

Abstract
Precise representation of the timing of sensory stimuli is essential for rapid motor coordination, a core function of the cerebellum. Feedforward inhibition has been implicated in precise temporal signaling in several regions of the brain, but little is known about this type of inhibitory circuit within the input layer of the cerebellar cortex. We investigated the synaptic properties of feedforward inhibition at near physiological temperatures (35°C) in rat cerebellar slices. We establish that the previously uncharacterized mossy fiber–Golgi cell–granule cell pathway can act as a functional feedforward inhibitory circuit. The synchronous activation of four mossy fibers, releasing a total of six quanta onto a Golgi cell, can reset spontaneous Golgi cell firing with high temporal precision (200 μs). However, only modest increases in Golgi cell firing rate were observed during trains of high-frequency mossy fiber stimulation. This decoupling of Golgi cell activity from mossy fiber firing rate was attributable to a strong afterhyperpolarization after each action potential, preventing mossy fiber–Golgi cell signaling for ∼50 ms. Feedforward excitation of Golgi cells induced a temporally precise inhibitory conductance in granule cells that curtailed the excitatory action of the mossy fiber EPSC. The synaptic and cellular properties of this feedforward circuit appear tuned to trigger a fast inhibitory conductance in granule cells at the onset of stimuli that produce intense bursts of activity in multiple mossy fibers, thereby conserving the temporal precision of the initial granule cell response.
Keywords: cerebellum, synaptic integration, Golgi cell, mossy fiber, inhibition, interneuron
Introduction
The precision with which an action potential (AP) can be triggered by synaptic input sets the fidelity with which temporal events can be represented and the amount of information carried by an individual AP (Rieke et al., 1997). Precise AP timing is crucial for neural codes that represent physical variables as spike times relative to oscillating reference signals (Stopfer et al., 1997; Harris et al., 2002). Spike timing is also important in motor systems because encoding temporal sequences is essential for coordinating movements (Ivry and Keele, 1989; Medina and Mauk, 2000). Indeed, impairment of spike precision causes ataxia (Walter et al., 2006). Many of the neurons in motor systems, particularly in the cerebellum, are spontaneously active (Llinás, 1988; Häusser et al., 2004) and several of these participate in feedforward inhibitory loops (Mittmann et al., 2005), which are thought to preserve temporal signaling as sensory signals flow through networks (Pouille and Scanziani, 2001; Mittmann et al., 2005). However, relatively little is known about how processing is accomplished in the presence of intrinsic activity in feedforward interneurons.
Golgi cells (GoCs), which reside in the input layer of the cerebellar cortex, are spontaneously active and provide the sole source of inhibition for thousands of granule cells (GrCs) (Eccles et al., 1967). GoCs are important for normal cerebellar function because pharmacological ablation of this inhibitory interneuron leads to sever motor deficits (Watanabe et al., 1998). Although classical anatomical studies have indicated the presence of a feedforward inhibitory circuit in the granule cell layer (Eccles et al., 1967), little is known about its functional properties. Moreover, in vivo experimental evidence for precise temporal signaling in the input layer of the cerebellar cortex is scarce. We used patch-clamp recordings and imaging in acute cerebellar slices to unambiguously identify the feedforward mossy fiber (MF) input onto GoCs and investigate its properties. We show that the synchronous activation of a small number of MFs can reset the timing of spontaneously firing GoCs with high precision. However, a powerful afterhyperpolarization after each spike reduced the reliability of MF–GoC transmission as the frequency increased, decoupling GoC firing from MF excitation. An MF-evoked GoC spike produced a precisely timed feedforward inhibitory conductance in GrCs that cancelled slow MF–GrC EPSC components. These results establish that a functional feedforward MF–GoC–GrC pathway is present in the input layer of the cerebellar cortex. The synaptic properties of this circuit, together with intrinsic pacemaker currents of the GoC, suggest that this circuit could help preserve information on the precise timing of the start a discrete sensory stimulus by shaping the early GrC response.
Materials and Methods
Electrical recording conditions.
Parasagittal slices of cerebellum (200–220 μm thick) were prepared from postnatal day 25 (P25) Sprague Dawley rats as described previously (Silver et al., 1996). Brain slices were either prepared in artificial CSF (ACSF) (125 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 25 mm glucose, pH 7.3, equilibrated with 5%CO2/95%O2) or in 130 mm K-gluconate, 15 mm KCl, 0.05 mm EGTA, 20 mm HEPES, and 25 mm glucose, pH 7.4 with NaOH (Dugué et al., 2005) because this was found to increase GoC viability (Dugué et al., 2005; Forti et al., 2006) and incubated in ACSF at 30–31°C for 45 min. The tissue was then stored at room temperature for up to 6 h before use. In the recording chamber, brain slices were continuously perfused with ACSF at 34–36°C. The following pharmacological agents were added to the external perfusion solution when mentioned: 10 μm SR95531 [6-imino-3-(4-methoxyphenyl)-1(6H)-pyridazinebutanoic acid hydrobromide] (gabazine), 0.3 μm strychnine, 10–100 μm 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium salt (NBQX), and 10 μm d-([−])-2-amino-5-phosphonopentanoic acid (APV). All chemicals were obtained from either Sigma-Aldrich or Tocris Cookson. Release probability was lowered by reducing the Ca2+/Mg2+ ratio in the extracellular solution to 1 mm/5 mm in a subset of experiments.
Slices were visualized with a 60× (numerical aperture 0.9) water-immersion objective using infrared differential interference contrast optics (Olympus BX50W1) and a CCD camera. Patch pipettes were produced from borosilicate glass capillaries (Sutter Instruments) using a Sutter P97 horizontal puller (Sutter Instruments). We used an Axopatch 200B for whole-cell voltage-clamp and an Axoclamp 2B amplifier (Molecular Devices) for current-clamp recordings. High-quality voltage and current recordings were not possible in the same cell because of the limitations of these amplifiers. Some later recordings were also performed with an Axopatch 700B. Data was low-pass filtered at 10–30 kHz and digitized at 50–100 kHz. Recordings were acquired and analyzed with IgorPro (WaveMetrics) using NeuroMatic (http://www.physiol.ucl.ac.uk/research/silver_a/neuromatic/). All traces displayed in the figures were further digitally filtered to 7 kHz using a binomial smoothing function in IgorPro. MF inputs were activated via an ACSF-filled glass pipette (2–3 MΩ) placed in the white matter tract (WMT), typically 100–200 μm away from the recording site using a stimulus isolator (model DS2A; Digitimer) to generate 10–100 V pulses of 20–200 μs duration. Parallel fibers (PFs) were activated by stimulating the upper GrC layer or molecular layer, which led to qualitatively similar results.
Loose cell-attached recordings.
GoC firing behavior was monitored in loose cell-attached (LCA) voltage-clamp configuration with ACSF-filled pipettes (7–10 MΩ). APs were identified based on prominent upward and downward deflections in the current trace (Vos et al., 1999). Recording stability was monitored by observing the AP shape. AP latency was defined as the time point of half-maximal amplitude of the inward deflection. The effect of MF stimulation on spontaneous rhythmic GoC firing was assessed with a phase–response curve, calculated with Equation 1, which determines whether a perturbation advances or retards the phase of an oscillator (Rinzel and Ermentrout, 1998):
 |
where Δθ is the phase response, T0 is the median of interspike interval (ISI) before stimulation, and Tn is the ISI between APs of interest. Tn can depend on time or the phase at which the stimulus occurs (θ = Ts/T0, where Ts is the interval between the spontaneous AP before stimulation and time point of stimulation).
Whole-cell recordings.
Postsynaptic currents (PSCs) and postsynaptic potentials were measured with 5–7 MΩ patch electrodes filled with the following (in mm): 5 Na-MeSO3, 130 K-MeSO3, 10 HEPES, 5 MgCl2, 0.1 EGTA, 0.3 NaGTP, 4 MgATP, and 100 μm Alexa488 or Alexa594, pH 7.3 with KOH for GoC and 110 K-MeSO3, 40 HEPES, 6 NaOH, 3 MgCl2, 0.02 CaCl2, 0.15 BAPTA, 0.3 NaGTP, and 4 NaATP, pH 7.3 with KOH for GrC recordings. In some experiments, voltage-gated channel blockers QX314 [(N-(2,6-dimethylphenylcarbamoylmethyl)triethylammonium bromide] (5 mm) and tetraethylammonium (TEA) (17.5 mm) were included in the internal solution. Voltage-clamp recordings were made at holding potentials of −60 and −30 mV (GoCs), −70 and 0 mV (GrCs), and not corrected for the 8 mV (GoC internal) and 6.3 mV (GrC internal) liquid junction potential. Whole-cell capacitance and series resistance were determined by canceling the current transient evoked by a 50 ms, 5 mV hyperpolarizing voltage step and read from the amplifier dials. Cell input resistance was calculated from the steady-state current at the end of the voltage step, and the recording stability was accessed with a Spearman's rank correlation test. GoCs had a whole-cell capacitance of 37 ± 14 pF, and in patch-clamp recordings the electrode series resistance (16 ± 6 MΩ; n = 64) was compensated (60–70%, 7–10 μs lag) to ∼5 MΩ. This gave an effective filtering [1/(2πRCm)] of ∼1 kHz. GrCs had a whole-cell capacitance of <5 pF and were recorded with an uncompensated series resistance of 26 ± 10 MΩ (n = 19), which resulted in an effective filtering of 1.5 kHz.
GoC current-clamp recordings were initially performed in the absence of current injection to compare the spontaneous firing in LCA and whole-cell configuration. Because the whole-cell configuration affected the spontaneous firing in an unpredictable manner (Forti et al., 2006), we injected hyperpolarizing or depolarizing currents to match the firing frequency observed in LCA recordings. The spike threshold was defined as the membrane voltage when the derivative of the membrane voltage (dV/dt) was 5 SDs above the mean dV/dt for subthreshold regions. This gave a threshold criterion of ∼5 mV/ms (Forti et al., 2006), which reliably identified the rising phase of APs and closely matched the visually identified inflection point. The threshold potential was then corrected for the liquid junction potential.
PSCs were baseline subtracted using a 0.5 ms time window before the stimulus. In some cases, the stimulus artifact was removed by subtracting a double-exponential fit to the averaged artifact. Individual events were classified as failures when the mean amplitude in a 1 ms window around the peak of the PSC was smaller than 3× SD of the mean background current of a 1 ms time window before the stimulus. To further distinguish slowly rising spillover currents, we grouped all events with a rise time 5× SD above the mean rise time of successes with failures (DiGregorio et al., 2002). To describe the PSC kinetics, we measured the 20–80% rise time, calculated the weighted decay (normalized integral over a 20 ms time window) (DiGregorio et al., 2002), or fit the decay with a double-exponential function.
The time course of synaptic reversal potential was calculated as follows (Johnston and Wu, 1995):
 |
where ESyn is reversal potential of the combined excitatory and inhibitory conductances, Gexc is excitatory conductance, Eexc is reversal potential of excitatory conductance (estimated to be 0 mV), Ginh is inhibitory conductance, and Einh is reversal potential of inhibitory conductance (estimated to be −75 mV). When ESyn is below the voltage threshold for spike initiation, the combined synaptic conductances will not induce firing.
Response latencies were measured between the beginning of the stimulus artifact and the 20% rise time point of the PSC. Because monosynaptic MF–GrC EPSCs have a mean latency of 0.8 ms (Sargent et al., 2005), we excluded all GoCs responding to WMT stimulation with currents of latencies larger than 1.2 ms (5× SD above mean MF–GrC latency) from additional analysis. Disynaptic IPSC latency jitter in GrCs arises from the jitter of MF-evoked GoC APs and the jitter of GoC–GrC transmission. Because variances add when two Gaussian distributions are convolved, the expected jitter of disynaptic GrC inhibition is as follows:
Deconvolution analysis was performed as described previously (Sargent et al., 2005) without spillover correction. Uniquantal events were measured under low release probability conditions, aligned on their 20% rise time. To reduce the effects of noise, these were determined from fits of Equation 4 to individual currents and averaged:
 |
The resulting mean current was also fit with Equation 4 and deconvolved with a fit of the average stimulus-evoked EPSC under control conditions to yield the vesicular release rate as a function of time across all release sites at the connection. We verified this approach by directly determining the release time course from the first latencies of EPSCs in low release probability conditions for four cells. At high failure rates (>80%), the correction for the occurrence of multiquantal events (Barrett and Stevens, 1972) was negligible [Kolmogorov–Smirnov (KS) test, p > 0.2; n = 4].
The quantal size at the time of the peak of the evoked EPSC (Qp) was determined in two ways. First, we averaged stimulus-aligned quantal events recorded under low release probability conditions (failure rate >0.8) and corrected the measured amplitude for multiquantal events (∼10%) (Silver, 2003). We also calculated Qp from variance/mean analysis under low release probability conditions (Silver, 2003). We corrected this estimate of Qp for the variance arising from quantal amplitude variability and latency jitter, measured from variability of stimulus-aligned uniquantal events:
 |
where σ2 is peak variance, I is mean EPSC amplitude at the time of the peak under low release probability conditions, and CVT is coefficient of variation of total quantal variability measured from the peak of stimulus-aligned successes.
Image acquisition and analysis.
Golgi cell anatomy was visualized with the dyes Alexa488 or Alexa594 (100 μm; Invitrogen) using fluorescence microscopy. Imaging was performed using whole-field excitation (USH102D Mercury Lamp; Ushio) and image acquisition (in nm: excitation filters, 480/40 and 580/20; dichroic, 505 long pass and 595 long pass; emission filters, 535/50 and 630/60 for Alexa488 and Alexa594, respectively) with a cooled digital camera (ORCA100; Hamamatsu Photonics) and Prairie Technologies software. Several images of cell body and dendritic arborization were taken to identify the cell type patched.
For Ca2+ measurements, we substituted Alexa dye with 200 μm Oregon Green BAPTA1 (OGB1) (Invitrogen). Images (series of three to four) were taken of various parts of the cell before and during a 100 Hz (100 pulses) stimulation of the white matter. We used a custom-made image analysis routine in Matlab (MathWorks) to calculate the ΔF/F for each pixel by averaging the images before and during stimulation and relating the intensity difference before and during stimulation to the average pixel intensity before stimulation. The distribution of ΔF/F values was fitted with a Gaussian function. Only pixels with an intensity value 5 SDs above the mean pixel intensity value that formed a contiguous group in the image were considered to represent a significant increase in local calcium concentration. The large depth of focus of CCD imaging allowed the simultaneous observation of fluorescence changes in large parts of the cell and hence reliable detection and mapping of synaptically evoked calcium signals (ΔF/F >0.1) onto the cell morphology in fewer trials than for confocal imaging. During Ca2+ imaging experiments, whole-cell capacitance and series resistance compensation was disabled to induce poor space clamp because this increased the probability of detecting a Ca2+ transient during MF stimulation. The Ca2+ signal was fully inhibited by blockers of glutamatergic synaptic transmission, but NMDA receptors (NMDARs) contributed little to the synaptic response (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and were thus an unlikely source of the Ca2+ increase. Furthermore, 100 μm Ni2+, a nonselective voltage-dependent Ca2+ channel blocker, fully inhibited the Ca2+ signal (n = 3; data not shown). Activation of voltage-gated Ca2+ channels by synaptically induced local depolarization is therefore likely to be responsible for the Ca2+ signal, although we cannot exclude Ca2+-permeable AMPA receptors (AMPARs) or synaptically induced release from internal Ca2+ stores.
Statistics.
Data are presented as mean ± SD and sample means compared with paired or unpaired two-tailed Student's t test. For pairwise comparison of groups, we used single and two-factor ANOVA, and distributions were compared using a KS test. Mean values were considered significantly different at p < 0.05. Linear, exponential, and Gaussian functions were fit to data points using the least squares method.
Results
Identification of Golgi cells and the functional properties of their excitatory inputs
GoCs in acute slices of cerebellar cortex from P25 rats (see Materials and Methods) were initially identified on the basis of their location within the GrC layer and their large somata (>15 μm). In LCA recordings, using pipettes containing extracellular solution, most of these putative GoCs (83%) exhibited spontaneous rhythmic firing (9 ± 5 Hz; n = 38), consistent with previous studies (Mitchell and Silver, 2000; Forti et al., 2006). For whole-cell recordings, we included a fluorescent tracer dye in the intracellular solution and visualized the cell morphology with fluorescence microscopy. Only those cells with basal dendrites in the GrC layer and ascending dendrites in the molecular layer were confirmed as GoCs (Eccles et al., 1967; Dieudonne, 1998) and included in our dataset (64 of 68). The extent of the ascending and basolateral arborization was ∼200 and 100 μm, respectively, in agreement with previous morphological description of GoCs (Eccles et al., 1967; Palay and Chan-Palay, 1974; Dieudonne, 1998). Identified GoCs exhibited frequent spontaneous excitatory postsynaptic events, a larger cell capacitance (37 ± 14 pF; range, 10–74 pF; n = 64) than the surrounding GrCs (<5 pF) and a relatively low input resistance (Rin = 330 ± 70 MΩ).
Anatomical data suggests that GoCs receive three types of excitatory inputs. PFs (GrC axons) synapse onto their ascending dendrites within the molecular layer, MFs contact basolateral dendrites in the GrC layer, and climbing fibers (CFs) may make connections at the soma (Eccles et al., 1967). To date, only the PF to GoC input has been characterized functionally (Dieudonne, 1998; Bureau et al., 2000; Beierlein et al., 2007). To establish whether MFs make functional synapses onto GoCs, we electrically stimulated the WMT, which primarily comprises MFs. However, this could also potentially activate CF input monosynaptically and PF input disynaptically, via the activation GrCs. We therefore examined the functional properties (kinetics, pharmacology, and plasticity) and anatomical location of WMT-evoked synaptic inputs and compared them with the properties of PF (to GoC) and CF (to Purkinje cell) transmission to determine whether MFs could be activated in isolation. Figure 1 compares the properties of synaptic currents evoked in GoCs by WMT and PF stimulation (see Materials and Methods). Both WMT and PF stimulation induced inward currents at −60 mV, indicating an excitatory input (Cl− reversal potential = −70 mV). Bath application of GABA and glycine receptor blockers had no significant effect on the mean WMT-evoked EPSC waveform (n = 7; p > 0.5, t test), nor did the incorporation of the sodium and potassium channels blockers QX314 (5 mm) and TEA (17.5 mm) in the patch pipette (n = 10; p > 0.9, t test; data not shown). However, the evoked currents were blocked by NBQX (10–100 μm), confirming they were both glutamate receptor mediated (Fig. 1Ai,Bi). The time between stimulation and the 20% rise of the mean synaptic current was short for both WMT and PF inputs (1.0 ± 0.1 and 1.0 ± 0.4 ms, respectively), consistent with monosynaptic activation. WMT stimulation produced EPSCs in GoC with both a fast rise (20–80% rise time = 0.23 ± 0.04 ms) and weighted decay times (τWD = 1.6 ± 0.5 ms; n = 42) (Fig. 1Ai,Aii). In contrast, PF-evoked EPSCs had significantly slower rise (0.5 ± 0.2 ms; p < 0.05; n = 6) and decay kinetics (τWD = 3.4 ± 1.5 ms; n = 6; p < 0.05) (Fig. 1Bi,Bii), and EPSCs occurred over a longer time window (80% of events within 430 ± 270 μs compared with 190 ± 50 μs for WMT stimulation) (Fig. 1Aiii,Biii). To test the potential contribution of NMDARs to WMT-evoked EPSCs, we applied APV at depolarized holding potentials (−30 mV). APV did not change the EPSC waveform in three of seven cells, and, in the remaining cells, the overall EPSC charge was reduced by only 18 ± 11%. This indicates that NMDARs contribute little to the EPSC waveform and currents remain fast at depolarized potentials (τWD = 1.9 ± 0.7 ms at −30 mV; n = 7; p > 0.2, paired t test) (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). The distinct kinetics of short-latency EPSCs arising from WMT and PF stimulation indicate that they arise from different synaptic inputs.
Figure 1.
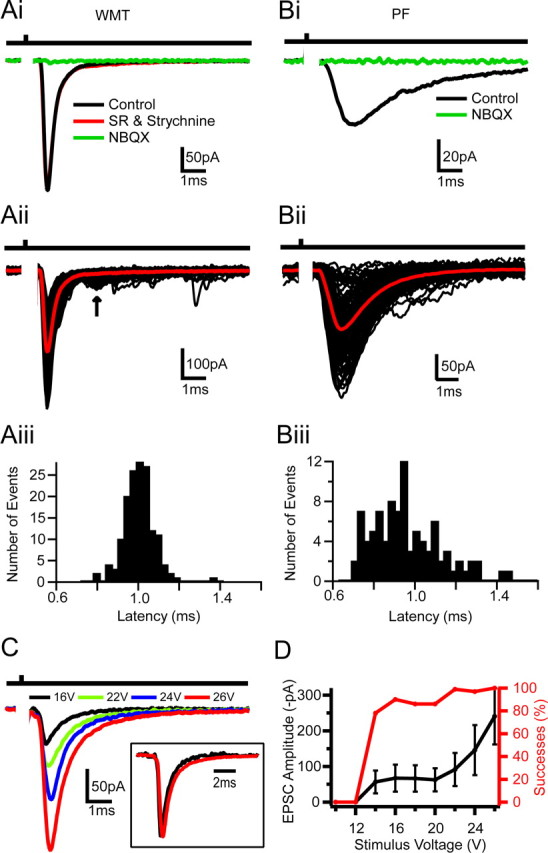
Comparison of Golgi cell EPSCs evoked by white matter tract and parallel fiber stimulation. Ai, Mean EPSCs recorded at −60 mV in response to WMT stimulation in control, 10 μm SR95531 (gabazine) and 0.3 μm strychnine, and in 100 μm NBQX. Bar above indicates time of stimulation. Aii, Individual EPSCs in response to WMT stimulation (black); mean current trace in red. Arrow indicates long-latency disynaptic EPSCs. Aiii, Latency distribution histogram of evoked events shown in Aii (mean latency = 1.00 ± 0.07 ms). Bi–Biii, Same as Ai–Aiii but for PF-evoked EPSCs activated by electrical stimulation in the upper GrC layer. The mean latency was for this cell 1.0 ± 0.2 ms. C, Mean EPSC waveforms elicited at various WMT stimulation intensities. Higher stimulus voltages led to increased EPSC amplitudes. Inset, Normalized current waveform for 16 and 26 V. D, Relationships between mean peak amplitude and stimulus voltage and EPSC successes and stimulus voltage.
We next examined whether the properties of WMT-evoked EPSCs depended on the stimulation voltage. The mean EPSC amplitude increased and the fraction of failures decreased as the stimulus intensity was increased (Fig. 1C,D). However, the EPSC waveform (Fig. 1C, inset) remained the same, indicating that the increase in EPSC size was attributable to the recruitment of inputs with similar kinetics. The initial step-like relationship between EPSC amplitude and stimulus voltage suggests that, at low voltages, a single fiber was activated, as observed previously for MF–GrC synaptic connections (Silver et al., 1996). Single-fiber activation exhibited a 22 ± 13% failure probability and a mean EPSCs amplitude of −66 ± 26 pA (n = 42). In 5 of these 42 recordings we observed slowly rising EPSCs similar to spillover currents described at the MF–GrC synapse (DiGregorio et al., 2002). In these cells, spillover currents contributed 32 ± 17% (n = 5) of the EPSC charge (supplemental Fig. 1Bi,Bii, available at www.jneurosci.org as supplemental material). During high-voltage WMT stimulation, when multiple fibers are activated, a second longer-latency (>2 ms) EPSC component was occasionally observed (22% of recordings) (Fig. 1Aii, arrow). The kinetics of this late component were similar to PF-evoked events (20–80% rise time = 0.4 ± 0.1; τWD = 2.3 ± 0.03 ms; p > 0.3; n = 3), consistent with the recruitment of a disynaptic PF input.
Synaptic connections made by PFs (onto GoCs and Purkinje cells), CFs (onto Purkinje cells), and MFs (onto GrCs) exhibit distinct short-term plasticity (STP) characteristics. WMT-evoked EPSCs onto GoCs exhibited little STP during five-pulse, 25 Hz stimulus trains (amplitude ratio EPSC5/EPSC1 = 0.95 ± 0.27; n = 14) (Fig. 2Ai). Even when the stimulation frequency was increased to 100 Hz, the EPSC only depressed by 22 ± 17% during 10 stimuli (n = 10) (Fig. 2Aii). This STP behavior contrasted with the 50% facilitation observed for five-pulse, 25 Hz PF stimulation (n = 6; p < 0.05, two-factor ANOVA on 25 Hz WMT vs PF) (Fig. 2B), consistent with previous studies of the PF input (Bureau et al., 2000). The STP characteristics of WMT EPSCs are also distinct from CF to Purkinje cell transmission, which depresses profoundly (Llano et al., 1991; Silver et al., 1998). Although the MF–GrC synapse also depresses moderately, this is predominantly attributable to postsynaptic AMPAR desensitization. Indeed, lack of plasticity in the WMT input onto GoC is similar to the presynaptic component of MF–GrC transmission, which depresses little during high-frequency stimulation (Saviane and Silver, 2006). The functional properties of WMT-evoked EPSCs are therefore consistent with MF rather than PF or CF input.
Figure 2.

Short-term plasticity of EPSCs evoked by white matter tract and parallel fiber stimulation. Ai, Left shows averaged EPSCs evoked with a five-pulse WMT stimulus with a 40 ms interval (25 Hz). Right shows normalized EPSC amplitudes for individual cells (dotted) and population mean (n = 14; solid line). The peak amplitudes of evoked events were normalized to the first peak. Aii, Same as Ai for 10 stimuli at 100 Hz. B, PF EPSCs evoked by GrC layer stimulation. Five pulses at 25 Hz as for Ai. Right shows normalized PF-EPSC amplitude for individual cells (dotted) and population mean (n = 6; solid line). Error bars denote SD.
Synaptic location of putative mossy fiber inputs
Anatomical studies have shown that excitatory inputs onto GoCs are spatially segregated (Eccles et al., 1967). We therefore examined the spatial location of putative MF–GoC synapses by calcium imaging during low-intensity WMT stimulation, which activated one or two fibers (see Materials and Methods). Figure 3A shows a two-dimensional projection of a GoC manually reconstructed from a montage of fluorescence images taken with a cooled CCD camera at different focal planes (Fig. 3B). The three fluorescent images in Figure 3B show part of the ascending dendrite (top), soma, and basal dendritic arbor (below). Figure 3C shows the fluorescent change during WMT stimulation for the same regions. In this cell, the fluorescence (ΔF/F), and therefore calcium concentration, increased in one of the basal dendrites. In cells in which calcium changes were detectable (7 of 11), WMT stimulation induced local calcium changes (ΔF/F = 0.31 ± 0.29) in descending or lateral dendrites of GoCs within the GrC layer but never in the soma or in the dendrites ascending into the molecular layer. Perfusion of NBQX (100 μm) and APV (10 μm) fully and reversibly blocked both the WMT-evoked EPSCs and the calcium signal, confirming their synaptic origin (n = 7). The lack of any calcium signals from the ascending dendrites was not attributable to an inability of the ascending dendrites to respond, because robust calcium signals were observed in the ascending dendrites during molecular layer stimulation (n = 3) (supplemental Fig. 2A–C, available at www.jneurosci.org as supplemental material).
Figure 3.

Calcium imaging of Golgi cells during white matter tract stimulation. A, Drawing of a Golgi cell reconstructed from a montage of fluorescent images acquired in various focal planes (examples are illustrated in B). The blue line indicates the border between the granular and molecular layer. B, Fluorescent images (1 focal plane) of the same Golgi cell loaded with the calcium indicator OGB1. C, ΔF/F (pseudocolor scale on right) was calculated for each pixel from images taken before, and during, 100 Hz WMT stimulation. D, WMT stimulation evoked EPSCs for same cell. Individual currents shown in black and mean in red. Current kinetics and latency distribution were indistinguishable from putative MF EPSCs described in Figure 1. E, EPSC amplitude, normalized to the first EPSC, during a 25 Hz, five-pulse WMT stimulation (individual cells indicated as dotted lines) from cells that exhibited Ca2+ signal on descending dendrites (n = 7). Error bars indicate SD. F, Dependence of mean EPSC rise time on distance between the center of the soma and location of the peak calcium change on the dendrite. Solid line shows linear regression (Pearson's r = 0.67; p < 0.05).
The EPSCs associated with the calcium signal in the basolateral dendrites were similar in latency (1.0 ± 0.1 ms; n = 7; p > 0.2), kinetics (20–80% rise time = 0.26 ± 0.1 ms, p > 0.3; τWD = 1.8 ± 0.7 ms, p > 0.2) and STP behavior (amplitude ratio EPSC5/EPSC1 = 1.02 ± 0.12; p > 0.9) to WMT-evoked EPSCs described previously (Fig. 3D,E). The mean distance between the peak of the fluorescence change and the center of the cell body was 56 ± 25 μm (range, 20–90 μm; n = 7). The fast rise time of the EPSCs and its weak distance dependence (Fig. 3F) suggest that basolateral dendrites introduce relatively little filtering of the synaptic current. The location of synaptic inputs on the basolateral dendrites and the short latency and lack of STP of the EPSCs confirm that WMT stimulation activates MFs rather than PF of CF inputs.
Mossy fiber input resets Golgi cell firing
To investigate the effects of MF excitation on GoC firing, we used the non-invasive LCA recording method to prevent disruption of the internal milieu. Figure 4A shows an LCA recording from a GoC that exhibited spontaneous rhythmic firing at 12 Hz. Single-pulse stimulation of the MF input induced an AP soon after the onset of the stimulus. APs induced by WMT stimulation were blocked by NBQX (10 μm), confirming their synaptic origin and ruling out direct activation of the GoC axon. The raster plot in Figure 4B shows that MF stimulation induced an AP that was time-locked to the stimulus across multiple trials. MF-evoked APs occurred in a narrow time window, but entrainment of subsequent spikes (Fig. 4C) was variable from cell to cell. To understand how the MF input affected GoC rythmicity as a function of stimulus timing, we calculated the phase–response curve for each cell (see Materials and Methods). Figure 4D shows the phase response averaged over 21 cells for the first (solid line) and second (broken line) AP after the stimulus as a function of stimulus phase. A phase response of 0 indicates that the stimulus had no effect on the spike timing of subsequent spikes, as seen for the second AP after stimulation. A value >0 implies a phase advance, which is observed for the first AP after stimulation when the stimulation phase was >0.2. The phase–response curves are statistically significantly different (p < 0.0001, single-factor ANOVA). This implies that the phase of spontaneous firing can be reset for stimulation phases >0.2.
Figure 4.

Effect of mossy fiber input on spontaneous Golgi cell firing. A, Loose cell-attached voltage-clamp recording from a Golgi cell during single-shock MF stimulation. The bar above indicates time of MF stimulation. T0 indicates a representative ISI (equal to the mode) before stimulation, T1 the ISI between spikes before and after stimulation, and T2 the ISI between the first and second spike after stimulation. Ts denotes the interval between stimulation and preceding action potential. Calibration bars: 20 pA, 50 ms. B, Spike raster plot from same cell as A. Dots represent spike times measured at the 50% time point of the AP inward deflection. C, Peristimulus spike time histogram for same recording. The frequency of AP occurrences in a 1 ms bin after the stimulation was significantly increased above the prestimulus baseline (p < 0.0001, single-factor ANOVA). D, Phase reset curve averaged across 21 cells. Black solid curve shows phase of first AP after stimulation, and black broken graph shows behavior of the second AP as a function of the phase of stimulation. The datasets were significantly different (p < 0.0001, single-factor ANOVA), indicating that a phase reset was induced by stimulation. E, Averaged cell-attached recordings during a paired-pulse MF stimulation protocol. A conditioning MF stimulation pulse that triggered an AP was followed by a second test pulse at various time intervals. The reduction in current amplitude indicates increased failures in triggering an AP. F, Relationship between percentage of trials that resulted in an AP on the second pulse as a function of the interpulse interval for the cell in E.
The probability of the MF input eliciting a spike depended on the time since the preceding spike. To quantify this, we used a paired-pulse protocol to assay the probability of triggering a spike at different times after an initial AP (Fig. 4E). On average, it took 52 ± 29 ms after the first spike (range, 20–120 ms; n = 9 cells) for the spike probability to recover to 50% (Fig. 4E,F). This value was similar to the time during which MF stimulation was ineffective in producing an AP after a spontaneous spike (p > 0.9, paired t test; n = 6). These results show that the feedforward excitatory MF input onto GoCs can reset the spike timing and thus the phase of the GoC firing pattern, but that the transmission reliability depends on the timing of the input with respect to the phase of the spontaneous activity.
Action potential precision and transmission reliability at low and high MF firing frequencies
APs evoked by low-frequency MF input had a mean latency across cells of 1.1 ± 0.4 ms (n = 37) and exhibited remarkably little temporal jitter (Fig. 5A,B). Their precision, defined as the SD of AP times, was 200 ± 120 μs (n = 37). This precision was unaffected by application of 10 μm gabazine (n = 4; p > 0.5, paired t test), which excluded the involvement of feedforward inhibition in setting the precision of GoC firing. Moreover, precision was only weakly dependent on stimulation strength because there was no correlation between SD and AP probability across cells (Fig. 5C). However, when comparing the precision at different AP probabilities in individual cells, a weak but significant correlation was evident (Pearson's r = −0.28; p < 0.05, t test on fractional change; n = 9). On average, APs were evoked in 52 ± 21% of trials across 37 cells. These results show that the MF–GoC synapse can achieve a greater precision in EPSP-spike coupling than has been reported for other inhibitory interneurons (Fricker and Miles, 2000; Galarreta and Hestrin, 2001; Pouille and Scanziani, 2001; Carter and Regehr, 2002).
Figure 5.

Precision and reliability of mossy fiber-evoked Golgi cell firing. A, MF-evoked APs recorded in LCA configuration at high temporal resolution. B, Spike latency histogram of APs during 1 Hz stimulation measured over a 3 ms window after the stimulus. C, Dependence of SD of AP times (precision) on AP probability for LCA recordings. Data points connected with lines show the AP precision at two different AP probabilities (stimulus intensities) for the same cell. Broken line indicates linear regression of all data points (Pearson's r = −0.14; p > 0.2). D, Spike raster plot showing Golgi cell firing in response to 10 Hz white matter tract stimulation. The distributions shown below illustrate the spike jitter for APs in response to the first and fifth stimulus (4 ms measurement window). E, Same as D for 100 Hz stimulation. F, Dependence of spike-time precision on stimulation frequency across cells. Spike-time precision deteriorates slightly but significantly with increasing input frequency (n = 5). G, Probability of generating MF-evoked spikes in Golgi cells during trains of five stimuli at 10 Hz (solid line) and 100 Hz (dashed line). H, Effect of MF stimulation frequency on Golgi cell mean firing rate. The moderate increase in firing rate was significant for stimulation frequencies ≥10 Hz (p < 0.05, pairwise t test).
We next examined AP precision during trains of MF activity, because some sensory inputs are sustained (van Kan et al., 1993). The spike raster plots in Figure 5, D and E, shows APs evoked by 10 and 100 Hz MF stimulation. GoCs fired in a brief time window after the stimulus. Analysis of the spike jitter in response to the first and consecutive stimuli showed that spike time distributions did not change during the train (KS test, p > 0.9 for 10 and 100 Hz; n = 6), indicating that spike precision was maintained. As stimulation frequencies increased from 10 to 100 Hz, the mean values of the SD of evoked AP times increased from 0.2 ± 0.2 to 0.4 ± 0.2 ms (Fig. 5F). We also observed an increase in the mean spike latency from 1.4 ± 0.2 to 1.8 ± 0.6 ms. This is likely to be attributable to a reduction in GoC excitability, because we observed a decline in spontaneous firing after high-frequency stimulation (0.4 ± 0.4 Hz after 100 Hz MF stimulation; p < 0.05, paired t test). The probability of an AP occurring on a particular trial depended on both the initial spike probability and on the time since the last spike, as expected from the relative refractory period. At stimulation frequencies of 1 and 10 Hz, the AP probability did not change during a train. However, when the frequency was increased to 100 Hz, the reliability of triggering an AP dropped from 51 ± 31% on the first spike to ∼20% during the rest of the train (single-factor ANOVA, p < 0.01) (Fig. 5G). Thus, low-frequency MF stimulation (1–10 Hz) produced little change in the GoC firing rate from the baseline firing rate of 6 ± 3 Hz (n = 5), but a stimulation frequency above 10 Hz led to sublinear increase in GoC firing to a saturated level of 17 ± 12 Hz (n = 5) at 100 Hz (Fig. 5H). The decoupling of GoC firing rate from MF rate shows that this connection transmits rate-based information poorly. However, timing information is reliably transmitted at low frequencies and at the onset of a high-frequency stimulus train.
Properties of EPSP-spike coupling
To examine how MF synapses induce precise firing and yet have little effect on GoC firing rate, we examined the membrane potential during synaptic integration. To do this, we established the MF stimulus intensity necessary to evoke an AP with ∼50% reliability in the LCA configuration (49 ± 18%, n = 5) and then re-patched with an electrode containing internal solution in the whole-cell current-clamp configuration. Figure 6A shows two voltage trajectories of a spontaneously firing GoC during MF stimulation. AP threshold was −55 mV, and APs produced a pronounced afterhyperpolarization to −80 mV, followed by a slower depolarizing phase leading to the next spike. These values were similar to the averages across cells (n = 5; −55 ± 6 and −80 ± 5 mV, respectively) and to previous findings (Forti et al., 2006). When MF input was stimulated soon after the spike, during the hyperpolarized phase, no AP was induced (Fig. 6A, black trace), but when it occurred later, during the depolarizing phase, an AP was triggered (Fig. 6A, red trace). This is also illustrated in Figure 6B, which plots the preceding membrane potential before an MF-induced event (AP in red, EPSP in black). On average, the minimum interval between an evoked AP and the proceeding spontaneous AP was 58 ± 17 ms (n = 5), similar to the relative refractory period determined in LCA recordings (Fig. 4F). These results suggest that the afterhyperpolarization prevents EPSPs reaching threshold and underlies the relative refractory period in MF–GoC signaling observed in LCA recordings (Fig. 4F).
Figure 6.

EPSP-spike coupling and efficacy of mossy fiber–Golgi cell transmission. A, Whole-cell current-clamp recordings from a Golgi cell exhibiting spontaneous rhythmic firing and pronounced afterhyperpolarization. The bar above indicates the timing of MF stimulation, which triggered an AP on one trial (red) but failed to cross threshold on the other (black). The stimulus intensity was set to give a 50% AP probability, which was determined in a previous LCA recording of the same cell. B, Membrane voltage during a 500 μs time window before the synaptic event. When MF EPSPs occurred late in the spike cycle at depolarized membrane potentials, they tended to trigger APs (red), but when membrane voltage was hyperpolarized, EPSPs remained subthreshold (black). C, Top, MF-evoked APs (red) and subthreshold EPSPs (black) for same cell on expanded scale. Bottom, Overlay of peak normalized spike latency histogram measured over 5 ms time window after the stimulus (red) and corresponding mean subthreshold EPSP (black) from the same cell. Latency was measured as the time between the stimulus and threshold crossing. The population mean EPSC waveform (dashed green line; 9 cells recorded with a different amplifier) (see Materials and Methods) was aligned to the foot of the uncompensated EPSC recorded in this cell. The fact that the peak of the EPSC and maximum rate of rise of the EPSP occur at the same time suggests that this alignment is correct. D, Dependence of EPSC amplitude on preceding membrane potential for subthreshold EPSPs. A linear regression was only fit to EPSP measurements with preceding potentials below −72 mV (black points) to avoid the sampling bias caused by the loss of EPSPs that generated APs from the population. The remaining EPSP measurements are shown in gray. E, Average amplitudes of mossy fiber EPSCs recorded at stimulus voltages that evoked APs (eAP) with 50% probability and EPSC amplitudes evoked with minimal stimulation (min) in the same cells.
Figure 6C (top) shows examples of successes and failures on an expanded timescale. On average, MF-evoked EPSPs had a rise time of 0.42 ± 0.07 ms (20–80%), a mean amplitude of 8.5 ± 2.2 mV, a coefficient of variation of 0.24 ± 0.1, and a rapid initial decay time constant (τdecay1 = 2 ± 1 ms; n = 5). The latency jitter of MF–GoC EPSPs was only 80 ± 40 μs (20% rise point; n = 5). MF-evoked APs tended to arise from the rising phase of the EPSP and had a narrow latency distribution (SD of 250 ± 40 μs; n = 5), consistent with LCA recordings (Fig. 5C). Figure 6C (bottom) compares the time courses of a typical spike latency histogram (measured as time to threshold crossing), corresponding mean subthreshold EPSP for that cell, and aligned population mean EPSC time course (see Materials and Methods). The average interval between the EPSP (20% rise time) and time of threshold crossing was 400 ± 200 μs (n = 5), confirming that many of the APs were triggered during the rising phase of EPSPs. Comparison of EPSP and the population mean EPSC time course shows that the EPSC rise and initial decay occur during the rising phase of the EPSP. Indeed, the shape of the spike latency distribution shared more similarity with the mean EPSC than EPSP waveform (Fetz and Gustafsson, 1983; Galarreta and Hestrin, 2001). Previous work in other cells has found that voltage-gated conductances in dendrites can contribute to rapid EPSP-spike coupling (Fricker and Miles, 2000; Martina et al., 2000). However, the lack of dependence of EPSP amplitude on preceding membrane potential (n = 5) (Fig. 6D) suggests that they do not play a major role in boosting the MF–GoC EPSP. These results show that EPSP-spike precision is achieved by triggering spikes predominantly during the rising phase of the EPSP, which is determined by the rapid initial components of the EPSC time course.
To investigate how many MF inputs were necessary to trigger an AP, we examined the mean EPSC peak amplitude under whole-cell voltage clamp at the same stimulus settings that gave a spike probability of 51 ± 14% (n = 9) in the LCA configuration. The EPSCs had a mean peak amplitude of −260 ± 172 pA (n = 9). Comparison with EPSCs arising from single MFs recorded in the same cells (−66 ± 29 pA; n = 9) revealed that, on average, four MF inputs are sufficient to trigger a GoC AP with 50% probability (Fig. 6E). These results show that the synchronous activation of a small number of MFs can trigger an AP in GoCs with high temporal precision.
Quantal determinants of the EPSC at mossy fiber–Golgi cell connections
At calycal and climbing fiber synapses, the synchronous release of several hundred quanta averages out fluctuations in the release time course, allowing precise temporal signaling with little trial-to-trial variability (Silver et al., 1998; Taschenberger et al., 2005). To investigate how MF–GoC synaptic connections achieve high EPSP-spike precision, we examined the quantal content and release time course at this connection. To do this, we reduced the release probability by changing the external Ca2+/Mg2+ concentration from 2/1 to 1/5 mm, which increased the failure probability from ∼0.2 to >0.8 at a single-fiber connection. Under these conditions, EPSC successes are predominately uniquantal regardless of the number of release sites present (Silver, 2003). Figure 7A shows a group of evoked uniquantal EPSCs and failures. Averaging successes and correcting for multiquantal contamination gave a mean quantal size across cells of −37 ± 11 pA (n = 9) (see Materials and Methods). This was similar to the estimate obtained by variance/mean analysis (Silver, 2003) (Qp = −34 ± 10 pA; n = 9). Surprisingly, the mean single-fiber EPSC amplitude under control conditions was often comparable with the quantal size (Fig. 7B). Dividing the mean EPSC amplitude under control conditions (−52 ± 14 pA; not significantly different from the average single fiber EPSC across all cells; p > 0.2) by the uniquantal size for these cells yielded a quantal content of 1.5 ± 0.4 (n = 9). Because the synchronous activation of four MFs triggered a GoC spike, our findings indicate that the release of only six quanta can generate spike output with high temporal precision.
Figure 7.
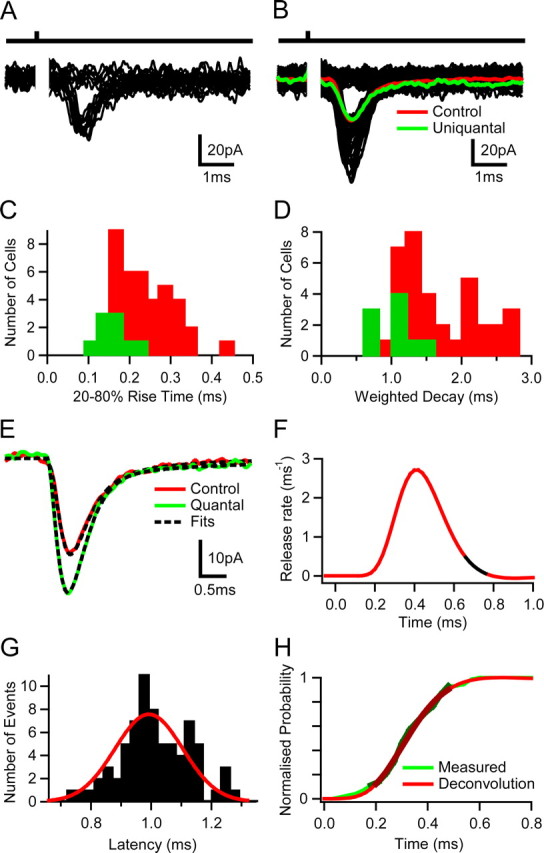
Release time course and quantal content at mossy fiber–Golgi cell synapses. A, Quantal currents and release failures in response to single mossy fiber (MF) stimulation under low release probability conditions (1 mm Ca2+ and 5 mm Mg2+). B, Individual MF EPSCs evoked under control conditions (2 mm Ca2+ and 1 mm Mg2+; black), the average current including failures (red) together with the average uniquantal current (green). C, Rise time (20–80%) distribution of mean uniquantal (green) and single fiber-evoked control currents (red) across cells. D, Same as C for weighted decay times. E, Mean quantal waveform obtained from averaging 20% rise time-aligned uniquantal events under low release probability conditions (green) with stimulus-aligned mean EPSC waveform under control conditions (red) for the same cell. The broken lines indicate fits used for deconvolution analysis. F, Vesicular release function obtained by deconvolution. The black segment represents a 100 μs time window at the peak of the mean EPSC. G, Latency distribution of uniquantal events recorded under low release probability conditions. The red line shows a fit with Gaussian function (SD of 110 μs). H, Comparison of cumulative release probability functions obtained by deconvolution and direct measurement.
The rise time distribution of mean quantal waveforms overlapped with the fastest mean stimulus-aligned evoked currents (Fig. 7C) in control conditions as did the weighted decay distribution of the quantal and control currents (Fig. 7D). Currents in control conditions with slowest decay kinetics typically exhibited a noticeable slow spillover-mediated current component (supplemental Fig. 1Bi,Bii, available at www.jneurosci.org as supplemental material). On average MF-evoked EPSCs in control conditions had a decay that could be fit by a two-exponential function (τ1 = 0.6 ± 0.4 ms, A1 = 79 ± 16%; τ2 = 4.4 ± 3.0 ms, A2 = 21 ± 16%; n = 38). The mean time course of rise-aligned quantal EPSCs, recorded in low calcium solution, was only marginally faster (20–80% rise time = 0.16 ± 0.04 ms and a two-exponential decay of τ1 = 0.4 ± 0.1 ms; A1 = 85 ± 11% and τ2 = 4.2 ± 2.2 ms; A2 = 15 ± 11%, giving a τw = 1.1 ± 0.3 ms; n = 9) (Fig. 7E). The similarity of the waveforms suggests that the time course of quantal release is brief, producing little temporal “smearing.”
We quantified the release time course under control conditions by deconvolving the mean evoked EPSC waveform with the mean quantal waveform measured from the same cell (see Materials and Methods) (Sargent et al., 2005). The time course of quantal release under normal conditions increased and decayed rapidly (Fig. 7F). Across cells, the peak release rate was 4.3 ± 1.2 ms−1, and the release function decayed with a time constant of 133 ± 99 μs (n = 9) (Fig. 7F). This is comparable with release at giant auditory synapses (Isaacson and Walmsley, 1995; Taschenberger et al., 2005) but slower than reported at the MF–GrC connection (τ = 75 μs) (Sargent et al., 2005). Although the 58 μs difference between MF–GrC and MF–GoC synapses could reflect differences in the vesicular release process, it could also be attributable to the lower temperature of the recordings (35°C vs 37°C) and subtle differences in deconvolution methods used (see Materials and Methods). To verify the results obtained by deconvolution analysis, we directly measured the time course of quantal release in four cells that had sufficient numbers of events, by examining the latency of uniquantal EPSCs under low probability conditions (Fig. 7G). Cumulative release functions for the direct measurement and deconvolution had similar shapes (p > 0.5, KS test; n = 4) (Fig. 7H), and their 10–90% rise times were 253 ± 145 μs (n = 4) and 255 ± 113 μs (n = 9), respectively. Vesicular release was therefore essentially over by the time of the peak of the EPSC (Fig. 7F, black section). These results show that highly synchronous vesicular release and a rapid time course of quantal currents allow MF inputs to reset GoC firing, precisely, with only few quanta.
Feedforward inhibition of granule cells
To investigate the downstream effects of feedforward excitation of GoCs, we made whole-cell voltage-clamp recordings from GrCs. IPSCs were recorded at an average holding potential of 3 ± 6 mV (n = 19), close to the reversal potential of AMPA and NMDARs. WMT stimulation evoked IPSCs in 16 of 19 GrCs (84%), and these often had two distinct latency components (Fig. 8A, red traces). To test whether the two latency components arose from direct stimulation of the GoC axon and disynaptic activation of the MF–GoC–GrC circuit, we blocked excitatory transmission with NBQX (10 μm). This eliminated the longer latency component, confirming its disynaptic origin and left only the short-latency monosynaptic IPSC (Fig. 8A, black traces). Figure 8B shows individual short-latency monosynaptic IPSCs (black traces) recorded in NBQX. Across cells, monosynaptic IPSCs had a latency of 1.2 ± 0.1 ms, a jitter of 160 ± 100 μs, and a success probability of 81 ± 20%. IPSCs had a mean amplitude of 33 ± 19 pA, with rise time of 0.4 ± 0.2 ms and a weighted decay of 8 ± 1 ms (n = 7). Application of gabazine blocked the outward current, confirming that the IPSCs were GABAA receptor mediated.
Figure 8.
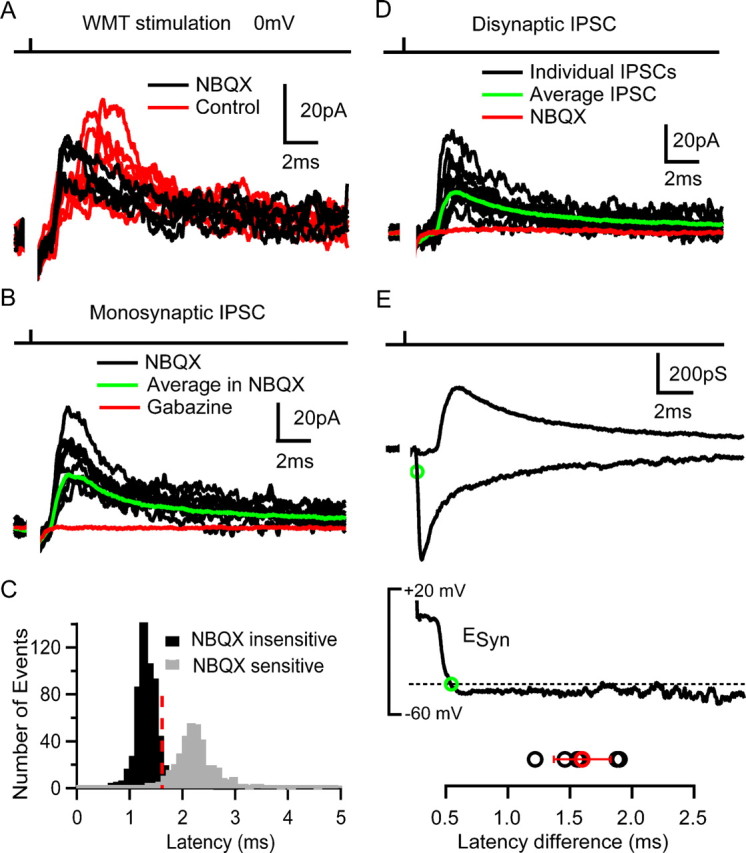
Feedforward inhibition of granule cells. A, IPSCs recorded in a GrC at 0 mV during WMT (100 V for 20 μs) stimulation in control conditions (red) and in the presence of 10 μm NBQX (black). B, Monosynaptic IPSCs in the presence of NBQX (black), the average IPSC (green), and its block by 10 μm gabazine (red). C, Latency distribution of NBQX-sensitive (gray) and -insensitive IPSC (black bars) pooled across cells. Red broken line indicates latency criterion used to distinguish GrCs receiving disynaptic inhibition. D, Group of long-latency IPSCs (in black) in response to WMT stimulation with average current (in green). IPSCs were fully blocked by 10 μm NBQX (red). E, Top, Average disynaptic inhibitory conductance (IPSG) recorded at 0 mV and average monosynaptic MF–GrC excitatory conductance (EPSG) recorded at −70 mV in the same cell at same stimulus intensity. Green circle indicates 20% rise time point of EPSG. Middle, Time course of calculated synaptic reversal potential (ESyn indicates reversal potential of the combined excitatory and inhibitory conductances), with the broken line indicating the spike threshold (Cathala et al., 2003). Green circle shows crossing of synaptic reversal potential with spike threshold (−40 mV), indicating a net inhibitory effect of the total synaptic conductance from that time point onward. Bottom, Latency difference between 20% rise time point of the EPSG and starting point of the net inhibitory effect for six cells. Mean value with SD is shown in red.
The pooled latency distribution of NBQX-insensitive (black) and NBQX-sensitive (gray) IPSCs is shown in Figure 8C. The two latency distributions were clearly distinguishable, allowing us to identify IPSCs arising from the MF–GoC–GrC feedforward circuitry using a latency criterion (>1.6 ms, 3 × SD above mean latency of monosynaptic IPSCs). IPSCs with latencies >1.6 ms were observed in 13 of 19 GrCs, suggesting that at least 68% of GrCs received feedforward inhibition. Of these, seven exhibited only the long-latency IPSCs component (average latency, 2.0 ± 0.3 ms; range, 1.6–2.2 ms) (Fig. 8D). These disynaptic IPSCs occurred with a success probability of 52 ± 29% (range, 18–91%) and had an average amplitude of 13.3 ± 10.2 pA (range, 4.8–31 pA). Their mean kinetics were very similar to monosynaptic IPSCs, with 20–80% rise of 0.4 ± 0.1 ms and a weighted decay of 7 ± 1 ms (p > 0.2, t test; n = 7). Application of 10 μm gabazine also blocked these IPSCs (n = 2), indicating that they were GABAA receptor mediated. Feedforward IPSCs had a latency jitter of 310 ± 140 μs (SD; n = 7) (Fig. 8C) as expected for the combined latency jitter of evoked GoC spikes and GoC–GrC transmission (p > 0.2, one-sample t test) (see Materials and Methods).
Cells that exhibited disynaptic inhibition in isolation were subsequently hyperpolarized to −70 mV to examine whether an excitatory MF–GrC input was also activated, as expected for an intact feedforward circuit. In six of the seven cells, we observed inward EPSCs with latency (0.8 ± 0.2 ms), kinetics (20–80% rise time of 0.20 ± 0.02 ms and weighted decay of 2.3 ± 1.1 ms) and mean peak amplitude (−41 ± 37 pA), similar to single-fiber-evoked MF–GrC EPSCs described previously (DiGregorio et al., 2002; Sargent et al., 2005). Because four MFs are required to fire the GoC, this suggests that more MFs contact a GoC than the average of four that contact a GrC. However, the exact number of MF synaptic inputs onto a GoC remains uncertain. Figure 8E shows the timing and average time courses of both the excitatory conductance (EPSG) and the inhibitory conductance (IPSG) for a representative cell. Although the EPSG peak amplitude was 1.5 ± 0.4 times larger than the IPSG (340 ± 230 vs 230 ± 130 pS), by the time the peak of the inhibitory conductance occurred, the EPSG had decayed by 30 ± 11% so that the inhibitory conductance was comparable in size (Fig. 8E, top panel). The time course of the net reversal potential of these two conductances (synaptic reversal potential) (see Materials and Methods) is plotted in the middle panel of Figure 8E (solid line). For this cell, the synaptic reversal potential drops below spike threshold (red broken line; −40 mV) (Cathala et al., 2003) 1.9 ms after the onset of the EPSG because of the inhibitory action of the IPSG. Thus, the IPSG restricts the excitatory action of the EPSG to a 1.6 ± 0.3 ms time window (n = 6) (Fig. 8E). These results confirm that a functional feedforward inhibitory network is present in the GrC layer and shows that the disynaptic IPSG truncates slow MF–GrC EPSG components, thereby narrowing the time window of synaptic integration.
Discussion
We have examined the functional properties of the previously uncharacterized feedforward MF input onto GoCs in the cerebellar cortex. Our results show that synchronous activation of four MFs can reset the spontaneous firing of GoCs with a precision of ∼200 μs. This precise EPSP-spike coupling is achieved by the highly synchronous release of few quanta. Spontaneous pacemaker conductances dominate GoC firing rate and introduce a relative refractory period for MF–GoC transmission. These features allow synchronously active MFs to entrain a GoC with high temporal precision at low frequencies but decouples GoC and MF firing during higher-frequency stimulation. Recordings from downstream GrCs confirm the presence of a functional feedforward inhibitory circuit. These results suggest that the MF–GoC–GrC pathway is tuned to transmit the precise timing of coincident MF activity preferentially at low frequencies and at the onset of a high-frequency stimulus train.
Properties of the MF–GoC EPSC
The MF–GoC synaptic connection has long been recognized from anatomical studies of the cerebellar cortex (Eccles et al., 1967; Palay and Chan-Palay, 1974), but its functional properties have not been examined. We used a number of functional and anatomical characteristics to identify MF inputs and distinguish them from CF and PF inputs. A characteristic feature of the MF–GoC connection is the rapid time course of the evoked EPSC. We show that this is generated by highly synchronized vesicular release and fast quantal currents. The time course of evoked EPSCs at the MF–GoC connection is significantly faster than MF-evoked EPSCs in cerebellar GrCs (τWD = 1.6 vs 2.9 ms, respectively) (DiGregorio et al., 2002), although they share the same presynaptic element. The prominent slow current components of the MF–GrC EPSC decay, which are mediated by glutamate spillover onto AMPARs (DiGregorio et al., 2002) and NMDARs (Cathala et al., 2000), are smaller or absent at MF–GoC synapses, although these two connections are made within the same glomerular structure (Eccles et al., 1967). A smaller spillover component could be attributable to differences in the anatomical arrangement (Cathala et al., 2005), a lower release probability (DiGregorio et al., 2002), an increased activity of glutamate transporters, or lower-affinity AMPARs at the MF–GoC synapse than the MF–GrC synapse (DiGregorio et al., 2007). The time courses of quantal and control evoked EPSCs at the MF–GoC synapse are comparable with those between hippocampal GrCs and basket cells (Geiger et al., 1997). Fast EPSCs have also been reported onto other interneurons (Galarreta and Hestrin, 2001; Carter and Regehr, 2002), suggesting that this is common to excitatory synapses in feedforward inhibitory circuits. Another characteristic feature of the MF–GoC synapse is the lack of STP. Resistance to frequency-dependent depression is likely to arise from rapid reloading of release-ready vesicles in the MF terminal (Saviane and Silver, 2006) and less AMPAR desensitization than at the MF–GrC synapse, which underlies the majority of the EPSC depression (Saviane and Silver, 2006; DiGregorio et al., 2007).
Properties of EPSP-spike coupling
GoC spikes are triggered predominantly during the rising phase of MF EPSPs, which is determined by the time course of synaptic charge injection. The fast time course of the MF–GoC EPSC is therefore of particular importance in determining AP precision (Fetz and Gustafsson, 1983; Galarreta and Hestrin, 2001). At synapses with few quanta and high quantal efficacy (Gulyás et al., 1993; Carter and Regehr, 2002), the stochastic nature of vesicular release can produce significant temporal jitter in the EPSC timing and shape from trial to trial (Sargent et al., 2005) and thus limit EPSP-spike coupling to ∼1 ms, even when the quantal EPSCs are fast (Miles, 1990; Galarreta and Hestrin, 2001; Carter and Regehr, 2002; Gabernet et al., 2005). Here we show that MF–GoCs synapses counteract this limitation with rapid vesicular release (τ = 130 μs) that produces a rapidly rising EPSP (20–80% rise time = 0.42 ms) and introduces little trial-to-trial jitter in the timing of the EPSP (SD of ∼80 μs), although the quantal content is low. These features allow as few as four synchronously active MFs releasing total of six quanta to reset spontaneous GoC firing, with high precision.
Our results show that the MF–GoC synapse is effective at signaling the precise timing of coincident MF activity at low input frequencies, close to or below the spontaneous firing rate of the GoC. Rapid EPSC kinetics in combination with active pacemaker currents, which lower the membrane resistance, produce the rapid rise and initial EPSP decay (τ = 2 ms), limiting the time window for AP generation. The temporal precision of spike generation that this produces in vitro (200 μs) is likely to represent an upper limit for signaling precision, because the perfectly synchronized activation of MFs used in this study is unlikely to occur in vivo. Precision and phase resetting may also be less precise in vivo because GoC are likely to experience greater synaptic drive than in the slice. Strong afterhyperpolarization, mediated by M and SK channels (Forti et al., 2006), reduces the efficacy of synaptic input immediately after an AP. Only after ∼50 ms is the hyperpolarization overcome sufficiently by depolarizing Ih and subthreshold Na+ conductances (Dieudonne, 1998; Forti et al., 2006) to allow MF inputs to trigger an AP reliably. Such prolonged membrane hyperpolarization has been shown to improve AP precision in a number of cell types by reducing jitter accumulation during trains (Berry and Meister, 1998; Schaefer et al., 2006) and is likely to contribute to the maintenance of GoC spike-time precision during trains of MF stimulation. However, the dominant afterhyperpolarization in GoCs make EPSP-spike coupling much less reliable at higher MF input frequencies, permitting only a weak modulation of GoC firing rate by MF inputs. This rejection of MF rate information contrasts with MF transmission onto GrCs in which slow EPSC components arising from spillover onto AMPARs produce a substantial tonic excitatory drive at high firing frequencies (Saviane and Silver, 2006). This, together with the wide dynamic range and non-accommodating nature of GrCs firing, promotes the efficient transmission of rate-coded information from MFs to GrCs (Mitchell and Silver, 2003). The distinct properties of the EPSCs and spike-generating mechanisms in GrC and GoCs could confer the ability to route information on the timing of a stimulus and more general rate information through separate synaptic “channels” in the cerebellar input layer.
Physiological implications
The cerebellar cortex coordinates movements and maintains balance by computing motor sequences from many types of sensory inputs on timescales from tens of milliseconds to several seconds (Eccles et al., 1967; Orlovsky, 1972; Rosen and Scheid, 1972; Yakusheva et al., 2007). How might the properties of the input layer feedforward inhibitory circuit aid these functions? Although it is difficult to extrapolate results from the in vitro slice preparation to in vivo function, the properties of the synaptic and cellular components of this circuit suggest that it is tuned to transmit the precise timing of coincident MF activity and then becomes refractory for tens of milliseconds. The size and timing of the inhibitory conductance produced in GrCs suggest that it could help shape the latency and precision of the early GrC response to sensory stimulation. However, because this circuit is only activated reliably for certain MF input patterns, feedforward inhibition is only likely to be effective in response to some sensory stimuli.
In vivo recordings from MFs and GoCs show a range of behaviors that depend on both the properties and modality of the sensory input. MFs that convey information about joint angle and head velocity are tonically active and show relatively slow modulations in firing rate during sensory stimulation in awake and anesthetized preparations (van Kan et al., 1993; Barmack and Yakhnitsa, 2008). GoCs respond to such sensory input with a modest and relatively slow modulation of their firing rate (Edgley and Lidierth, 1987; Barmack and Yakhnitsa, 2008). Under these conditions, the feedforward circuit may have little effect. In contrast, experiments in decerebrate and anesthetized animals show that tactile stimulation of whiskers and forelimbs evokes a barrage of EPSCs at hundreds of hertz in GrCs arising from one or multiple MFs, which exhibit low firing rates at rest (Jörntell and Ekerot, 2006; Rancz et al., 2007). At the onset of such sensory stimulation, some GoCs exhibit a temporally precise spike response followed by a pause in firing (Vos et al., 1999; Holtzman et al., 2006), consistent with feedforward excitation of GoCs, although pauses in firing in the absence of excitation were more commonly observed and had a larger receptive field (Holtzman et al., 2006). These diverse properties of MF inputs and the wide range of GoC responses make it difficult to identify unequivocally the role of feedforward excitation of GoC from in vivo experiments published to date.
Our results establish that most GrCs receive feedforward inhibition and suggest that activation of a small number of MFs onto a GoC can trigger phasic inhibition in >1000 GrCs innervated by its axon (Eccles et al., 1967). The feedforward inhibitory conductance produced is small but appropriately timed to cancel the slow component of the MF–GrC EPSC. Previous studies of feedforward inhibition in other systems suggest that these properties will enhance spike timing precision by reducing the time window for synaptic integration (Pouille and Scanziani, 2001). Feedforward inhibition could therefore influence the latency and precision of the first GrC spike at the onset of a stimulus, but later spikes are unlikely to be affected by this circuit because of the inability of GoCs to follow MF high-frequency activity. Curiously, fast stimulus-locked inhibition of GrCs has rarely been observed in vivo (Chadderton et al., 2004; Jörntell and Ekerot, 2006). This might be attributable to the small size of the inhibitory conductance, a developmental change in inhibitory transmission (Brickley et al., 1996; Wall and Usowicz, 1997), and the variability of GoC responses to sensory stimuli (Holtzman et al., 2006). Although additional in vitro and in vivo studies are required to determine the role of feedforward inhibition in the cerebellar input layer, our in vitro characterization of its synaptic and cellular properties suggests that GoCs can extract the precise timing of coincident MF activity and reduce the time window for synaptic integration in GrCs at the onset of certain sensory stimuli.
Footnotes
This work was supported by the Wellcome Trust, Medical Research Council Grant G0400598, and the European Union (EUSynapse LSHM-CT-2005-019055). R.A.S. is in receipt of a Wellcome Senior Research Fellowship. R.T.K. is supported by the Wellcome Trust Neuroscience Graduate Program. We thank Zoltan Nusser for helpful discussions during the project and Beverly Clark, David DiGregorio, Mark Farrant, Padraig Gleeson, Troy Margrie, Zoltan Nusser, and Koen Vervaeke for their comments on this manuscript.
References
- Barmack NH, Yakhnitsa V. Functions of interneurons in mouse cerebellum. J Neurosci. 2008;28:1140–1152. doi: 10.1523/JNEUROSCI.3942-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Stevens CF. The kinetics of transmitter release at the frog neuromuscular junction. J Physiol. 1972;227:691–708. doi: 10.1113/jphysiol.1972.sp010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beierlein M, Fioravante D, Regehr WG. Differential expression of posttetanic potentiation and retrograde signaling mediate target-dependent short-term synaptic plasticity. Neuron. 2007;54:949–959. doi: 10.1016/j.neuron.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry MJ, 2nd, Meister M. Refractoriness and neural precision. J Neurosci. 1998;18:2200–2211. doi: 10.1523/JNEUROSCI.18-06-02200.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Cull-Candy SG, Farrant M. Development of a tonic form of synaptic inhibition in rat cerebellar granule cells resulting from persistent activation of GABAA receptors. J Physiol. 1996;497:753–759. doi: 10.1113/jphysiol.1996.sp021806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bureau I, Dieudonne S, Coussen F, Mulle C. Kainate receptor-mediated synaptic currents in cerebellar Golgi cells are not shaped by diffusion of glutamate. Proc Natl Acad Sci U S A. 2000;97:6838–6843. doi: 10.1073/pnas.97.12.6838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AG, Regehr WG. Quantal events shape cerebellar interneuron firing. Nat Neurosci. 2002;5:1309–1318. doi: 10.1038/nn970. [DOI] [PubMed] [Google Scholar]
- Cathala L, Misra C, Cull-Candy S. Developmental profile of the changing properties of NMDA receptors at cerebellar mossy fiber-granule cell synapses. J Neurosci. 2000;20:5899–5905. doi: 10.1523/JNEUROSCI.20-16-05899.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Brickley S, Cull-Candy S, Farrant M. Maturation of EPSCs and intrinsic membrane properties enhances precision at a cerebellar synapse. J Neurosci. 2003;23:6074–6085. doi: 10.1523/JNEUROSCI.23-14-06074.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cathala L, Holderith NB, Nusser Z, DiGregorio DA, Cull-Candy SG. Changes in synaptic structure underlie the developmental speeding of AMPA receptor-mediated EPSCs. Nat Neurosci. 2005;8:1310–1318. doi: 10.1038/nn1534. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Dieudonne S. Submillisecond kinetics and low efficacy of parallel fibre-Golgi cell synaptic currents in the rat cerebellum. J Physiol. 1998;510:845–866. doi: 10.1111/j.1469-7793.1998.845bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGregorio DA, Nusser Z, Silver RA. Spillover of glutamate onto synaptic AMPA receptors enhances fast transmission at a cerebellar synapse. Neuron. 2002;35:521–533. doi: 10.1016/s0896-6273(02)00787-0. [DOI] [PubMed] [Google Scholar]
- DiGregorio DA, Rothman JS, Nielsen TA, Silver RA. Desensitization properties of AMPA receptors at the cerebellar mossy fiber granule cell synapse. J Neurosci. 2007;27:8344–8357. doi: 10.1523/JNEUROSCI.2399-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugué GP, Dumoulin A, Triller A, Dieudonné S. Target-dependent use of co-released inhibitory transmitters at central synapses. J Neurosci. 2005;25:6490–6498. doi: 10.1523/JNEUROSCI.1500-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eccles JC, Ito M, Szentagothai J. The cerebellum as a neuronal machine. Berlin: Springer; 1967. [Google Scholar]
- Edgley SA, Lidierth M. The discharges of cerebellar Golgi cells during locomotion in the cat. J Physiol. 1987;392:315–332. doi: 10.1113/jphysiol.1987.sp016782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fetz EE, Gustafsson B. Relation between shapes of post-synaptic potentials and changes in firing probability of cat motoneurones. J Physiol. 1983;341:387–410. doi: 10.1113/jphysiol.1983.sp014812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forti L, Cesana E, Mapelli J, D'Angelo E. Ionic mechanisms of autorhythmic firing in rat cerebellar Golgi cells. J Physiol. 2006;574:711–729. doi: 10.1113/jphysiol.2006.110858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricker D, Miles R. EPSP amplification and the precision of spike timing in hippocampal neurons. Neuron. 2000;28:559–569. doi: 10.1016/s0896-6273(00)00133-1. [DOI] [PubMed] [Google Scholar]
- Gabernet L, Jadhav SP, Feldman DE, Carandini M, Scanziani M. Somatosensory integration controlled by dynamic thalamocortical feed-forward inhibition. Neuron. 2005;48:315–327. doi: 10.1016/j.neuron.2005.09.022. [DOI] [PubMed] [Google Scholar]
- Galarreta M, Hestrin S. Spike transmission and synchrony detection in networks of GABAergic interneurons. Science. 2001;292:2295–2299. doi: 10.1126/science.1061395. [DOI] [PubMed] [Google Scholar]
- Geiger JR, Lübke J, Roth A, Frotscher M, Jonas P. Submillisecond AMPA receptor-mediated signaling at a principal neuron- interneuron synapse. Neuron. 1997;18:1009–1023. doi: 10.1016/s0896-6273(00)80339-6. [DOI] [PubMed] [Google Scholar]
- Gulyás AI, Miles R, Sík A, Tóth K, Tamamaki N, Freund TF. Hippocampal pyramidal cells excite inhibitory neurons through a single release site. Nature. 1993;366:683–687. doi: 10.1038/366683a0. [DOI] [PubMed] [Google Scholar]
- Harris KD, Henze DA, Hirase H, Leinekugel X, Dragoi G, Czurkó A, Buzsáki G. Spike train dynamics predicts theta-related phase precession in hippocampal pyramidal cells. Nature. 2002;417:738–741. doi: 10.1038/nature00808. [DOI] [PubMed] [Google Scholar]
- Häusser M, Raman IM, Otis T, Smith SL, Nelson A, du Lac S, Loewenstein Y, Mahon S, Pennartz C, Cohen I, Yarom Y. The beat goes on: spontaneous firing in mammalian neuronal microcircuits. J Neurosci. 2004;24:9215–9219. doi: 10.1523/JNEUROSCI.3375-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman T, Rajapaksa T, Mostofi A, Edgley SA. Different responses of rat cerebellar Purkinje cells and Golgi cells evoked by widespread convergent sensory inputs. J Physiol. 2006;574:491–507. doi: 10.1113/jphysiol.2006.108282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Counting quanta: direct measurements of transmitter release at a central synapse. Neuron. 1995;15:875–884. doi: 10.1016/0896-6273(95)90178-7. [DOI] [PubMed] [Google Scholar]
- Ivry RB, Keele SW. Timing functions of the cerebellum. J Cogn Neurosci. 1989;1:136–152. doi: 10.1162/jocn.1989.1.2.136. [DOI] [PubMed] [Google Scholar]
- Johnston D, Wu SM-S. Foundations of cellular neurophysiology. Cambridge, MA: MIT; 1995. [Google Scholar]
- Jörntell H, Ekerot CF. Properties of somatosensory synaptic integration in cerebellar granule cells in vivo. J Neurosci. 2006;26:11786–11797. doi: 10.1523/JNEUROSCI.2939-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. J Physiol. 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás RR. The intrinsic electrophysiological properties of mammalian neurons: insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Martina M, Vida I, Jonas P. Distal initiation and active propagation of action potentials in interneuron dendrites. Science. 2000;287:295–300. doi: 10.1126/science.287.5451.295. [DOI] [PubMed] [Google Scholar]
- Medina JF, Mauk MD. Computer simulation of cerebellar information processing. Nat Neurosci. 2000;3(Suppl):1205–1211. doi: 10.1038/81486. [DOI] [PubMed] [Google Scholar]
- Miles R. Synaptic excitation of inhibitory cells by single CA3 hippocampal pyramidal cells of the guinea-pig in vitro. J Physiol. 1990;428:61–77. doi: 10.1113/jphysiol.1990.sp018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Shunting inhibition modulates neuronal gain during synaptic excitation. Neuron. 2003;38:433–445. doi: 10.1016/s0896-6273(03)00200-9. [DOI] [PubMed] [Google Scholar]
- Mittmann W, Koch U, Häusser M. Feed-forward inhibition shapes the spike output of cerebellar Purkinje cells. J Physiol. 2005;563:369–378. doi: 10.1113/jphysiol.2004.075028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlovsky GN. Activity of rubrospinal neurons during locomotion. Brain Res. 1972;46:99–112. doi: 10.1016/0006-8993(72)90008-x. [DOI] [PubMed] [Google Scholar]
- Palay SL, Chan-Palay V. Cerebellar cortex. New York: Springer; 1974. [Google Scholar]
- Pouille F, Scanziani M. Enforcement of temporal fidelity in pyramidal cells by somatic feed-forward inhibition. Science. 2001;293:1159–1163. doi: 10.1126/science.1060342. [DOI] [PubMed] [Google Scholar]
- Rancz EA, Ishikawa T, Duguid I, Chadderton P, Mahon S, Häusser M. High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature. 2007;450:1245–1248. doi: 10.1038/nature05995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieke F, Warland D, de Ruyter van Steveninck RR, Bialek W. Spikes. Exploring the neural code. Cambridge, MA: MIT; 1997. [Google Scholar]
- Rinzel J, Ermentrout B. Analysis of neural excitability and oscillations. In: Koch C, Segev I, editors. Methods in neuronal modeling: from ions to networks. Ed 2. Cambridge, MA: MIT; 1998. [Google Scholar]
- Rosén I, Scheid P. Cerebellar surface cooling influencing evoked activity in cortex and in interpositus nucleus. Brain Res. 1972;45:580–584. doi: 10.1016/0006-8993(72)90486-6. [DOI] [PubMed] [Google Scholar]
- Sargent PB, Saviane C, Nielsen TA, DiGregorio DA, Silver RA. Rapid vesicular release, quantal variability and spillover contribute to the precision and reliability of transmission at a glomerular synapse. J Neurosci. 2005;25:8173–8187. doi: 10.1523/JNEUROSCI.2051-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saviane C, Silver RA. Fast vesicle reloading and a large pool sustain high bandwidth transmission at a central synapse. Nature. 2006;439:983–987. doi: 10.1038/nature04509. [DOI] [PubMed] [Google Scholar]
- Schaefer AT, Angelo K, Spors H, Margrie TW. Neuronal oscillations enhance stimulus discrimination by ensuring action potential precision. PLoS Biol. 2006;4:e163. doi: 10.1371/journal.pbio.0040163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA. Estimation of nonuniform quantal parameters with multiple-probability fluctuation analysis: theory, application and limitations. J Neurosci Methods. 2003;130:127–141. doi: 10.1016/j.jneumeth.2003.09.030. [DOI] [PubMed] [Google Scholar]
- Silver RA, Cull-Candy SG, Takahashi T. Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol. 1996;494:231–250. doi: 10.1113/jphysiol.1996.sp021487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple- probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol. 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopfer M, Bhagavan S, Smith BH, Laurent G. Impaired odour discrimination on desynchronization of odour-encoding neural assemblies. Nature. 1997;390:70–74. doi: 10.1038/36335. [DOI] [PubMed] [Google Scholar]
- Taschenberger H, Scheuss V, Neher E. Release kinetics, quantal parameters and their modulation during short-term depression at a developing synapse in the rat CNS. J Physiol. 2005;568:513–537. doi: 10.1113/jphysiol.2005.093468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kan PL, Gibson AR, Houk JC. Movement-related inputs to intermediate cerebellum of the monkey. J Neurophysiol. 1993;69:74–94. doi: 10.1152/jn.1993.69.1.74. [DOI] [PubMed] [Google Scholar]
- Vos BP, Volny-Luraghi A, De Schutter E. Cerebellar Golgi cells in the rat: receptive fields and timing of responses to facial stimulation. Eur J Neurosci. 1999;11:2621–2634. doi: 10.1046/j.1460-9568.1999.00678.x. [DOI] [PubMed] [Google Scholar]
- Wall MJ, Usowicz MM. Development of action potential-dependent and independent spontaneous GABAA receptor-mediated currents in granule cells of postnatal rat cerebellum. Eur J Neurosci. 1997;9:533–548. doi: 10.1111/j.1460-9568.1997.tb01630.x. [DOI] [PubMed] [Google Scholar]
- Walter JT, Alviña K, Womack MD, Chevez C, Khodakhah K. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–397. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Watanabe D, Inokawa H, Hashimoto K, Suzuki N, Kano M, Shigemoto R, Hirano T, Toyama K, Kaneko S, Yokoi M, Moriyoshi K, Suzuki M, Kobayashi K, Nagatsu T, Kreitman RJ, Pastan I, Nakanishi S. Ablation of cerebellar Golgi cells disrupts synaptic integration involving GABA inhibition and NMDA receptor activation in motor coordination. Cell. 1998;95:17–27. doi: 10.1016/s0092-8674(00)81779-1. [DOI] [PubMed] [Google Scholar]
- Yakusheva TA, Shaikh AG, Green AM, Blazquez PM, Dickman JD, Angelaki DE. Purkinje cells in posterior cerebellar vermis encode motion in an inertial reference frame. Neuron. 2007;54:973–985. doi: 10.1016/j.neuron.2007.06.003. [DOI] [PubMed] [Google Scholar]