

Abstract
In human brain the flavoprotein d-amino acid oxidase (hDAAO) is responsible for the degradation of the neuromodulator d-serine, an important effector of NMDA-receptor mediated neurotransmission. Experimental evidence supports the concept that d-serine concentration increase by hDAAO inhibition may represent a valuable therapeutic approach to improve the symptoms in schizophrenia patients. This study investigated the effects on hDAAO conformation and stability of the substrate d-serine (or of the pseudo-substrate trifluoro-d-alanine), the FAD cofactor, and two inhibitors (benzoate, a classical substrate-competitive inhibitor and the drug chlorpromazine (CPZ), which competes with the cofactor). We demonstrated that all these compounds do not alter the interaction of hDAAO with its physiological partner pLG72. The ligands used affect the tertiary structure of hDAAO differently: benzoate or trifluoro-d-alanine binding increases the amount of the holoenzyme form in solution and stabilizes the flavoprotein, while CPZ binding favors a protein conformation resembling that of the apoprotein, which is more sensitive to degradation. Interestingly, the apoprotein form of hDAAO binds the substrate d-serine: this interaction increases FAD binding thus increasing the amount of active holoenzyme in solution. Benzoate and CPZ similarly modify the short-term cellular d-serine concentration but affect the cellular concentration of hDAAO differently. In conclusion, the different alteration of hDAAO conformation and stability by the ligands used represents a further parameter to take into consideration during the development of new drugs to cope schizophrenia.
Keywords: d-serine, schizophrenia, flavoproteins, ligand binding, folding
Introduction
d-Amino acid oxidase (DAAO, EC 1.4.3.3) is a FAD-dependent flavoenzyme that catalyzes the stereospecific oxidative deamination of d-amino acids to the corresponding α-keto acids, hydrogen peroxide, and ammonia.1 Experimental evidence shows that, in brain, this flavoenzyme degrades the gliotransmitter d-serine, the only neutral d-amino acid present at significant concentration in this tissue: d-serine binds the glycine modulatory site on the NMDA receptor, allowing the activation of the receptor by glutamate, for a review see Refs. 2 and 3. The glutamatergic hypothesis of schizophrenia, the most severe human psychiatric disorder, is based on hypofunction of NMDA receptors. This theory is supported by observations that glycine abolishes the psychotomimetic effect of ketamine and improves neuroleptic therapy and that the most interesting susceptibility genes for schizophrenia act on glutamatergic transmission mediated by NMDA receptors.4,5 Among these, the genes encoding for human DAAO (hDAAO) and its putative binding partner pLG72 have been linked to schizophrenia susceptibility through genetic analysis of different human populations.6 We recently demonstrated that in vitro hDAAO specifically interacts with pLG72, yielding a ≈ 200-kDa complex constituted by 2 hDAAO homodimers and 2 pLG72 monomers.7–9 This interaction results in a time-dependent loss of hDAAO activity which is mainly due to alteration of the tertiary structure of hDAAO. Furthermore, we confirmed in vivo the hDAAO-pLG72 interaction and demonstrated that the cellular concentration of d-serine decreases in U87 glioblastoma cells transfected with a plasmid encoding for hDAAO but is not modified in those simultaneously transfected with cDNAs encoding both pLG72 and hDAAO.9 Therefore, we proposed a molecular mechanism by which hDAAO and pLG72 are involved in schizophrenia susceptibility: an anomalous increase in hDAAO activity (e.g., related to pLG72 hypoexpression) will result in a decrease in the synaptic concentration of d-serine, thus causing hypofunctionality of NMDA receptor-mediated neurotransmission. Importantly, and in agreement with the aforementioned model, hDAAO is now considered the target for a new class of drugs (hDAAO inhibitors) to treat schizophrenia.10–13
Human DAAO shows the main properties of the dehydrogenase-oxidase class of flavoproteins and shares a low turnover number and a ternary complex (sequential) kinetic mechanism with pig kidney DAAO (pkDAAO, ≈85% sequence identity).7 Indeed, this human flavoenzyme can be distinguished from the other known DAAOs because it is a stable homodimer even in the apoprotein form, because the binding of the FAD cofactor is the weakest among the known DAAOs (Kd value is in the micromolar range)1,7 and because it specifically interacts with pLG72 (which is expressed on primates only).6 These features raise a question concerning the actual in vivo activity of hDAAO: based on the weak cofactor binding (FAD concentration in brain is estimated to be ≈5 μM)14 hDAAO might be largely present in the inactive apoprotein form. It is noteworthy that a 20-fold tighter interaction between the FAD cofactor and the protein moiety is observed in the presence of the competitive inhibitor benzoate.7 Furthermore, we have recently shown that the apoprotein of hDAAO can be distinguished from the corresponding holoenzyme form by the “more open” tertiary structure and larger exposure of hydrophobic surfaces; it is also a slightly more sensitive to denaturation by urea or temperature.15
In this work we analyze the consequences of ligand binding on conformation and stability of hDAAO alone and in complex with pLG72. To reach this goal, the substrate d-serine or the pseudo-substrate trifluoro-d-alanine (CF3-d-Ala), benzoate (the most studied substrate–competitive inhibitor of hDAAO, Kd = 7 μM),1,9,16 and the drug chlorpromazine (CPZ) were employed. The aliphatic phenothiazine CPZ is a dopamine D2 receptor antagonist that was introduced in the treatment of schizophrenia in the early 1950s; it also acts as a FAD-competitive inhibitor of hDAAO (Kd = 5 μM).9,17 We here demonstrate that the different ligands affect the conformation and stability of the holoenzyme and apoprotein forms of the human flavoenzyme as well as the cellular concentration of hDAAO differently. We also provide evidence that these ligands do not alter the interaction between hDAAO and pLG72 and that affect the time course of cellular d-serine concentration similarly.
Results
Effect of ligand binding on the conformation of hDAAO
Spectral properties (and quaternary structure)
Because of the weak interaction with the FAD cofactor (Kd = 8 ± 2 μM), the recombinant hDAAO exists in solution as an equilibrium of holo- and apoprotein forms (at 1 mg protein/mL the solution contains similar amounts of the two protein species). The holoenzyme shows the typical absorbance spectrum of the FAD-containing flavoproteins, with maxima at 454, 372, and 278 nm.7 The interaction of hDAAO with the competitive inhibitor benzoate results in a typical perturbation of the flavin absorbance spectrum (formation of a shoulder at ≈500 nm) from which a Kd of 7 ± 2 μM was estimated.7 Interestingly, in the presence of a saturating concentration of benzoate the Kd for FAD binding decreases to 0.3 ± 0.1 μM and, therefore, a 1-mg hDAAO/mL solution contains ≥ 90% of the corresponding holoenzyme form. We also demonstrated that CPZ binds to the apoprotein form of hDAAO with an affinity similar to that estimated for FAD.9 Furthermore, and differently to that observed for FAD binding, the presence of benzoate does not modify the Kd for CPZ binding to hDAAO apoprotein.
The observation that benzoate increases the binding of FAD to hDAAO apoprotein9 suggested that a similar effect might also be given by the physiological substrate d-serine. This hypothesis cannot be directly verified because the substrate reduces the flavin cofactor generating the enzyme-product complex and altering the enzyme spectral properties. Similarly, l-serine cannot be used since it is neither substrate or inhibitor of hDAAO. CF3-d-Ala is a pseudo-substrate of DAAO that was previously used to deep insight the mode of substrate binding in the yeast enzyme:18 it possesses all the structural and steric requirements to be a substrate of DAAO but dehydrogenation is not feasible because of the inductive effect due to the —CF3 side chain. In fact, the addition of CF3-d-Ala under anaerobic conditions does not convert the oxidized hDAAO into the reduced enzyme form, as instead observed using a classical substrate such as d-alanine.1,7,9 CF3-d-Ala acts as competitive inhibitor of hDAAO: the Ki value (3.0 ± 0.5 mM) is close to the Km,app value determined for d-serine (7.5 mM).7 A further demonstration of CF3-d-Ala binding to hDAAO was achieved measuring the Kd for FAD binding in the presence of the pseudo-substrate: following the change in protein fluorescence a Kd = 0.5 ± 0.1 μM was determined, a value in good agreement with that observed for the hDAAO-benzoate complex (see previous).
Tryptophan fluorescence (following excitation at 280 nm) shows an emission maximum at 332 nm for the holoenzyme and at 338 nm for the apoprotein of hDAAO; the emission intensity being fivefold higher in the apo- than in the holoprotein, which indicates that the relevance of quenching interactions between tryptophans and nearby side chains is different in the two protein forms.15 The addition of 0.1 mM benzoate yields a significant quenching of hDAAO holoenzyme fluorescence [the emission at 338 nm drops from 210 to 165 a.u., Fig. 1(A)], while the change is marginal in the presence of 40 μM free FAD. Most significant alterations in protein fluorescence are observed using CF3-d-Ala: a 160 a.u. decrease in fluorescence intensity is observed at saturating concentration of the pseudo-substrate (Kd = 3.9 ± 0.1 mM), while FAD fails to give additional modification [Fig. 1(B)]. These results indicate that the change in protein fluorescence following benzoate or CF3-d-Ala binding to hDAAO is related to the acquisition of a protein conformation resembling that of the holoenzyme form (while at 0.1 mg hDAAO/mL concentration and in the absence of a ligand the solution contains ≈80% of the corresponding apoprotein form). Under the same experimental conditions, the addition of 0.1 mM CPZ yields a quenching of protein fluorescence of hDAAO similar to that observed following benzoate binding when the holoenzyme is used [Fig. 1(C)]. Indeed, the addition of 0.1 mM CPZ in the presence of 40 μM FAD alters the emission spectrum more significantly than when benzoate is used, thus pointing to a different alteration of hDAAO conformation as compared to adding benzoate or CF3-d-Ala.
Figure 1.
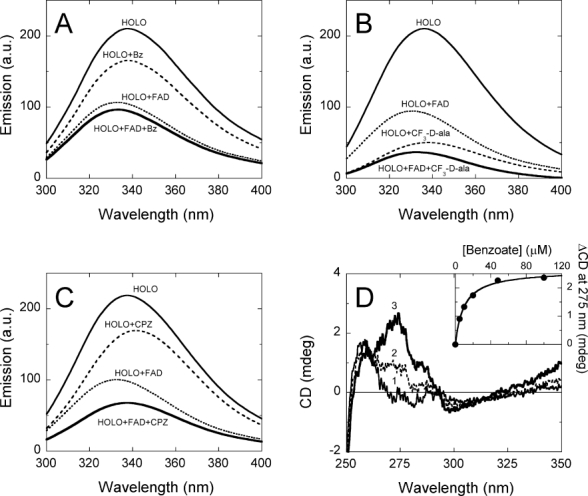
Effect of adding benzoate, CF3-d-Ala or CPZ on the spectral properties of hDAAO holoenzyme. (A) Comparison of protein fluorescence (excitation at 280 nm) of 2.5 μM (= 0.1 mg protein/mL) hDAAO before ( , in the absence and
, in the absence and  , in the presence of 40 μM FAD) and after adding 0.1 mM benzoate (
, in the presence of 40 μM FAD) and after adding 0.1 mM benzoate ( , in the absence and
, in the absence and  , in the presence of 40 μM FAD). (B) Comparison of protein fluorescence of hDAAO before and after adding 30 mM CF3-d-Ala (conditions as in panel A). (C) Comparison of protein fluorescence of hDAAO before and after adding 0.1 mM CPZ (conditions as in panel A). (D) Effect of benzoate addition on the near-UV CD spectrum of hDAAO (10 μM = 0.4 mg protein/mL): (1, —) no benzoate; (2,
, in the presence of 40 μM FAD). (B) Comparison of protein fluorescence of hDAAO before and after adding 30 mM CF3-d-Ala (conditions as in panel A). (C) Comparison of protein fluorescence of hDAAO before and after adding 0.1 mM CPZ (conditions as in panel A). (D) Effect of benzoate addition on the near-UV CD spectrum of hDAAO (10 μM = 0.4 mg protein/mL): (1, —) no benzoate; (2,  ) 4.8 μM benzoate; (3,
) 4.8 μM benzoate; (3,  ) 48 μM benzoate. Inset: change in CD signal at ≈275 nm at increasing benzoate concentrations.
) 48 μM benzoate. Inset: change in CD signal at ≈275 nm at increasing benzoate concentrations.
A larger change in protein fluorescence intensity (from 295 up to 95 a.u.) is apparent using the apoprotein form of hDAAO following d-serine or CF3-d-Ala addition (Kd = 3.0 ± 0.3 and 4.6 ± 0.2 mM, respectively): this also demonstrates that the apoprotein is competent in substrate binding. A similar fluorescence change is apparent adding 0.1 mM CPZ while no modification is observed using benzoate.
The far-UV CD spectra of apo- and holoprotein of hDAAO are identical, revealing a similar amount of secondary structure (not shown). In agreement with this observation, adding 0.1 mM FAD, benzoate, CF3-d-Ala or CPZ does not modify the far-UV CD spectrum of the protein. Differences are instead apparent in the near-UV CD spectra of the apo- and holoprotein forms, further indicating a slight alteration of the tertiary structure after cofactor binding.15 The near-UV CD features of hDAAO holoenzyme are to some extent modified by adding benzoate and CF3-d-Ala. At increasing ligand concentrations an higher intensity in the ≈ 270-nm signal is apparent, from which Kd values of 9.5 ± 0.7 μM and 3.1 ± 0.6 mM are determined for benzoate and CF3-d-Ala, respectively [see Fig. 1(D) for benzoate]. Indeed, the addition of CPZ decreases the CD signal at 270 nm, a change that is more evident in the presence of 40 μM FAD and that is not observed using the apoprotein form of hDAAO (not shown). These results reveal a different alteration of hDAAO tertiary structure following the binding of the ligands used.
We previously reported that both the holo- and apoprotein forms of hDAAO elute in size-exclusion chromatography as a single peak corresponding to an 80-kDa homodimer in the 0.1- to 10-mg protein/mL concentration range.7 Under the same conditions, the addition of 40 μM FAD in the elution buffer does not modify the elution volume of either the holo- or apoprotein forms of hDAAO or alter the presence of 0.1 mM benzoate, CF3-d-Ala or CPZ (not shown).
Limited proteolysis studies
Limited proteolysis of proteins in a native-like state is a strong tool to test changes in the higher-order structure in response to ligand binding.19 SDS-PAGE Analysis during limited proteolysis experiments of the hDAAO apoprotein form (using 10% (w/w) trypsin and at 25°C) clearly shows that the 40-kDa band corresponding to the intact protein is fully degraded in a monophasic process in ≈10 minutes (kobs1 = 0.27 min−1, Fig. 2). On the other hand, proteolysis of the holoenzyme (in the absence of exogenous FAD) is a biphasic and slower process: first there is a rapid phase that accounts for the degradation of ≈68% of the protein (reasonably corresponding to the amount of apoprotein in solution, kobs1 = 0.30 min−1) which is followed by a slow phase (kobs2 = 7 × 10−3 min−1, Fig. 2 and Table I). Adding 0.1 mM FAD largely protects hDAAO from trypsin cleavage: more than 75% of the intact enzyme is still present after 30 min of incubation (Fig. 2). Indeed, the apoprotein, too, is largely protected from proteolysis by previous incubation for 5 minutes with the cofactor: the sensitivity of the reconstituted holoprotein to trypsin resembles that of the native hDAAO holoenzyme (Table I).
Figure 2.
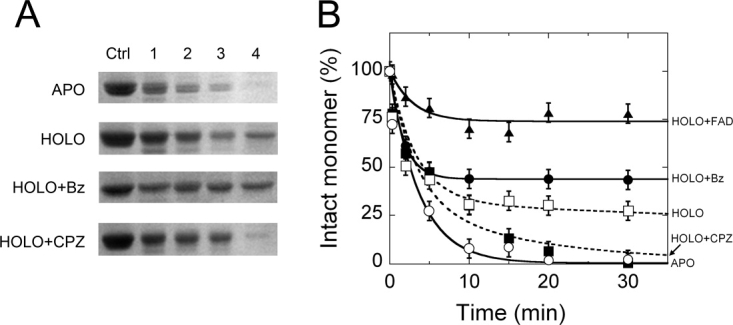
Time course of trypsin digestion of different hDAAO forms (10 μM = 0.4 mg protein/mL) with 10% (w/w) trypsin at 25°C in 20 mM Tris-HCl pH 8.5, 150 mM NaCl, 5% glycerol, 0.06% NLS, and 5 mM 2-mercaptoethanol. Panel A) SDS-PAGE Analysis of samples at different times: (lane Ctrl) control before adding trypsin; (lane 1) immediately after adding trypsin (30 s); (lanes 2–4) 2, 5, and 30 min after adding trypsin. APO = Apoprotein; HOLO = holoenzyme; HOLO+Bz = holoenzyme to which 1 mM benzoate was added; HOLO+CPZ = holoenzyme to which 0.1 mM CPZ was added. B) Time course of the intensity of the 40-kDa band (corresponding to the intact hDAAO monomer) as determined by densitometric analysis of gels such as those reported in panel A. HOLO+FAD is the sample obtained following the addition of 100 μM free FAD to the hDAAO holoenzyme. The intensity of the 40-kDa band of the sample before adding trypsin (sample in lane Ctrl, panel A) is indicated as 100%. Bars indicate S.E. as determined for at least three independent experiments.
Table I.
Comparison of the Kinetics of Trypsin Cleavage of 10 μM hDAAO in the Presence of Various Ligands
| holo-hDAAO |
apo-hDAAO |
hDAAO-pLG72 complex |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A1 (%) | k1 (min−1) | A2 (%) | k2 (min−1) | A1 (%) | k1 (min−1) | A1 (%) | k1 (min−1) | A2 (%) | k2 (min−1) | |
| No ligands | 68 ± 3 | 0.30 ± 0.04 | 32 ± 3 | 0.7 ± 0.2 ×10−2 | 100 | 0.27 ± 0.04 | 58 ± 8 | 0.55 ± 0.18 | 42 ± 8 | 6.0 ± 1.1 × 10−2 |
| + 0.1 mM FAD | 26 ± 4 | 0.32 ± 0.12 | 74 ± 4 | (≈ stable) | 66 ± 5 | 0.34 ± 0.08 | 23 ± 3 | 0.55 ± 0.10 | 77 ± 3 | 3.3 ± 0.4 × 10−2 |
| + 1 mM benzoate | 56 ± 3 | 0.48 ± 0.12 | 44 ± 3 | (≈ stable) | 100 | 0.27 ± 0.04 | 56 ± 5 | 0.60 ± 0.09 | 44 ± 6 | (≈ stable) |
| + 30 mM CF3-d-Ala | 56 ± 4 | 0.32 ± 0.09 | 44 ± 1 | (≈ stable) | 100 | 0.27 ± 0.04 | 49 ± 6 | 0.7 ± 0.23 | 51 ± 4 | (≈ stable) |
| + 0.1 mM CPZ | 68 ± 2 | 0.30 ± 0.02 | 32 ± 2 | 6.0 ± 1.8 ×10−2 | 100 | 0.27 ± 0.04 | 100 | 0.14 ± 0.01 | — | — |
| + 10 μM pLG72 | 42 ± 9 | 0.55 ± 0.18 | 58 ± 8 | 6.8 ± 1.1 ×10−2 | 100 | 0.30 ± 0.04 | ||||
| + 20 μM pLG72 | 58 ± 6 | 3.6 ± 0.7 | 42 ± 5 | 0.16 ± 0.03 | 100 | 0.27 ± 0.02 | ||||
The presence of 1 mM benzoate or of 30 mM CF3-d-Ala increases the amount of hDAAO that is resistant to protease (Fig. 2). In contrast, the addition of 0.1 mM CPZ increases the sensitivity of hDAAO holoenzyme to trypsinolysis: a 10-fold increase in the rate constant for the second phase of degradation is observed (kobs2 = 6 × 10−2 min−1, Fig. 2). However, adding CPZ, benzoate or CF3-d-Ala to the hDAAO apoprotein does not modify the time course of proteolysis (Table I).
Taken together, data from limited proteolysis experiments indicate that the amplitude of the observed first phase of proteolysis of the holoenzyme correlates with the amount of apoprotein present in solution; the second phase is related to the slow FAD dissociation from the remaining holoenzyme form. The ligands benzoate and CF3-d-Ala increase the amount of holoenzyme species in solution (thus decreasing the amplitude of the first phase) and prevent FAD dissociation from the remaining holoenzyme form (thus slowing down/abolishing the second phase). In contrast, binding of CPZ increases the susceptibility of hDAAO holoenzyme by accelerating the second phase of proteolysis (see Table I).
Effect of ligand binding on the thermal stability of hDAAO
A comparison of the temperature sensitivity of specific structural features of hDAAO showed higher midpoint transition temperatures for the holo- than for the apoprotein form.15 We now performed temperature ramp experiments in the presence of the ligands by following the change in protein fluorescence (Table II). The addition of 0.1 mM benzoate or 30 mM CF3-d-Ala increases the Tm value of the holoenzyme form of hDAAO (ΔTm ≈ 5°C), while the presence of 0.1 mM CPZ destabilizes the same enzyme form. In contrast, the stability of the apoprotein of hDAAO is not altered by benzoate (there is no evidence of benzoate binding to hDAAO apoprotein), while CPZ and CF3-d-Ala binding moderately stabilizes this protein form (ΔTm ≈ 2°C).
Table II.
Effect of Ligand Binding on the Tm Values for hDAAO Unfolding as Determined by Protein Fluorescence
|
Tm (°C) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Apoprotein |
Holoenzyme |
|||||||||
| +Bz | +CF3-d-Ala | +CPZ | +FAD | +Bz | +Bz +FAD | +CF3-d-Ala | +CF3-d-Ala +FAD | +CPZ | ||
| 50.2 ± 1.5 | 50.1 ± 1.7 | 52.8 ± 0.2 | 52.0 ± 1.3 | 50.1 ± 0.2 | 51.8 ± 0.2 | 55.2 ± 0.2 | 55.4 ± 0.2 | 54.7 ± 0.2 | 55.7 ± 0.2 | 48.0 ± 0.7 |
Fluorescence experiments were performed at an identical heating rate (0.5°C/min) in the absence and in the presence of 0.1 mM benzoate, 0.1 mM CPZ, 30 mM CF3-d-Ala, and/or 40 μM FAD. Protein concentration was 0.1 mg/mL. Tm values were obtained by calculating the first derivative of the spectroscopic signals and are uncorrected for delay effects.15
Effect of benzoate (and CF3-d-Ala) on the FAD-binding process to hDAAO apoprotein
By using the time courses of flavin and protein fluorescence and activity recovery, we recently demonstrated15 that the overall hDAAO holoenzyme reconstitution process starting from apoprotein and free FAD can be described by the series of steps reported in Eq. (1):
| (1) |
FAD binding to the apoprotein moiety is a reversible process which yields a catalytically competent Holo* form (steps k1/k-1); the k2 rate constant (= 0.0034 ± 0.0002 s−1) pertains to a sequential rearrangement of the reconstituted holoenzyme (and it is only apparent following the protein fluorescence). We now analyzed the kinetics of reconstitution of the hDAAO apoprotein-FAD complex under the same experimental conditions (pseudo-first order conditions, at 15°C) and in the presence of 1 mM benzoate (in the presence of benzoate the recovery of enzyme activity cannot be used to test holoenzyme reconstitution). The time course of flavin fluorescence change is monophasic and depends on apoprotein concentration: in the absence of benzoate, the amplitude of the fluorescence change increases with the amount of apoprotein used and corresponds to the amount of reconstituted holoenzyme. In contrast, the amplitude of fluorescence change is always maximal when 1 mM benzoate is present [Fig. 3(A)], thus indicating that a large part of the holoprotein is reconstituted in the presence of the ligand at all protein concentrations used. The observed first-order rate constant for the change in flavin fluorescence shows a linear dependence on the apoprotein concentration in both the absence and presence of the ligand: the only difference is that a clear y-intercept is manifest in the former [Fig. 3(B)]. Such a behavior is indicative of a reversible second-order process, with k1 rate constants of 0.7 and 1.0 mM−1s−1 and k-1 values of 0.0074 and ≤ 0.0005 s−1 in the absence and in the presence of benzoate, respectively. The Kd value for FAD binding to hDAAO apoprotein estimated from the kinetic data is 10 and 0.5 μM in the absence and in the presence of benzoate, respectively, which is in good agreement with the values obtained from static titrations.7 Noteworthy, the time course of fluorescence change observed using a saturating concentration of CF3-d-Ala is identical to that determined using benzoate as hDAAO ligand (not shown).
Figure 3.
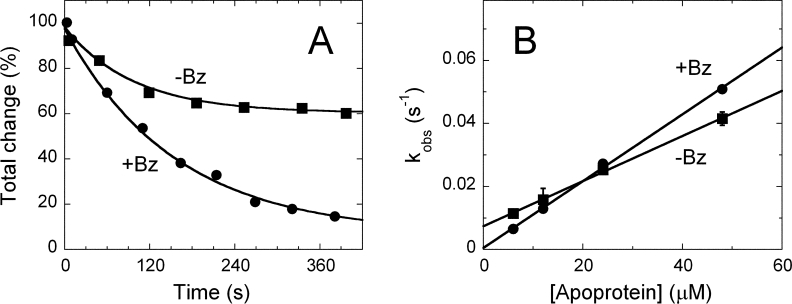
Effect of benzoate on the kinetics of hDAAO apoprotein reconstitution with FAD. (A) Overlay of time course of flavin fluorescence quenching in the absence (▪) and in the presence (•) of 1 mM sodium benzoate after mixing a 10-fold excess of apoprotein (0.6 μM FAD to which 6 μM apoprotein solution was added) at pH 8.3 and 15°C. The fits through the data points are obtained according to a single exponential decay equation. (B) Effect of apoprotein concentration on the observed first-order rate constants (kobs) of the reaction of FAD with hDAAO apoprotein. The values of kobs were determined from experiments such as those reported in panel A.
Effect of ligand binding on the hDAAO-pLG72 complex formation
hDAAO binds pLG72 yielding an ≈200-kDa complex constituted by 2 hDAAO homodimers and 2 pLG72 monomers.9 The presence of 40 μM free FAD and 0.1 mM CPZ or benzoate in the elution buffer does not affect the amount of protein complex formed as judged by size exclusion chromatography and measuring the area of the corresponding peak at ≈12.8 mL.9 A comparable result is also apparent using the apoprotein form of hDAAO (and in the absence of free FAD in the elution buffer, data not shown). In both cases a saturation in the peak's area corresponding to the hDAAO-pLG72 complex is attained at a 2:1 molar ratio, as previously assessed in the absence of CPZ.9
Real-time interaction analyses between hDAAO and pLG72 as well as between its different ligands were performed by SPR method. Both pLG72 and hDAAO were individually immobilized by standard amine coupling chemistry on a CM5 sensor chip. The signals describing the interaction of a ligand-free hDAAO solution flushed over a sensor with immobilized pLG72 in the absence and in the presence of 40 μM FAD [Fig. 4(A,B)] are threefold higher for the holoenzyme than for the apoprotein form of hDAAO. The sensorgrams were analyzed by means of Biacore 1.1.1 software and showed similar apparent affinities (Kd values were 5.3 vs. 3.1 × 10−7M with and without free FAD, respectively). The apparent affinity is higher than that determined previously using a different buffer composition (Kd = 8 μM).9 Indeed, no signal was observed using control proteins (human serum albumin or acylase), thus confirming the specificity of recognition. Comparative analyses of sensorgrams were carried out using hDAAO-immobilized sensors and flushing different concentrations of pLG72 with or without 20 μM free FAD [Fig. 4(C,D)]. Under these conditions, the ka association rate constants are higher than with immobilized pLG72 and the traces are not affected by the presence of FAD [Fig. 4(C,D); Kd value is 0.8 ± 0.1 × 10−7M].
Figure 4.
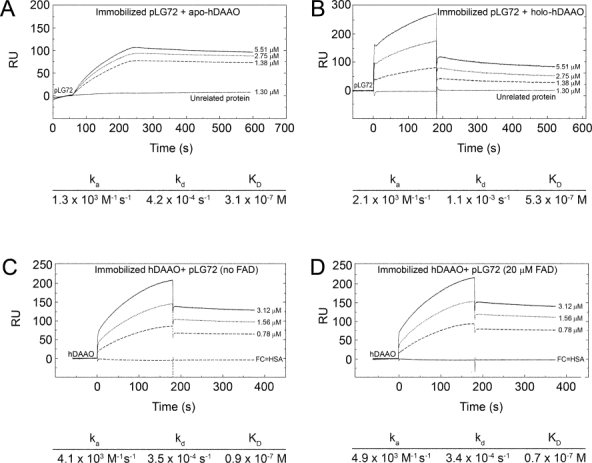
Sensorgrams (Biacore analysis) of the interaction between pLG72 and hDAAO. Panels (A,B) Kinetic evaluation of hDAAO apoprotein and holoenzyme binding to immobilized pLG72. Apo- (panel A) and holoprotein of hDAAO (panel B) diluted in HBS EP+ buffer at concentration of 1.38, 2.75, and 5.51 μM was injected at a flow rate of 20 μL/min over pLG72 previously immobilized on a CM5 sensor chip. The “on” and “off” rates were calculated with Biacore T100 BIAevaluation software. An unrelated protein at a concentration of 1.3 μM was injected under the same conditions. Panels (C,D) Kinetic evaluation of pLG72 binding to immobilized hDAAO. pLG72 diluted in HBS EP+ buffer at concentrations of 0.78, 1.56, and 3.12 μM was injected at a flow rate of 20 μL/min over hDAAO previously immobilized on a CM5 sensor chip (panel C). To investigate FAD effect on interaction, the same experiment was performed by diluting pLG72 in the presence of different concentrations of FAD (20 μM in panel D). As negative control a flow cell with previously immobilized human serum albumin was used. The rate constants for binding (ka) and dissociation (kd) as well as the Kd (= kd/ka) values for each single experimental are reported below each single panel.
Under similar experimental conditions, the ka and kd values for pLG72 binding and dissociation to immobilized hDAAO are unchanged in the presence of 20 μM CPZ, of 35 μM benzoate (with or without 40 μM free FAD), or of 50 mM d-serine (not shown). Indeed, from these experimental time courses binding data for the small ligand interaction to hDAAO were determined.20 The corresponding values are reported in Table III and correspond well with Kd values previously determined by static titration or from Michaelis-Menten kinetics (for the substrate d-serine) for hDAAO alone. The most significant discrepancy (≈ 40-fold) is observed for the binding of the FAD cofactor, for which the interaction with hDAAO apoprotein is known to be affected by the experimental conditions used but not by pLG72 binding.20 Otherwise, the latter result might also arises from slight alterations in hDAAO conformation/dynamics following the immobilization procedure.
Table III.
Evaluation of the Kinetics of Interaction of Different Ligands as Estimated by Means of SPR Analysis During pLG72 Binding to Immobilized hDAAO (at 25°C, See Legend of Fig. 4 for Details)
| Ligand | ka (M−1 s−1) | kd (×10−3 s−1) | Kd (M) | Known value (M) | |
|---|---|---|---|---|---|
| d-Serine | 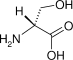 |
0.36 | 4.07 | 1.1 ×10−2 | (1.3 ×10−2)a |
| FAD | 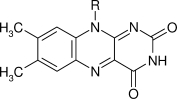 |
900 | 0.17 | 1.9 ×10−7 | (8 ×10−6)b |
| CPZ | 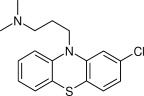 |
345 | 0.55 | 1.6 ×10−6 | (5 ×10−6)b |
| Benzoate | 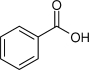 |
92.5 | 1.10 | 12 ×10−6 | (7 ×10−6)c |
As determined form stopped-flow kinetic analysis.7
As determined following the protein fluorescence quenching during apoprotein titration with increasing FAD7 and CPZ9 concentration.
As determined following absorbance changes at ≈ 500 nm following titration of hDAAO with increasing benzoate concentration.7
Limited proteolysis studies
Limited proteolysis experiments were used to probe ligand-induced conformational changes in the hDAAO-pLG72 complex. At first the effect of increasing amounts of pLG72 on the time course of degradation of the 40-kDa band of hDAAO (corresponding to the intact monomer) was investigated. As shown in Figure 5, the presence of a stoichiometric amount of pLG72 increases the rate constant for the second phase of holoenzyme degradation; furthermore, the destabilization of hDAAO was most evident when a twofold excess of pLG72 was added (Table I). On the other hand, the time course of proteolysis of the apoprotein form is not affected by the addition of pLG72 (cf. Figs. 2 and 5).
Figure 5.
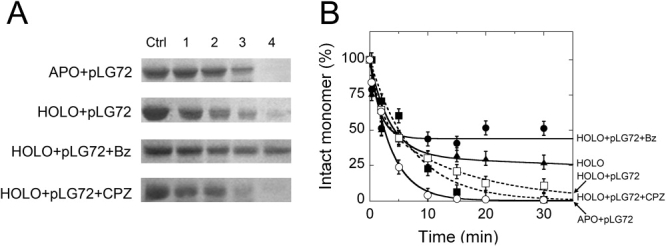
Time course of trypsin digestion of the hDAAO-pLG72 complex (10 μM = 0.4 mg protein/mL of hDAAO and 10 μM = 0.18 mg protein/mL of pLG72) with 10% (w/w) trypsin at 25°C in 20 mM Tris-HCl pH 8.5, 150 mM NaCl, 5% glycerol, 0.06% NLS, and 5 mM 2-mercaptoethanol. Panel (A) SDS-PAGE Analysis of samples at different times (see legend to Fig. 2 for details). APO+pLG72 = apoprotein + pLG72; HOLO+pLG72 = holoenzyme + pLG72; HOLO+pLG72+Bz = holoenzyme + pLG72 to which 1 mM benzoate was added; HOLO+pLG72+CPZ = holoenzyme + pLG72 to which 0.1 mM CPZ was added. (B) Time course of the intensity of the 40-kDa band (corresponding to the intact hDAAO monomer) as determined by densitometric analysis of gels such as those reported in panel (A). The intensity of the 40-kDa band of the sample before adding trypsin (sample in lane Ctrl, panel A) is indicated as 100%. Bars indicate S.E. as determined for at least three independent experiments.
Concerning the effect of small ligands on the trypsin susceptibility of the hDAAO-pLG72 complex (obtained mixing stoichiometric amounts of hDAAO and pLG72) the presence of 0.1 mM free FAD halves the amount of protein degraded in the first (rapid) phase and slows down the second phase of proteolysis of the hDAAO holoenzyme (this latter phase is absent when pLG72 is missing, see Table I). The addition of 0.1 mM CPZ produces a hDAAO-pLG72 complex which is fully sensitive to proteolysis, similarly to that observed for the free apoprotein (Fig. 5 and Table I). However, the holoenzyme is partially protected from degradation in the presence of 0.1 mM benzoate or 30 mM CF3-d-Ala (≈ 45% of the enzyme is resistant to proteolysis, Figure 5, analogously to that observed in the absence of pLG72). Taken together, these results suggest that pLG72 binding destabilizes the holoenzyme form of hDAAO, an effect which is counteracted by FAD, CF3-d-Ala and benzoate binding.
Effect of benzoate and CPZ on cellular D-serine and hDAAO concentrations
To investigate the effect of hDAAO ligands at the cellular level, we produced stably transfected U87 human glioblastoma cells expressing EGFP-hDAAO.9 The fusion of the GFP protein at the N-terminus of hDAAO does not alter the main properties of the flavooxidase. The recombinant EGFP-hDAAO is active (kcat is 2.5 s−1 and Km is 4 mM vs. 3.0 s−1 and 7 mM for the wild-type on d-serine) and each protein monomer (≈69 kDa) interacts with one molecule of FAD (with a Kd of 7 μM vs. 8 μM as determined for wild-type hDAAO).7 Furthermore, EGFP-hDAAO interacts with pLG72 yielding a complex which molecular mass is about twice that of the native dimeric enzyme (in agreement with the formation of a complex constituted by two hDAAO homodimers and two pLG72 molecules) and which time course of inactivation strictly resembles that observed using the native hDAAO.9 U87 Transfected cells were treated with 10 or 100 μM benzoate (these two concentrations yielded similar results) or 10 μM CPZ and the d- and l-serine concentrations were determined by HPLC chromatography (these concentrations of hDAAO inhibitors do not affect cell viability while massive cell death was observed at 100 μM CPZ). Both compounds yield a statistically significant increase in D/(D+L) serine concentration ratio as compared to untreated controls after 24 hours, a change that is cleared 48 hours later [Fig. 6(A)]. No significant differences were observed in the amount of cellular hDAAO, as determined by means of Western blot analysis and activity assay, with the only exception being CPZ-treatment after 48 hours [20% decrease, Fig. 6(B)]. The same experiment was carried out using U87 glioblastoma cells stably producing EGFP-hDAAO and transiently expressing pLG72. Under these conditions, CPZ treatment significantly decreases the amount of active hDAAO while benzoate slightly increases the cellular hDAAO in cotransfected cells [see values at 48 hours in Fig. 6(C)]. These latter results resemble the effects exerted by the two model ligands on hDAAO susceptibility as observed in limited proteolysis experiments on the hDAAO-pLG72 complex (Fig. 5). The aggregated data show that benzoate and CPZ affect hDAAO stability in the cell differently (thus resembling the effects observed on the pure protein, see previous) but similarly affect the time course for the cellular D/(D+L) serine concentration ratio due to hDAAO inhibition.
Figure 6.
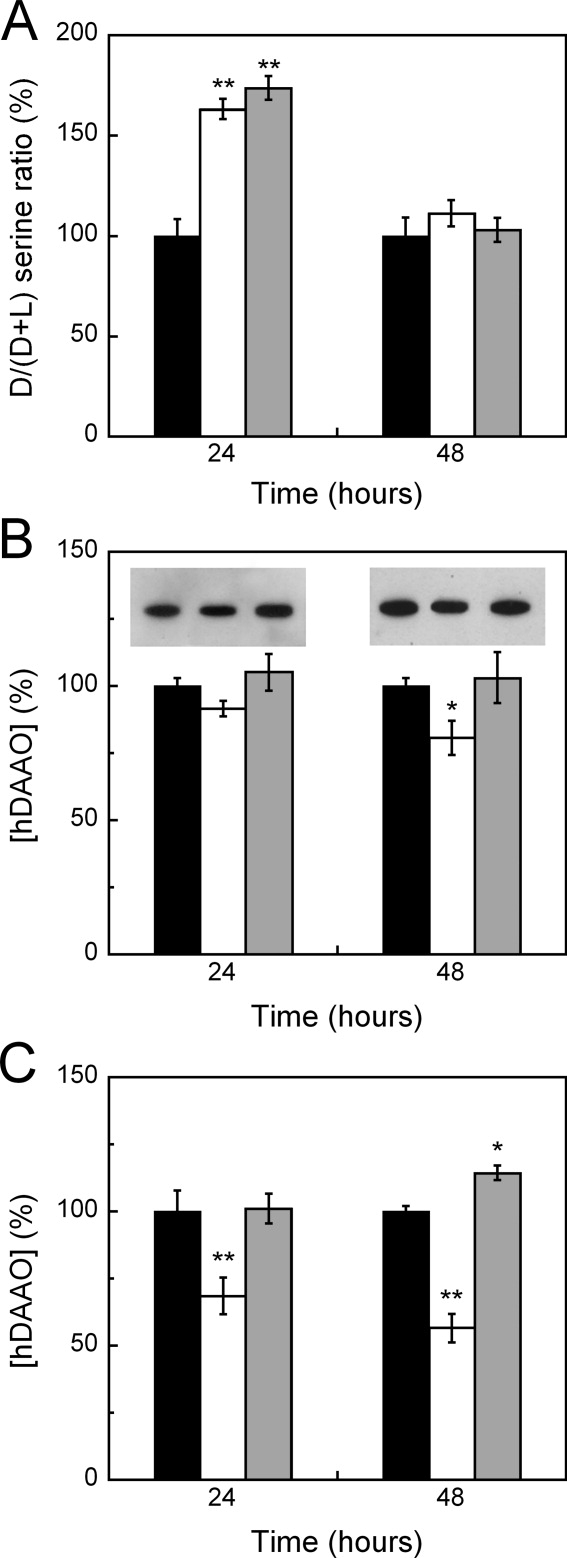
Dependence of d-serine and EGFP-hDAAO concentrations on treatment with benzoate or CPZ in U87 glioblastoma cells stably transfected with pEGFP-hDAAO-C3 expression vector. (A) Histogram reporting the time course of the D/(D+L) serine concentration ratio in U87 cells transfected with EGFP-hDAAO and treated without (control, black bars) or with 10 μM CPZ (white bars) or with 10–100 μM benzoate (gray bars). The values are expressed as a percentage, taking as 100% the ratio determined for the control at each time point (0 hours: 2.40 ± 0.34; 24 hours: 1.89 ± 0.16; 48 hours: 1.93 ± 0.18). The change in D/(D+L) serine ratio was demonstrated to be significant for CPZ and benzoate treatments at 24 hours (P < 0.001, see double asterisk) and not significant at 48 hours. (B) Histogram reporting the time course of the hDAAO concentration in the same cell samples as in panel A, as determined by means of Western blot analysis (the bands detected using anti-GFP antibodies are shown at the top) and activity assay using the Amplex UltraRed system (see “Materials and Methods” section). The values are expressed as a percentage, taking as 100% the amount of EGFP-hDAAO determined for the control at each time point (24 hours: 0.059 μg hDAAO/5 x 104 cells and 40 mU hDAAO/mg protein; 48 hours: 0.058 μg hDAAO/5 x 104 cells and 43 mU hDAAO/mg protein). The change in hDAAO concentration with respect to the control was demonstrated to be statistically significant for CPZ-treated samples at 48 hours (P = 0.017, see asterisk) and not statistically significant for all the remaining samples (P > 0.05). (C) Same analysis as reported in panel B performed on U87 glioblastoma cells stably expressing EGFP-hDAAO and transiently transfected with pLG72. The change in hDAAO concentration with respect to the control is statistically significant for CPZ-treated samples at 24 and 48 hours (P < 0.007, see double asterisk) and for the benzoate-treated sample at 48 hours (P = 0.017, see asterisk). The data are reported as means ± S.E.; for each point at least six independent determinations were performed.
Discussion
Medical evidence supporting the role of NMDA receptor hypofunction in schizophrenia has prompted clinical trials of agents that enhance NMDA receptor functionality.4,21 For example, schizophrenia patients receiving d-serine (an allosteric activator of NMDA-type glutamate receptor) together with antipsychotics showed significant improvement in their positive, negative, and cognitive symptoms.22 In human brain hDAAO is the main perpetrator of oxidation of the gliotransmitter d-serine, for a review see Refs. 1–3. A number of recent studies focused on the development of new and effective hDAAO inhibitors as a treatment strategy for schizophrenia by modulating d-serine concentrations in the brain.10–13 These studies reported on the synthesis and identification of new hDAAO inhibitors (mainly aromatic compounds that bind in the hDAAO active site, for a structural comparison see Ref. 13) and on their effect on rat brain d-serine concentration. However, the investigations did not focus on the effects of these ligands on hDAAO conformation and stability and on its interaction with the modulator pLG72 (which is absent in rats since it is a primate-specific protein).
We here demonstrated that the presence of ligands profoundly affects hDAAO conformation as indicated by protein fluorescence, near-UV CD spectroscopy, and limited proteolysis studies. Benzoate and CF3-d-Ala binding protect the holoenzyme from thermal denaturation and trypsinolysis (Tables I and II). Both ligands exert a stabilizing effect by increasing the amount of hDAAO holoenzyme present in solution as the result of a 20-fold decrease in the Kd for FAD binding,7 mainly due to a similar decrease in k-1 [Eq. (1)]. Indeed, these two ligands modify to a different extent the protein conformation (as made apparent by protein fluorescence): this difference might be ascribed to the two alternative positions occupied by Tyr224 side chain in the 3D structure of hDAAO in complex with benzoate or with iminoserine (PDB ID 2DU8 and 2E49).1,16 Furthermore, the large change in protein fluorescence of the apoprotein in the presence of d-serine or CF3-d-Ala and the thermal stabilization following the binding of the pseudo-substrate clearly shows that the site for substrate binding is formed in the apoprotein form of hDAAO. Limited proteolysis studies demonstrate that FAD binding to the apoprotein quickly produces a compact protein whose conformation is similar to that of the hDAAO holoenzyme.15 Susceptibility of hDAAO to trypsinolysis is instead enhanced by CPZ binding, which also alters the protein fluorescence and the near-UV CD signal of the flavoenzyme differently than in benzoate binding: the binding of CPZ appears to favor a protein conformation resembling that of the apoprotein form.
Importantly, none of the ligands tested -CPZ, FAD, benzoate, and d-serine- modified the hDAAO-pLG72 complex formation, as judged by SPR and gel permeation analyses although CPZ binding increased the susceptibility to proteolysis.
Understanding the modulation of hDAAO enzymatic activity and stability in vivo (by its cofactor and/or ligands) is a crucial physiological and pathological issue that has not been clarified yet. At an estimated in vivo FAD concentration of ≈5 μM, hDAAO should be mainly in the apoprotein inactive form due to the weak cofactor binding. We here demonstrate that the amount of hDAAO holoenzyme is largely augmented in the presence of the substrate d-serine (or the pseudo-substrate CF3-d-Ala). All together, we conclude that hDAAO folding takes place first, and the cofactor binds to the already folded apoprotein where the active site is also formed: substrate binding increases FAD-apoprotein interaction and in turn the holoenzyme active form. Substrate or benzoate binding also increases hDAAO stability (Fig. 2), while the stability of the flavoenzyme is negatively affected by the binding of the protein pLG72 or the drug CPZ. In agreement with these results benzoate and CPZ affect the cellular concentration of hDAAO differently [Fig. 6(B,C)] although they modulate the short-term cellular d-serine concentration similarly. An increase in the D/(D+L) serine concentration ratio at 24 hours from treatment with both compounds is apparent (a similar result was also observed for plasma d-serine levels of rats treated with the hDAAO inhibitor pyrrole carboxylic acids),11 which is abolished at 48 hours [Fig. 6(A)]. Furthermore, the different effect exerted by benzoate and CPZ on hDAAO stability and cellular concentration might affect the in vivo levels of the gliotransmitter d-serine differently over time. This represents a further parameter to take into consideration during the development of new drugs to treat schizophrenia.
Materials and Methods
Enzymes and activity assay
Recombinant hDAAO and pLG72 proteins were expressed in E. coli cells and purified as reported.7,8 The EGFP-hDAAO protein form (see later) was also expressed in E. coli cells by using the pET20 plasmid: the recombinant enzyme was purified with a 35% yield and >85% purity as reported in.7 The final hDAAO preparation was in 20 mM Tris-HCl buffer, pH 8.0, 100 mM NaCl, 5% glycerol, 5 mM 2-mercaptoethanol, and 40 μM FAD and diluted in plain buffer without FAD before use. Apoprotein of hDAAO was prepared by overnight dialysis of the holoprotein against 1 M KBr as detailed in.7 The final pLG72 preparation was stored in 20 mM Tris-HCl, pH 8.5, 100 mM NaCl, 5% glycerol, and 5 mM 2-mercaptoethanol and contained ≈ 0.1% N-lauroylsarcosine (NLS).
Spectral measurements
Flavin and protein fluorescence measurements were performed in a Jasco FP-750 instrument. The kinetics of FAD binding to hDAAO apoprotein were investigated by using a BioLogic SFM-300 stopped-flow apparatus interfaced to the same Jasco fluorimeter.15 Protein emission spectra were taken from 300 to 400 nm (excitation at 280 nm); flavin emission spectra were recorded from 475 to 600 nm (excitation at 450 nm). Steady-state fluorescence measurements were performed at 0.1 mg/mL protein concentration and corrected for buffer contributions. Fixed wavelength protein and flavin fluorescence measurements were taken at 335 and 530 nm, respectively. Temperature ramp experiments were carried out by using the same instrumentation equipped with a software-driven Peltier-based temperature controller (temperature gradient of 0.5°C/min); data were analyzed by means of Jasco software.15 Circular dichroism (CD) spectra were recorded on a J-815 Jasco spectropolarimeter: cell pathlength was 1 cm for measurements above 250 nm (0.4 mg protein/mL) and 0.1 cm for measurements in the 190–250 nm region (0.1 mg protein/mL). All spectral measurements were carried out in 20 mM Tris-HCl pH 8.5, 150 mM sodium chloride, and 5% glycerol (a different buffer as compared to15) and at 15°C.
Limited proteolysis
Holo- and apoprotein samples of hDAAO (0.4 mg protein/mL, corresponding to ≈10 μM) were incubated at 25°C in 20 mM Tris-HCl, pH 8.5, 150 mM NaCl, 5% glycerol, 0.06% NLS, and 5 mM 2-mercaptoethanol, with 10% (w/w) trypsin as reported previously15 and in the presence of different compounds: pLG72, FAD, benzoate, CF3-d-Ala and CPZ. At various times after adding trypsin, aliquots (containing 7 μg of hDAAO) were diluted in sample buffer for SDS-PAGE, heated at 100°C for 3 min, and analyzed electrophoretically. The intensity of the protein bands was determined by densitometric analysis using the QuantityOne software (BioRad Lab.). Kinetic data for protein fragment formation/degradation were fit to a single or a double exponential decay equation by using KaleidaGraph™ (Sinergy Software).
Size-exclusion chromatography
Size-exclusion chromatography was performed at room temperature on a Superdex 200 column by means of an Äkta chromatographic system (Amersham Pharmacia Biotech), using 20 mM Tris-HCl, pH 8.5, 150 mM sodium chloride, 5% glycerol, 5 mM 2-mercaptoethanol as elution buffer and the following compounds added: 40 μM FAD, 0.1 mM CPZ, or 0.1 mM benzoate. The column was calibrated with suitable standard proteins.
hDAAO-pLG72 complex formation and oligomeric state were also determined by gel-permeation chromatography using the aforementioned buffer to which 0.06% NLS and 40 μM FAD, 0.1 mM benzoate or CPZ was added as the elution buffer (the detergent is required because the solubility and the oligomeric state of pLG72 strongly depends on the presence of NLS).9 The area of each peak was estimated by nonlinear curve fitting of the elution profile using PeakFit software (Systat Software, Germany); the error was <10% as estimated using known amounts of hDAAO as standard protein (5–100 nmoles/run). The amount of pLG72 and hDAAO present in the peak corresponding to the protein complex (≈12.8 mL) was estimated by means of the intensity of their bands following SDS-PAGE, as obtained using the program QuantityOne and known amounts of purified hDAAO and pLG72 as standard.9
SPR analysis
All experiments were performed on a Biacore T100 instrument (GE Healthcare) and materials were purchased from the same supplier if not otherwise specified. All the analyses were carried out at 25°C, employing 10 mM Hepes, 150 mM NaCl, 3.4 mM EDTA, 0.05% P20, and 0.01% NLS at pH 7.4 (HBS EP+ buffer) as running buffer. For protein immobilization, pLG72 or hDAAO diluted at 20 and 25 μg/mL, respectively, in 10 mM sodium acetate, pH 4.5, were covalently linked to two flow cells of a CM5 sensor chip through standard amine coupling chemistry. The pLG72 sensor chip was less stable and the coupling procedure was less reproducible than that for hDAAO, probably because of the low solubility of pLG72.7,9 For kinetic experiments, hDAAO was diluted in running buffer at concentrations ranging from 1 to 10 μM with or without 40 μM FAD (to evaluate the binding of both the apo- and the holoenzyme forms of hDAAO to immobilized pLG72). Analytes were flushed over the immobilized pLG72 for 120 s at a flow rate of 20 μL/min and the dissociation phase was followed for 300 s after shifting to running buffer. Otherwise, pLG72 diluted at concentrations ranging from 5 to 0.5 μM in running buffer was flushed over the immobilized hDAAO sensor chip for 180 s at a flow rate of 20 μL/min followed by a dissociation phase of 300 s. Surface regeneration was achieved with a 30-s pulse of a solution containing 0.5 M NaCl and 0.5% NLS. The evaluation of the effect of different compounds on the hDAAO-pLG72 interaction was performed by flushing pLG72 over a sensor chip with immobilized hDAAO in the presence of d-serine (50 mM), sodium benzoate (35 μM), CPZ (from 5 to 40 μM), or FAD (from 5 to 40 μM). For each analysis an empty flow cell was used as a blank reference and a flow cell with immobilized human serum albumin was used as negative control. The unrelated acylase and human serum albumin proteins were employed as negative control analytes.
Kinetic traces were analyzed by Biacore T100 evaluation software 1.1.1 using a 1:1 Langmuir model for hDAAO-pLG72 interaction. For interactions performed in the presence of small compounds, kinetic traces were evaluated by means of a model accounting for heterogeneous analyte interactions.20
Effect of benzoate and CPZ on cellular d-serine and DAAO concentrations
The cDNA fragment coding for hDAAO was inserted into the pEGFP-C3 (Clontech Laboratories) vector as reported in,9 in frame with the gene coding for EGFP (pEGFP-hDAAO-C3 plasmid). The U87 human glioblastoma cells (ATCC) were transfected by using the Fugene HD Transfection Reagent (Roche) as suggested by the supplier. The expression of the fusion protein was monitored by detecting GFP fluorescence. U87 Clones stably producing EGFP-hDAAO were then selected by adding 0.4 mg/mL G418 to the culture media. The same cells were then transiently transfected with a plasmid encoding for pLG72.9 The cellular d- and l-serine levels were determined using transfected cells treated with 10 or 100 μM benzoate, 10 μM CPZ, or plain buffer for up to 72 hours. At 0, 24, and 48 hours, 5 × 105 cells were suspended in 1 mL of ice-cold 5% trichloroacetic acid, sonicated, and centrifuged for 30 min at 13000 rpm. The soluble fraction was extracted and derivatized with o-phthaldialdehyde/N-acetyl-l-cysteine: d- and l-serine were resolved by HPLC chromatography on a 5-μm Waters C8 (4.6 x 250 mm) reversed-phase column.9
The amount of cellular hDAAO on the same U87 transfected cells was determined by two assays: a) by SDS-PAGE and Western blot analysis using anti-hDAAO antibodies raised against its N-terminal sequence (Davids Biotechnologie) and quantification by densitometric analysis, see previous and Ref. 9. The intensity of the band recognized by a goat anti-α-tubulin antibody (Santa Cruz Biotechnology) was used as internal control; and b) by DAAO activity measurements on transfected U87 cell crude extracts using the Amplex UltraRed assay kit (Invitrogen) based on the detection of H2O2 by the peroxidase-mediated oxidation of the fluorogenic dye, as reported in.9 To measure the entire hDAAO cellular content, the cell extracts were incubated in the presence of a large excess of FAD (10 μM) and d-serine (50 mM); for each sample a control without the substrate d-serine was also analyzed. DAAO activity was expressed as the difference in fluorescence emission between sample and control assay mixtures and reported as specific activity (units/mg protein). Protein content was determined using the Bradford reagent (Sigma).
The measurements were replicated at least six times for each condition and statistical analyses were performed using Kaleidagraph software (Synergy Software). Variation between groups was evaluated by one-way ANOVA, and post hoc significance tests were performed using a Student's t test. Significance was assessed at P < 0.05.
Acknowledgments
The authors thank Consorzio Interuniversitario per le Biotecnologie and the Centro di Ricerca in Biotecnologie per la Salute Umana (Università degli studi dell'Insubria) and for the technical help of Dr. M. G. Bernasconi.
Glossary
Abbreviations:
- Bz
benzoate
- CPZ
chlorpromazine
- CF3-d-Ala
trifluoro-d-alanine
- hDAAO
human d-amino acid oxidase (EC 1.4.3.3)
- NLS
N-lauroylsarcosine
- pkDAAO
pig kidney d-amino acid oxidase
- SPR
surface plasmon resonance
- Tm
melting temperature.
References
- 1.Pollegioni L, Piubelli L, Sacchi S, Pilone MS, Molla G. Physiological functions of D-amino acid oxidases: from yeast to humans. Cell Mol Life Sci. 2007;64:1373–1394. doi: 10.1007/s00018-007-6558-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Oliet SH, Mothet JP. Regulation of N-methyl-d-aspartate receptors by astrocytic D-serine. Neuroscience. 2009;158:275–283. doi: 10.1016/j.neuroscience.2008.01.071. [DOI] [PubMed] [Google Scholar]
- 3.Pollegioni L, Sacchi S. Metabolism of the neuromodulator D-serine. Cell Mol Life Sci. doi: 10.1007/s00018-010-0307-9. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 5.Ross CA, Margolis RL, Reading SA, Pletnikov M, Coyle JT. Neurobiology of schizophrenia. Neuron. 2006;52:139–153. doi: 10.1016/j.neuron.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 6.Chumakov I, Blumenfeld M, Guerassimenko O, Cavarec L, Palicio M, Abderrahim H, Bougueleret L, Barry C, Tanaka H, La Rosa P, Puech A, Tahri N, Cohen-Akenine A, Delabrosse S, Lissarrague S, Picard FP, Maurice K, Essioux L, Millasseau P, Grel P, Debailleul V, Simon AM, Caterina D, Dufaure I, Malekzadeh K, Belova M, Luan JJ, Bouillot M, Sambucy JL, Primas G, Saumier M, Boubkiri N, Martin-Saumier S, Nasroune M, Peixoto H, Delaye A, Pinchot V, Bastucci M, Guillou S, Chevillon M, Sainz-Fuertes R, Meguenni S, Aurich-Costa J, Cherif D, Gimalac A, Van Duijn C, Gauvreau D, Ouellette G, Fortier I, Raelson J, Sherbatich T, Riazanskaia N, Rogaev E, Raeymaekers P, Aerssens J, Konings F, Luyten W, Macciardi F, Sham PC, Straub RE, Weinberger DR, Cohen N, Cohen D. Genetic and physiological data implicating the new human gene G72 and the gene for D-amino acid oxidase in schizophrenia. Proc Natl Acad Sci USA. 2002;99:13675–13680. doi: 10.1073/pnas.182412499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Molla G, Sacchi S, Bernasconi M, Pilone MS, Fukui K, Pollegioni L. Characterization of human d-amino acid oxidase. FEBS Lett. 2006;580:2358–2364. doi: 10.1016/j.febslet.2006.03.045. [DOI] [PubMed] [Google Scholar]
- 8.Molla G, Bernasconi M, Sacchi S, Pilone MS, Pollegioni L. Expression in Escherichia coli and in vitro refolding of the human protein pLG72. Protein Expr Purif. 2006;46:150–155. doi: 10.1016/j.pep.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 9.Sacchi S, Bernasconi M, Martineau M, Mothet JP, Ruzzene M, Pilone MS, Pollegioni L, Molla G. pLG72 modulates intracellular D-serine levels through its interaction with D-amino acid oxidase: effect on schizophrenia susceptibility. J Biol Chem. 2008;283:22244–22256. doi: 10.1074/jbc.M709153200. [DOI] [PubMed] [Google Scholar]
- 10.Adage T, Trillat AC, Quattropani A, Perrin D, Cavarec L, Shaw J, Guerassimenko O, Giachetti C, Gréco B, Chumakov I, Halazy S, Roach A, Zaratin P. In vitro and in vivo pharmacological profile of AS057278, a selective D-amino acid oxidase inhibitor with potential anti-psychotic properties. Eur Neuropsychopharmacol. 2008;18:200–214. doi: 10.1016/j.euroneuro.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 11.Sparey T, Abeywickrema P, Almond S, Brandon N, Byrne N, Campbell A, Hutson PH, Jacobson M, Jones B, Munshi S, Pascarella D, Pike A, Prasad GS, Sachs N, Sakatis M, Sardana V, Venkatraman S, Young MB. The discovery of fused pyrrole carboxylic acids as novel, potent D-amino acid oxidase (DAO) inhibitors. Bioorg Med Chem Lett. 2008;18:3386–3391. doi: 10.1016/j.bmcl.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 12.Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, Hashimoto K, Tsukamoto T. Synthesis and biological evaluation of D-amino acid oxidase inhibitors. J Med Chem. 2008;51:3357–3359. doi: 10.1021/jm800200u. [DOI] [PubMed] [Google Scholar]
- 13.Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, Hu LY, El-Kattan A, James LC, Liu S, Lu J, Maklad N, Mansour MN, Mente S, Piotrowski MA, Sakya SM, Sheehan S, Steyn SJ, Strick CA, Williams VA, Zhang L. Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent D-amino acid oxidase (DAAO) inhibitors. J Med Chem. 2009;52:3576–3585. doi: 10.1021/jm900128w. [DOI] [PubMed] [Google Scholar]
- 14.Spector R. Riboflavin homeostasis in the central nervous system. J Neurochem. 1980;35:202–209. doi: 10.1111/j.1471-4159.1980.tb12507.x. [DOI] [PubMed] [Google Scholar]
- 15.Caldinelli L, Molla G, Sacchi S, Pilone MS, Pollegioni L. Relevance of weak flavin binding in human D-amino acid oxidase. Protein Sci. 2009;18:801–810. doi: 10.1002/pro.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawazoe T, Tsuge H, Pilone MS, Fukui K. Crystal structure of human D-amino acid oxidase: context-dependent variability of the backbone conformation of the VAAGL hydrophobic stretch located at the si-face of the flavin ring. Protein Sci. 2006;15:2708–2717. doi: 10.1110/ps.062421606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iwana S, Kawazoe T, Park HK, Tsuchiya K, Ono K, Yorita K, Sakai T, Kusumi T, Fukui K. Chlorpromazine oligomer is a potentially active substance that inhibits human D-amino acid oxidase, product of a susceptibility gene for schizophrenia. J Enzyme Inhib Med Chem. 2008;23:901–911. doi: 10.1080/14756360701745478. [DOI] [PubMed] [Google Scholar]
- 18.Umhau S, Pollegioni L, Molla G, Diederichs K, Welte W, Pilone MS, Ghisla S. The x-ray structure of D-amino acid oxidase at very high resolution identifies the chemical mechanism of flavin-dependent substrate dehydrogenation. Proc Natl Acad Sci USA. 2000;97:12463–12468. doi: 10.1073/pnas.97.23.12463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochim Biophys Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 20.Karlsson R, Jendeberg L, Nilsson B, Nilsson J, Nygren PA. Direct and competitive kinetic analysis of the interaction between human IgG1 and a one domain analogue of protein A. J Immunol Methods. 1995;183:43–49. doi: 10.1016/0022-1759(95)00030-e. [DOI] [PubMed] [Google Scholar]
- 21.Lang UE, Puls I, Muller DJ, Strutz-Seebohm N, Gallinat J. Molecular mechanisms of schizophrenia. Cell Physiol Biochem. 2007;20:687–702. doi: 10.1159/000110430. [DOI] [PubMed] [Google Scholar]
- 22.Tsai G, Van Kammen DP, Chen S, Kelley ME, Grier A, Coyle JT. Glutamatergic neurotransmission involves structural and clinical deficits of schizophrenia. Biol Psychiatry. 1998;44:667–674. doi: 10.1016/s0006-3223(98)00151-6. [DOI] [PubMed] [Google Scholar]