platform for rapidly and comprehensively surveying binding affinity. Glass7,21 and cellulose20 -based histone peptide arrays were recently reported, but have been limited to one to two modifications per peptide. It remains a significant challenge to represent the combinatorial complexity of histones in a format that is amenable to facile analysis with ‘histone code readers’. We have addressed this fundamental problem through the development of resin-bound PTM-randomized histone tail libraries22. Here, we report the design and use of an unbiased 5000-member PTM-randomized combinatorial peptide library based on the N-terminus of histone H3 for examination of the binding specificity of a diverse panel of PHD fingers. Specifically, the PHD fingers from the ING2, RAG2, BHC80 and AIRE proteins, as well as the double tudor domain (DTD) of a jumonji family demethylase (JMJD2A) were analyzed. While all of these proteins interpret the modification status at K4, we discovered that three additional PTM sites (R2, T3 and T6) dramatically influenced the binding affinity. Phosphorylation of T3 resulted in a severe attenuation of interaction with N-terminal H3 peptides among all five modules analyzed. However, the PTM status of R2 and T6 was differentially recognized. Our finding that phosphorylation at T6 had variable effects on interaction with histone-binding domains suggests that this mark may represent an important regulatory mechanism for readers of the H3 tail. This possibility led us to examine whether this previously undiscovered modification exists within native chromatin. Indeed, using mass spectrometry and immunological methods H3T6ph was detected in histones extracted from HeLa cell nuclei. NMR analysis of the ING2- PHD finger revealed the structural basis for differential binding of H3 peptides containing H3K4me3 and H3K4me3T6ph. Together, our results reveal new insights into the multi-site PTM crosstalk interpreted by a collection of proteins that recognize the H3 tail.
Results
PTM-randomized combinatorial H3 library
To probe PTM specificity of chromatin binding modules, a PTM-randomized combinatorial peptide library based on the first ten amino acids of H3 was developed. Split-pool methodology23 was used to randomize PTMs at seven positions of the histone H3 tail (Fig. 1a) (Full methods available in Supplementary Methods). Randomization at lysine residues included unmodified (K), acetylated lysine (Kac), monomethylated lysine (Kme1), dimethylated lysine (Kme2), or trimethylated lysine (Kme3); at arginine included unmodified (R), monomethylated arginine (Rme1), asymmetrically dimethylated arginine (Rme2a), symmetrically dimethylated arginine (Rme2s) or citrulline (Cit); at threonine included unmodified threonine (T) or phosphorylated (Tph); and at serine included unmodified (S) or phosphorylated (Sph). Prior to split-pool synthesis, an appropriate linker was installed, containing a PEG segment (a hydrophilic spacer), methionine (a cyanogen bromide cleavage point), and arginine (provides extra charge for MS-based sequencing) (Fig. 1a).
Figure 1. PTMs included in the 5000- member combinatorial H3 tail peptide library.
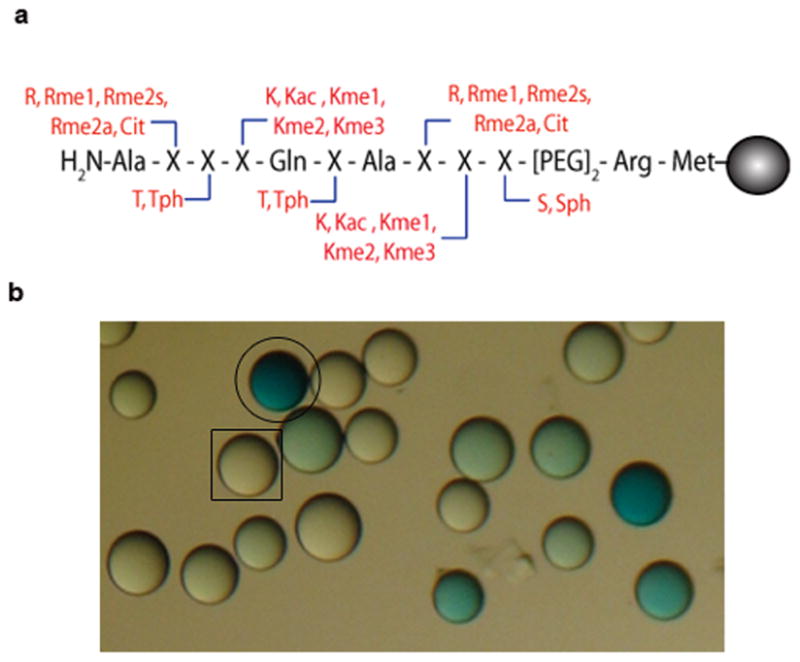
(a) Positions of randomization are annotated by ‘X’. Arginines 2 and 8 can be unmodified, monomethylated, symmetrically or asymmetrically dimethylated or citrullinated. Lysines 4 and 9 can be unmodified, mono-, di-, or trimethylated or acetylated. Threonines 3 and 6 as well as serine 10 can be unmodified or phosphorylated. Peptides are tethered to a solid-support via a linker comprised of methionine, arginine and a PEG spacer. (b) A digital image of the library screen for the JMJD2A double tudor domain. Dark blue beads are marked with a circle (○) and colorless beads with a rectangle (□).
To distinguish isomeric/nearly isobaric residues and to assign PTMs to the correct position during MS sequencing of individual beads, we employed a chemical capping strategy during library synthesis24, which results in the formation of a mass ladder by which the peptide PTM pattern can be deduced using MALDI-TOF MS (Supplementary Fig. 1). Ac-Ala (1.5–3 %) and Boc-Ala (3–5 %) capping reagents were incorporated at positions of arginine (2 and 8) and lysine (4 and 9). Ac-Ala was used in the case of K, Kme1, Kme2, Kme3, R, Rme1, Rme2a while a combination of Ac-Ala and Boc-Ala was used for Kac, Rme2s and Cit. In instances where both capping reagents were used, a signature “doublet” was obtained in the mass spectrum, which facilitated differentiation of amino acids of similar or identical mass. For the first capping step (i.e. position 9), only a single Boc-Ala cap was necessary for distinguishing acetylated lysine from trimethylated lysine. Capping ratios were optimized at each position since the various H3 fragments ionize with different efficiencies. To ensure the quality of each library synthesized, 100 random beads were selected for peptide sequencing (Supplementary Table 1).
On-bead screening strategy
To thoroughly and rapidly delineate chromatin-binding module specificity, an on-bead screening assay was developed (Supplementary Fig. 2a). In this ‘on-bead Western’ assay, the library is incubated first with a GST-tagged version of the protein of interest, second with a GST-specific primary antibody, third with a biotinylated secondary antibody and finally with streptavidin-conjugated alkaline phosphatase (SAAP). 5-Bromo-4-chloro-3-indolyl phosphate (BCIP) was used as a substrate for SAAP, resulting in formation of a turquoise precipitate on beads bearing sequences that bind to the target protein (Fig. 1b)25. Use of a GST tag in the assay eliminated the need for direct biotinylation or the use of antibodies specific to each target protein. To ensure that the general procedure did not produce false positives, the assay was performed with GST alone. No color development for up to forty minutes was noted in the GST sample (Supplementary Fig. 2b). These results indicated that the occurrence of false positives would be extremely low during the screening of GST-fused chromatin-binding modules.
Combinatorial screening of PHD finger specificity
To explore the histone code interpreted by H3 binding modules, we selected a diverse panel of PHD fingers for screening the H3 library. These PHD fingers were derived from ING2, RAG2, BHC80 and AIRE (PHDs 1 and 2) proteins. For comparison, we also evaluated the DTD of JMJD2A, which is known to recognize H3K4me326,27. For the screening assay, color development was allowed to proceed for ~10 minutes but varied slightly for each domain and correlated with binding affinity. Approximately 30 dark blue beads and 30 colorless beads (basis for selection in Fig. 1b) were selected from each protein screen, cleaved with CNBr, and evaluated for PTM patterns with MALDI-TOF analysis (full sequences can be found in Supplementary Tables 2–6). Sequences from the colorless beads reflect PTM patterns that lead to low affinity binding and represent the inverse trends of the dark blue beads. The choice to select ~30 beads/category allowed us to perform statistically significant chi-squared (χ2) analyses of whether the modification state of a particular position was important for binding (Fig. 2 labeled on the x-axis)28. Residues in which the PTM were particularly important for binding are demarcated with an asterisk (*) or double asterisk (**) depending on the level of significance. To determine which PTMs were important at a particular position, discrimination factors were calculated by taking the frequency of that particular PTM among the intensely blue population (positive hits) divided by the frequency of that PTM observed in a random pool of 100 beads (full sequences can be obtained in Supplementary Table 1). The magnitude of the discrimination factor correlates with binding preferences at each variable position, and corrects for any synthetic bias in library construction.
Figure 2. Graphical depiction of discrimination factors obtained from H3 library screens.

Values for discrimination factors were obtained by dividing the percent frequency of each modification observed in the intensely blue pool for a given screen by the percent frequency of each corresponding modification from a random group of 100 library members. Discrimination factors represent the fold-likelihood of observing a particular modification in a protein screening experiment relative to random chance. Chi-squared values for each residue are reported along the x-axis. Serine and threonine residues allow for 1 degree of freedom (DF) while lysine and arginine allow for 4 DF. Values above the 99% confidence level for statistical significance are marked with an asterisk (*) (for 1 degree of freedom (DF) = 6.63 and 4 DF = 13.28). A double asterisk (**) is used to denote positions with notably high chi-squared values (above 99.9% confidence level for statistical significance where 1 DF = 10.83 and 4 DF = 18.47).
BHC80- PHD finger
The ability of the BHC80- PHD finger to bind unmodified H3K4 is coupled to LSD1-mediated gene silencing9. To evaluate the detailed binding preferences of the BHC80- PHD finger, a GST-fusion of the BHC80-PHD finger was screened against the H3 library. Peptides from 33 intensely blue beads and 28 non-colored beads were sequenced (Supplementary Table 2). A strong preference for unmodified H3K4 was observed among the intensely blue beads (32/33 sequences; discrimination factor 4.6) (Fig. 2). At H3R2, most of the sequences from the intensely blue pool were either unmodified or monomethylated (27/32). To validate this trend, ITC (isothermal titration calorimetry) binding assays were performed using soluble peptides (Supplementary Fig. 3). Consistent with the library results, a slight decrease in binding affinity was measured for an H3R2me2s- modified peptide (Kd = 57.2 μM) relative to an unmodified H3 peptide (Kd = 19.6 μM) (all Kd values summarized in Supplementary Table 7). The fact that neither phosphorylation of H3T3 nor H3T6 appeared in the intensely blue pool suggested hat these modifications abrogated binding. Chi-squared (χ2) values of 24.78 (H3T3) and 21.70 (H3T6) underscore the importance of modification state at these positions. H3S10ph did not exhibit an appreciable effect on interaction of the BHC80- PHD finger with H3 (χ2 = 0.32). In agreement with the library screen, ITC analysis revealed no detectable binding of the BHC80- PHD finger with peptides containing either T6ph or T3ph (Supplementary Table 7).
PHD finger 1 and PHD finger 2 of AIRE
PHD1 of AIRE is implicated in activation of gene expression via association with unmodified H3K48. To explore the PTM-dependent specificity of this interaction, we screened a GST- fusion of AIRE- PHD1 against the H3 library and selected 30 dark blue beads and 33 colorless beads for analysis (Supplementary Table 3). Chi-squared analysis revealed the modification states at H3T3, H3T6, and H3K4 to be particularly significant for binding (Fig. 2). The modification state at H3R2 and H3S10 were less significant and the modification state at H3R8 and H3K9 did not impact recognition. As predicted, unmodified H3K4 was well represented in the intensely blue pool (18/30 sequences; discrimination factor of 2.8). Unlike the BHC80- PHD finger, AIRE- PHD1 was more tolerant to H3K4me1 (12/30 sequences in the intensely blue pool; discrimination factor of 1.6). Preference for unmodified H3R2 in the intensely blue pool (15/30 sequences; discrimination factor of 2.3) was also noted, and is consistent with a recent study29. It is noteworthy that H3K4me1 is only observed in the intensely blue pool when H3R2 is unmodified (Supplementary Table 3). As with the BHC80- PHD, the AIRE- PHD1 was extremely intolerant to phosphorylation of H3T3 (1/30 in the dark blue pool) and of H3T6 (0/30 in the dark blue pool). Consistent with the screening results, we were unable to detect appreciable binding of the AIRE- PHD1 for an H3 peptide containing either phosphorylation at T3 or T6 (Supplementary Table 7). The H3S10ph mark attenuated but did not prohibit binding (21/30 sequences in the intensely blue pool were unmodified; discrimination factor of 1.8). A GST- fusion of the AIRE- PHD2 was also screened against the H3 library but no binding was detected, consistent with a previous observation8.
ING2- PHD finger
Prior work suggested that the ING2- PHD finger is linked to gene repression by a mechanism that involves association with trimethylated H3K413. To evaluate the complete binding preferences of the ING2- PHD finger in an unbiased fashion, the H3 library was screened with a GST-tagged version of this module. Beads were selected and peptide sequences were obtained from 32 intensely blue beads and 31 colorless beads (Supplementary Table 4). A strong preference for H3K4me3 was observed in the intensely blue bead pool (29/32 sequences; discrimination factor of 4.1) (Fig. 2). By chi-squared analysis, the modification state at H3R2, H3T3 and H3K4 had a significant impact on binding, while the modification state at H3T6 was borderline significant. In accordance with a recent study14, more potent binding was observed when H3R2 was unmodified (21/32 sequences in the intensely blue pool; discrimination factor of 3.0). A ~20-fold decrease in binding affinity was measured for an H3 peptide bearing K4me3 and R2me2s marks (Kd = 17.3 μM) relative to an H3K4me3 peptide lacking modification at R2 (Kd = 0.98 μM). Unphosphorylated versions of H3T3 (29/32 sequences; discrimination factor of 1.7) and H3T6 (25/32 sequences; discrimination factor of 1.4) were favored in the intensely blue pool (Fig. 2). For K4me3-modified peptides, ITC analysis supports the finding that the presence of H3T3ph strictly prohibits binding, while H3T6ph dampens the affinity relative to the unphosphorylated peptide (~20-fold difference; (Supplementary Table 7)).
RAG2- PHD finger
The RAG2- PHD finger plays an instrumental role in triggering V(D)J recombination7. Recent studies have established that the RAG2-PHD finger preferentially associates with H3K4me37,15 in combination with H3R2me2s/a14,15. Using the H3 library, we sought to determine whether this binding specificity could be resolved within our library screen and whether the RAG2- PHD finger binding is modulated by other H3 PTMs. We selected 32 dark blue beads and 31 colorless beads for analysis (Supplementary Table 5). In agreement with previous analysis of the RAG2- PHD finger7,15, we noted a strong affinity for H3K4me3 (31/32 sequences; discrimination factor of 4.4), but little preference for the modification state at R2 (χ2 = 12.71), which is unique among the PHD fingers tested in this study. Interestingly, the RAG2- PHD finger was extremely intolerant to phosphorylation of H3T3 and/or H3T6 (discrimination factors of 1.9 and 1.8 respectively) (Fig. 2). Phosphorylation of these residues was observed only once in the intensely blue pool. Consistent with the screening analysis, an unphosphosphorylated H3K4me3 peptide bound efficiently (Kd = 17.8 μM), while no binding was detected by ITC in the presence of either T3ph or T6ph (Supplementary Table 7). Phosphorylation of H3S10 also had a significantly detrimental impact on binding (discrimination factor of 2.1).
JMJD2A double tudor domain
To determine whether the trends uncovered in the PHD finger screens are common to other H3-binding modules, we explored the H3 PTM histone code interpreted by the JMJD2A double tudor domain (DTD), which is known to recognize H3K4me3 26,27. JMJD2A is a lysine demethylase associated with transcriptional repression30 and transcriptional coactivation31 in a context-dependent manner. Peptides from 28 intensely blue beads and 27 non-colored beads were chosen for analysis (Supplementary Table 6). The JMJD2A- DTD displayed strong selectivity for H3K4me3 (26/28 intensely blue sequences; discrimination factor of 4.2) (Fig. 2). Among the intensely blue beads, unmodified H3T3 was strongly favored (27/28 sequences; discrimination factor of 1.9). In contrast to the PHD fingers surveyed in this study, the JMJD2A DTD was insensitive to phosphorylation at H3T6 (15/28 sequences in the intensely blue pool; χ2 = 0.81). Consistent with the library screen, ITC analysis revealed similar binding affinities for H3K4me3 (Kd = 1.1 μM) and H3K4me3T6ph (Kd = 1.8 μM) (Supplementary Table 7). The modification states at H3R8, H3K9 and H3S10 exhibited minimal impact on affinity toward the JMJD2A DTD (Fig. 2).
Identification of Phosphorylated H3T6 from HeLa cell nuclei
For chromatin binding proteins that gauge the H3K4 methylation status, we found that PTMs at other positions—most notably R2, T3 and T6—affect binding. Generally, phosphorylation of T3 caused a >100-fold decrease in binding affinity among the modules analyzed, while phosphorylation at T6 was differentially recognized. Specifically, T6 phosphorylation abolished detectable interaction with the BHC80-, AIRE - and RAG2- PHD fingers, but resulted in only a ~20-fold loss in affinity for ING2- PHD finger, and had a negligible effect on JMJD2A DTD binding. The context-specific effects of the T6ph mark suggested that this modification might represent an important regulatory mechanism for readers of the H3 tail. This possibility led us to examine whether this previously undiscovered modification exists on native H3.
To determine the physiological existence of the H3T6ph mark, we utilized a recently developed commercial antibody for this modification. Epitope mapping was performed using six H3 peptides immobilized on cellulose to examine the specificity of this antibody in greater detail. The peptides were derived from the first eleven residues of H3, and contained the following modifications: H3T3ph; H3T6ph; H3T11ph; H3K4me3T6ph; H3S10ph and no modification. The antibody displayed strong immunoreactivity for H3T6ph and weak reactivity for unmodified H3 and H3T11ph (Fig. 3a). No significant reactivity was observed for H3T3ph, H3S10ph or H3K4me3T6ph-containing peptides. Collectively, this peptide mapping analysis indicated that H3T6ph was immunodominant, and that epitope recognition involves K4 and S10 as well. Next, we investigated whether H3T6ph could be detected in native chromatin. Histones extracted from asynchronously grown HeLa cells were analyzed by Western blot (complete methods available in Supplementary Methods). The α-H3T6ph antibody revealed an immunoreactive band that corresponded to histone H3 (Fig. 3b). To demonstrate that the α-H3T6ph antibody specifically recognized phosphorylated H3T6, a competition assay with the H3T6ph (1–11) peptide demonstrated reduced Western signal to near background levels under identical conditions (Fig. 3c).
Figure 3. Detection of H3T6ph by Western blot analysis.

(a) Spot blot Western with six distinct H3 peptides residues 1-11 (1:H3T3ph; 2:H3unmod; 3:H3T11ph; 4:H3T6ph; 5:H3K4me3T6ph; 6:H3S10ph). (b) H3T6ph-antibody recognizes H3T6ph from native histones extracted from HeLa cell nuclei (left panel). (c) A 1 μM H3T6ph (1-11) peptide competition diminishes the signal to background levels. The black arrow indicates the band corresponding to H3. The asterisk (*) most likely pertains to H3 C-terminal degradation products.
To confirm the existence of H3T6ph on native chromatin, we employed mass spectrometry. Acid extracted histones from HeLa cells were subjected to propionic anhydride derivatization (pr), trypsin digestion, methyl esterification (OMe) and immobilized metal affinity chromatography (IMAC) for phosphopeptide enrichment prior to analysis with an Orbitrap mass spectrometer. The extracted ion chromatogram is shown (Fig. 4a) for the ions at 455.724 m/z, which correspond to either H3T3 or H3T6 phosphorylation. A major peak elutes at 16.1 minutes, while a clearly chromatographically distinct minor peak with the same m/z elutes at 15.8 minutes. The MS/MS spectrum of the major 455.724 m/z peak at 16.1 minutes (Fig. 4b) reveals fragments which indicate a sequence of pr-TphosKprQTAR-OMe. The y-type ions at 361, 489, 673 m/z are indicative of unmodified H3T6, while the b1 ion at 238 and other b-type ions (some with loss of phosphate) indicate H3T3 phosphorylation. Inspection of the MS/MS spectrum taken from the minor 455.724 m/z peak eluting at 15.8 minutes (Fig. 4c) reveals a distinctly different MS/MS pattern than that observed in Fig. 4b, although the primary sequence is still TKQTAR. A notable difference between the two spectra is a shift from the [M+2H-H3PO4]2+ ion (Fig. 4b) to the [M+2H-H2O]2+ ion (Fig. 4c) being the most intense species. Additionally, many of the critical ions observed in Fig. 4b supporting H3T3ph are absent or at around background levels in the spectrum shown in Fig. 4c. For example, the key y-type ions at 361, 489 and 673 or b-type ions at 238 and 550 m/z are substantially reduced or absent. Observed ions at 471 (y4-H3PO4) and 655 (y 5-H3PO4) suggest H3T6 phosphorylation. The partial chromatographic separation of the phosphorylated isomer peptides along with distinctive MS/MS spectra indicate that H3T6ph is the minor peak eluting at 15.8 minutes and H3T3ph is the major peak eluting at 16.1 minutes. Collectively, the immunological and mass spectral results underscore the physiological existence of the H3T6ph mark.
Figure 4. Detection of H3T6ph using mass spectrometry.

(a) Extracted ion chromatogram of the [M+2H]2+ ion at 455.724 m/z, the expected mass of both H3T3phos and H3T6phos from HeLa cells. This sample was subjected to propionic anhydride derivatization, methyl esterification and immobilized metal affinity chromatography (IMAC) for facilitated analysis and phosphopeptide enrichment. As can be see, a major peak is observed at 16.1 minutes, while a second minor resolved peak is clearly visible at 15.8 minutes. Mass accuracy was found to be ~2ppm on either peak as recorded on an Orbitrap mass spectrometer. (b) MS/MS spectrum of the species eluting at 16.1 minutes. The MS/MS fragments show that the sequence is from the 3-8 residues of histone H3 containing T3 phosphorylation. (c) MS/MS spectrum of the species eluting at 15.8 minutes. The MS/MS fragments show that the sequence is from the 3–8 residues of histone H3 containing T6 phosphorylation. Note pr = propionyl amide (56 Da), phos = phosphorylation (80 Da), and —OMe = methyl ester (14 Da).
NMR analysis reveals structural basis for H3T6 discrimination
Having demonstrated that H3T6ph is a newly discovered PTM, we next sought to provide structural evidence for the physical discrimination of H3T6ph by a PHD finger. We chose the ING2- PHD finger because it bound tightly to an H3 peptide containing K4me3 (Kd = 0.98 μM), while the additional incorporation of T6ph yielded an appreciable decrease in affinity (Kd = 19.9 μM) (Supplementary Table 7). Importantly, this difference was within the range to capture binding by NMR titration experiments. Gradual addition of peptides with either H3K4me3 or H3K4me3T6ph marks caused significant resonance perturbations in 1H, 15N HSQC spectra of the PHD finger (Fig. 5a, b). Importantly, the binding affinities measured by NMR were in close agreement with those determined by ITC (Supplementary Fig. 4). While the pattern and magnitude of chemical shift perturbations observed for the majority of the amide resonances in the PHD finger was similar for both peptides, the cross peaks of V221, S222, Y223 and G224 shifted differentially. To a lesser extent, the cross peak at E225 shifted upon binding to H3K4me3T6ph relative to H3K4me3 (Fig. 5a, b). These data demonstrate that the V221, S222, Y223 and G224 residues experience different chemical environments in the two complexes, which is indicative of the location of the phosphate moiety. Mapping the V221, S222, Y223 and G224 residues onto the surface of the H3K4me3-bound PHD finger reveals a well-defined shallow pocket situated directly underneath the side chain of H3T6 (Fig. 5d). The S222 and G224 residues that interact with the hydroxyl group of H3T6 in the PHD-H3K4me3 complex were particularly sensitive to the presence of the phosphate moiety. The inability of the ING2- PHD finger to incorporate a phosphate group into this shallow pocket and the disruption of hydrogen bonding contacts with the H3T6 hydroxyl group reveal the mechanistic details for an observed decrease in binding upon phosphorylation at H3T6.
Figure 5. Identification of the H3K4me3T6ph-binding site of the ING2 PHD finger.

The histograms show normalized 1H,15N chemical shift changes in backbone amides of the ING2 PHD finger induced by the H3K4me3T6ph (a) and H3K4me3 (b) peptides. The protein:peptide ratio is 1:5, which represents saturation for both interactions. (c) Superimposed 1H,15N heteronuclear single quantum coherence (HSQC) spectra of the ligand-free (black), H3K4me3T6ph-bound (purple) and H3K4me3-bound (green) ING2 PHD finger (0.2 mM). (d) Residues that show a unique pattern of chemical shift perturbations in (c) are colored in red on the surface of the ING2 PHD-H3K4me3 complex and labeled. The residues that exhibit large but parallel chemical shift changes upon addition of either H3K4me3T6ph or H3K4me3 are colored in gray.
Discussion
Combinatorial histone tail libraries represent a powerful means for dissecting interactions between histone-binding modules and a multitude of histone modification states22. This study describes the design and use of a directed, PTM-randomized histone H3 tail library of 5000 unique members, providing the means to systematically explore how combinatorial N-terminal H3 modification patterns influence affinity and recognition by chromatin-binding proteins. Importantly, the library is not restricted to known modifications or modification patterns and therefore permits an unbiased survey of combinatorial space. Moreover, the chemical cap sequencing strategy eliminates the need for tandem mass spectrometry, and the GST-screening method allows facile detection of binding interactions without the need for chemical modification of the target protein.
Use of the combinatorial H3 library not only revealed the complex binding properties of chromatin-binding modules, but screening results suggested a previously unappreciated PTM that exists in native chromatin. The H3 library was utilized to interrogate multi-site specificity of five chromatin-binding modules, which read the methylation status of H3K4. We found that T3 phosphorylation, R2 methylation, and T6 phosphorylation are critical additional PTMs that modulate the ability of these proteins to recognize and bind histone H3. Notably, H3T6ph yielded the most varied effect on binding affinity, suggesting that the phosphorylation state of T6 may represent an important regulatory mechanism for readers of the H3 tail. The novel observation that H3T6ph affects recognition by H3-binding proteins spurred us to examine whether this modification exists on native histones. Both mass spectral and immunological analyses provided evidence that H3T6ph exists on H3 extracted from HeLa cells. Furthermore, recently reported H3 mutational analysis revealed that H3T6D (Thr to Asp substitution) but not H3T6A (Thr to Ala substitution) caused a loss of rDNA silencing in yeast, while mutations at H3T3 showed no phenotype32. These genetic data indicate that H3T6 is mutationally sensitive and suggests that replacement of Thr with Asp may mimic phosphorylated H3T6, adding provocative evidence that H3T6ph is a physiologically relevant PTM.
Using the PTM-randomized H3 library, we explored the binding selectivity of six protein modules. As expected, a marked preference for H3K4me3 was noted in the cases of the ING2 and RAG2 PHD- fingers as well as the JMJD2A DTD. In contrast, the AIRE PHD- finger 1 and the BHC80 PHD- finger were specific for unmodified H3K4. Consistent with previous studies14,15, we found that H3R2 methylation had a deleterious effect on the binding of the ING2- PHD finger but not that of RAG2- PHD finger. In the ING2- PHD finger, H3R2 is sequestered into a binding pocket adjacent to those of H3K4me3 by an interdigitating tryptophan33. The ING2- PHD finger contacts the H3R2 guanidinium group with D230 and E237. Methylation of H3R2 decreases its hydrogen bond donating capacity and may result in steric exclusion from the binding pocket. From structural analysis of the RAG2- PHD finger bound to H3 peptide, H3R2 is occluded from an analogous groove and is not engaged in a salt bridge/hydrogen bond, consistent with our observation that citrullination/methylation at this position had little effect on binding.
Unlike modification of H3R2, phosphorylation of H3T3 impinged upon the interaction of all assayed protein modules. Interestingly, a RAG2- PHD finger-H3K4me3 peptide cocrystal structure reveals that the hydroxyls of H3T3 and H3T6 participate in an intramolecular hydrogen bond, which imposes a kinked peptide conformation (Supplementary Fig. 5)15. Our results suggest that phosphorylation of either of these residues disrupts this interaction and may serve as a conformational switching mechanism. Intriguingly, phosphorylation of H3T6 did not hamper binding to the JMJD2A DTD and only partially impeded binding to the ING2 PHD- finger. Examination of cocrystal structures with H3K4me3 peptides reveals that while H3T6 participates in hydrogen bonding contacts in the ING233 and RAG215 complexes, H3T6 meanders off the protein surface in the JMJD2A complex26 (Supplementary Fig. 5). In the case of the ING2 PHD- finger, our NMR titration data suggest that the moderate attenuation of binding caused by H3T6ph binding to the ING2 PHD- finger can be mapped to perturbation of V221, S222, Y223, G224 and E225 (Fig. 5d). It is also noteworthy that H3T6ph completely abrogates association with the AIRE and BHC80 PHD- fingers. Evidence for the existence of the H3K4unmod modification state in combination with H3T6ph is provided in the current study; this PTM state represents a form of the H3 tail that precludes AIRE and BHC80 interaction and may represent an important regulatory mechanism for their interaction with chromatin.
In 2003, it was proposed that some aspects of a histone code might manifest as binary switches, controlling the binding activities of histone-binding proteins34. An apparent example of this type of control is the ejection of HP1 from H3K9me3 as a result of H310 phosphorylation during mitosis16. Using our unbiased PTM-randomized H3 tail screening strategy, we conclude that chromatin-binding activity involves multi-site PTM recognition and has both binary switch and rheostat- like properties. These features reveal a more complex model than initially proposed. We noted that while phosphorylation of H3T3 acts like a binary switch for all proteins examined in this study, the binding perturbations from PTMs at R2 and T6 are graded and protein-specific. To a first approximation, we consider a PTM that causes a change in Kd values of ≥ 100-fold to display switch-like behavior, whereas a change of <100-fold to display rheostat-like effects. For the RAG2-, AIRE-, and BHC80- PHD fingers, phosphorylation of H3T6 acts as an off-switch for binding to the preferred H3K4 methylated tail. However, the same modification (H3T6ph) modestly decreases binding affinity of the ING2- PHD finger, and negligibly affects the JMJD2A DTD. In the case of ING2 and JMJD2A, these changes can be considered rheostat-like, as the PTM is expected to modulate binding within a range that permits weaker, physiologically relevant binding. The differential effect of this mark may allow certain proteins to retain H3 binding (JMJD2A), weaken binding for some (ING2), while inhibiting others (RAG2). Although this library was limited to the first ten amino acids of H3 for practical reasons, it is possible that distal modifications may also impact binding interactions.
Histone PTMs are often considered to promote the tight binding of chromatin binding proteins; however, the results of this work demonstrate that overall affinity and PTM recognition may involve a combination of both repulsive and attractive marks. The results of this investigation emphasize the importance of considering PTM crosstalk when examining histone-binding-module specificity. A continuum of binding affinities exists among chromatin-binding proteins in which multi-site PTM recognition appears to involve both switch- and rheostat-like properties, yielding graded effects that depend on the inherent ‘reader’ specificity.
Methods
General Methods
All chemical and biochemical reagent were purchased from commercial suppliers. Plasmids for GST fusions of the AIRE PHD fingers 1 (293–354) and 2 (426–485) were furnished by Prof. Giovanna Musco. Plasmids for the GST fusions of the PHD fingers of BHC80 (486–543) and RAG2 (414–487) were obtained from Prof. Yang Shi and Dr. Wei Yang, respectively. The plasmid for a GST fusion of the double tudor domain of JMJD2A (895–1011) was provided by Prof. Rui-Ming Xu. Peptides were manually synthesized. Analytical gradient HPLC was performed on a Shimadzu series 2010C HPLC with a Vydac C18 column (10 μm, 4.6 × 250 mm). Mass spectrometry was conducted on an Applied Biosystems 4800 instrument, and isothermal titration calorimetry was performed on a MicroCal VP-ITC MicroCalorimeter (full methods in supplementary methods).
On-bead assays
Assays were performed on ~ 5 mg portions of the H3 tail combinatorial peptide library. Briefly, the resin was washed 5 × 1 mL with each of the following: DCM, MeOH, doubly deionized water and HBST (30 mM Hepes pH 7.5, 150 mM NaCl, 0.1 % Tween 20). Next, the library was swelled for 1 hr in HBST and blocked for an additional hour with 1 % BSA in HBST. The library was then incubated with 600 μL of the GST-fusion protein (50 nM) of interest for 1 hour in 1 % BSA in HBST. Afterwards, the resin was washed 3 × 600 μL HBST. This was followed by a 1-hour incubation with a GST-specific primary antibody (1:5000 dilution), three washes with 600 μL of HBST, and a 1-hour incubation with a biotinylated goat-anti-rabbit secondary antibody (1:5000 dilution). After another washing step (3 × 600 μL HBST), the resin was incubated with 600 μL of a 1:800 dilution of streptavidin alkaline phosphatase (SAAP) in SAAP buffer (30 mM Tris pH 7.6, 1 M NaCl, 10 mM MgCl2, 70 μM ZnCl2, 20 mM KPi) for 10 minutes. The resin was then washed 1 × 350 μL SAAP buffer, 1 × 350 μL HBST and 1 × 350 μL staining buffer (30 mM Tris pH 8.5, 100 mM NaCl, 5 mM MgCl2, 20 μM ZnCl2). The resin was transferred to a Petri dish with 1 mL of straining buffer. Following addition of 30 μL of 5 mg/mL 5-Bromo-4-chloro-3- indolyl phosphate (BCIP) in staining buffer, color development was followed under a low power microscope. Color development was typically quenched after ~12 minutes with 3 mL 8 M guanidinium hydrochloride. Afterwards beads were thoroughly washed with water and selected based on color intensity.
Peptide sequencing
Beads were selected by micropipette under a low power microscope. Individual beads were placed in a microcentrifuge tube and peptides were cleaved with 20 μL of a solution of 40 mg/mL cyanogen bromide in 70 % TFA overnight in the dark. Cleavage solutions were removed by evaporation, peptides were resuspended in 2 μL of 0.1 % TFA, and analyzed by MALDI-TOF MS. Posttranslational modification patterns were inferred from the resulting mass ladders.
Statistical analysis
Statistical analysis was conducted with the chi-square test 28. The chi-square test was performed at each position of PTM-randomization when comparing two populations. The equation used to execute the chi-square test is:
Here, “Observed” refers to the number of one type of modification (e.g. acetylation) observed at a particular position in a population of selected beads. “Expected” refers to the expected number for that modification at that particular position and is determined by taking a weighted average of occurrences between the two populations (randomly selected beads vs. positively selected beads from a library screen) being compared. The sum of the square of the “Observed” minus the “Expected” divided by the “Expected” for each modification type at a single position gives the chi-square value for that position.
Spot blot assays
Residues 1-11 of histone H3 were synthesized (Intavis ResPep SL) on functionalized cellulose paper35. The peptides included the following modifications: H3T3ph, H3T6ph, H3unmod, H3K4me3T6ph, H3T11ph, and H3S10ph. Blots were blocked overnight in 5% BSA-TBST (0.05%) at 4°C. They were then incubated with 1:7,500 or 1:10,000 anti-histone H3 phospho T6 (Abcam ab14102) in blocking solution for 30 minutes at room temperature, followed by three five-minute washes in TBST (0.05%). Spot blots were incubated with 1:10,000 HRP-conjugated goat anti-rabbit (Santa Cruz) in blocking solution for 30 minutes at room temperature, and washed three times for five minutes in TBST (0.05%). All antibody solutions were pre-equilibrated in batch to insure uniformity. Detection was performed using SuperSignal West Pico Substrate (Pierce) and signal was captured using an Epi Chemi II Darkroom (UVP Laboratory Products).
NMR spectroscopy
NMR spectra were collected at 25°C on a Varian INOVA 500 MHz spectrometer. 1H,15N heteronuclear single quantum coherence (HSQC) spectra of the ING2 PHD finger (0.2 mM) were recorded in the presence of increasing concentrations (up to 1 mM) of the H3K4me3T6ph and H3K4me3 histone tail peptides. The normalized chemical shift change was calculated using the equation [(ΔδH)2 +(ΔδN/5)2]0.5, where δis the chemical shift in parts per million (ppm). The synthetic histone peptides were: NH2-ARTK(X)QT(Y)ARKSTG-CONH2, where X = me3 and Y = H or PO32−.
Supplementary Material
Acknowledgments
We thank Greg Barrett-Wilt and Greg Sabat (University of Wisconsin Biotechnology Center) for their help with MALDI-TOF mass spectrometry. We are grateful to Gary Case (University of Wisconsin Peptide Synthesis Facility) for helpful conversations and advice on peptide synthesis. We thank Giovanna Musco (Dulbecco Telethon Institute), Yang Shi (Harvard Medical School), Wei Yang (National Institute of Health), Rui-Ming Xu (New York University) for expression plasmids used in this study. This work was supported by the grants from the US National Institutes of Health (GM059785 to J.M.D.) and an American Society for Mass Spectrometry research award (B.A.G.).
Footnotes
Competing financial interests
J.M.D and A.L.G have a patent pending (No. 11/585,625) that describes the construction and uses of PTM peptide libraries.
Author Contributions
A.L.G., S.S.O. and J.M.D. designed the library, screened the proteins and analyzed the results. S.S.O. executed the ITC validation experiments and E.K.G. performed and analyzed the western blots. C.M. and T.K. carried out the NMR analysis. B.A.G. and G.L. performed the mass spectrometry experiments for H3T6ph identification. Initial manuscript drafts were written by A.L.G., S.S.O. and J.M.D. All authors participated in writing the final document.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 2.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–40. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 5.Bienz M. The PHD finger, a nuclear protein-interaction domain. Trends Biochem Sci. 2006;31:35–40. doi: 10.1016/j.tibs.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 6.Baker LA, Allis CD, Wang GG. PHD fingers in human diseases: Disorders arising from misinterpreting epigenetic marks. Mutat Res. 2008 doi: 10.1016/j.mrfmmm.2008.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matthews AG, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–10. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Org T, et al. The autoimmune regulator PHD finger binds to non-methylated histone H3K4 to activate gene expression. EMBO Rep. 2008;9:370–6. doi: 10.1038/embor.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lan F, et al. Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression. Nature. 2007;448:718–22. doi: 10.1038/nature06034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wysocka J, et al. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006;442:86–90. doi: 10.1038/nature04815. [DOI] [PubMed] [Google Scholar]
- 12.Vermeulen M, et al. Selective Anchoring of TFIID to Nucleosomes by Trimethylation of Histone H3 Lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 13.Shi X, et al. ING2 PHD domain links histone H3 lysine 4 methylation to active gene repression. Nature. 2006;442:96–9. doi: 10.1038/nature04835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Iberg AN, et al. Arginine methylation of the histone H3 tail impedes effector binding. J Biol Chem. 2008;283:3006–10. doi: 10.1074/jbc.C700192200. [DOI] [PubMed] [Google Scholar]
- 15.Ramon-Maiques S, et al. The plant homeodomain finger of RAG2 recognizes histone H3 methylated at both lysine-4 and arginine-2. Proc Natl Acad Sci U S A. 2007;104:18993–8. doi: 10.1073/pnas.0709170104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischle W, et al. Regulation of HP1-chromatin binding by histone H3 methylation and phosphorylation. Nature. 2005;438:1116–22. doi: 10.1038/nature04219. [DOI] [PubMed] [Google Scholar]
- 17.Flanagan JF, et al. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–5. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 18.Couture JF, Collazo E, Trievel RC. Molecular recognition of histone H3 by the WD40 protein WDR5. Nat Struct Mol Biol. 2006;13:698–703. doi: 10.1038/nsmb1116. [DOI] [PubMed] [Google Scholar]
- 19.Guccione E, et al. Methylation of histone H3R2 by PRMT6 and H3K4 by an MLL complex are mutually exclusive. Nature. 2007;449:933–7. doi: 10.1038/nature06166. [DOI] [PubMed] [Google Scholar]
- 20.Nady N, Min J, Kareta MS, Chedin F, Arrowsmith CH. A SPOT on the chromatin landscape? Histone peptide arrays as a tool for epigenetic research. Trends Biochem Sci. 2008;33:305–13. doi: 10.1016/j.tibs.2008.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Shi X, et al. Proteome-wide analysis in Saccharomyces cerevisiae identifies several PHD fingers as novel direct and selective binding modules of histone H3 methylated at either lysine 4 or lysine 36. J Biol Chem. 2007;282:2450–5. doi: 10.1074/jbc.C600286200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Garske AL, Craciun G, Denu JM. A combinatorial H4 tail library for exploring the histone code. Biochemistry. 2008;47:8094–102. doi: 10.1021/bi800766k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lam KS, et al. A new type of synthetic peptide library for identifying ligand-binding activity. Nature. 1991;354:82–4. doi: 10.1038/354082a0. [DOI] [PubMed] [Google Scholar]
- 24.Youngquist RS, Fuentes GR, Lacey MP, Keough T. Generation and Screening of Combinatorial Peptide Libraries Designed for Rapid Sequencing by Mass Spectrometry. J Am Chem Soc. 1995;117:3900–3906. [Google Scholar]
- 25.Wavreille AS, Garaud M, Zhang Y, Pei D. Defining SH2 domain and PTP specificity by screening combinatorial peptide libraries. Methods. 2007;42:207–19. doi: 10.1016/j.ymeth.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM. Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science. 2006;312:748–51. doi: 10.1126/science.1125162. [DOI] [PubMed] [Google Scholar]
- 27.Kim J, et al. Tudor, MBT and chromo domains gauge the degree of lysine methylation. EMBO Rep. 2006;7:397–403. doi: 10.1038/sj.embor.7400625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clarke GM, Cooke D. A Basic Course in Statistics. Arnold; London: 2004. [Google Scholar]
- 29.Chignola F, et al. The solution structure of the first PHD finger of autoimmune regulator in complex with non-modified histone H3 tail reveals the antagonistic role of H3R2 methylation. Nucleic Acids Res. 2009 doi: 10.1093/nar/gkp166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gray SG, et al. Functional characterization of JMJD2A, a histone deacetylase-and retinoblastoma-binding protein. J Biol Chem. 2005;280:28507–18. doi: 10.1074/jbc.M413687200. [DOI] [PubMed] [Google Scholar]
- 31.Shin S, Janknecht R. Diversity within the JMJD2 histone demethylase family. Biochem Biophys Res Commun. 2007;353:973–7. doi: 10.1016/j.bbrc.2006.12.147. [DOI] [PubMed] [Google Scholar]
- 32.Huang H, et al. HistoneHits: A database for histone mutations and their phenotypes. Genome Res. 2009;19:674–81. doi: 10.1101/gr.083402.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pena PV, et al. Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature. 2006;442:100–3. doi: 10.1038/nature04814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischle W, Wang Y, Allis CD. Binary switches and modification cassettes in histone biology and beyond. Nature. 2003;425:475–9. doi: 10.1038/nature02017. [DOI] [PubMed] [Google Scholar]
- 35.Frank R. The SPOT-synthesis technique. Synthetic peptide arrays on membrane supports--principles and applications. J Immunol Methods. 2002;267:13–26. doi: 10.1016/s0022-1759(02)00137-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.