

Abstract
Over the past decade, the development and application of chemical genomic assays using the model organism Saccharomyces cerevisiae has provided powerful methods to identify the mechanism of action of known drugs and novel small molecules in vivo. These assays identify drug target candidates, genes involved in buffering drug target pathways and also help to define the general cellular response to small molecules. In this review, we examine current yeast chemical genomic assays and summarize the potential applications of each approach.
Keywords: yeast, chemical genomics, chemical biology, drug target, drug action
1. Introduction
Current approaches to drug discovery are typically target-oriented, making use of validated targets as the starting point for discovery and development efforts. In practice, promising targets are selected based on several criteria including: 1) prior knowledge of a targets biological role(s) and potential for therapeutic intervention 2) proven value based on approved drugs (i.e. “me too” targets) 3) a target’s essentiality for cell growth and 4) druggability (Hopkins & Groom, 2002). As a consequence of these constraining criteria, the selection of targets is biased toward well-characterized proteins or pathways. Once a target has been selected in this manner, biochemical assays are developed so the target can be screened in a high-throughput assay. Because these assays are performed in vitro using purified components, once an identified lead compound is assessed for cellular activity, the contributions and consequences of other potential protein-compound interactions are not predictable.
During the past two decades, target-based approaches to drug discovery have produced novel lead compounds and therapeutic candidates, yet the overall approval rate for new chemical entities has remained flat despite the exponential increase in research development costs (Higgins & Graham, 2009). Due in part to this lack of increased productivity, cell-based phenotypic screens have gained renewed interest. Advantages of cell-based screens include 1) identified compounds are cell-permeable and 2) sophisticated tools are available to screen a wide range of desired phenotypes. However, a major challenge for cell-based assays is that once a compound producing the desired phenotype is identified, the cellular target of the compound must be determined (Chan, Nislow, & Emili, 2009). New technologies and experimental approaches for identifying drug targets have been developed including in silico docking approaches (Teotico, et al., 2009), computational predictions (Keiser, et al., 2009; Song, Lim, & Tong, 2009), novel compound derivation strategies (Schreiber, 2000; Stockwell, 2004), chemical proteomics (Rix & Superti-Furga, 2009) and many others which have been the subject of several recent reviews (E. C. Butcher, Berg, & Kunkel, 2004; J. W. Li & Vederas, 2009; Mandal, Moudgil, & Mandal, 2009; Quon & Kassner, 2009; Wagner & Clemons, 2009). However, most of these approaches are not yet amenable to genome-wide approaches to identify targets in vivo. This review focuses on the in vivo chemical genomic assays developed in the yeast Saccharomyces cerevisiae and how these tools allow the relative sensitivity of all potential drug targets to be measured simultaneously, to identify candidate drug-target interactions.
The model organism Saccharomyces cerevisiae has been a test bed for the development of virtually all “omics” techniques (Bader, et al., 2003; Costanzo, Giaever, Nislow, & Andrews, 2006; Dixon, Costanzo, Baryshnikova, Andrews, & Boone, 2009; Provart & McCourt, 2004; Rual, Hill, & Vidal, 2004; Sidhu, Bader, & Boone, 2003; Snyder & Gallagher, 2009). The S. cerevisiae genome and proteome is extremely well-characterized (Pena-Castillo & Hughes, 2007) due to its rapid generation time, inexpensive cultivation and facile genetics. Recent molecular genetics efforts have produced a complete molecular-barcoded gene deletion collection (Giaever, et al., 2002; Winzeler, et al., 1999). Because of these experimental attributes, S. cerevisiae will continue to be a major player in biological studies aimed at understanding proteins and pathways that can be modulated to ameliorate disease (Dixon & Stockwell, 2009). Yeast can also be used to model processes in metazoans, e.g. approximately 45% of the genes in yeast are homologous to mammalian genes (BLAST e-value <10-10) (Hughes, 2002), encouraging efforts aimed at translating assays and results from yeast to metazoans (Chervitz, et al., 1998).
Despite its numerous advantages, yeast assays are not without limitations for the purposes of drug discovery. Principal among these is the high concentration of compound often required to produce a biological response, likely due to the barrier presented by the cell wall, and the presence of numerous active efflux pumps and detoxification mechanisms (Cowen & Steinbach, 2008; Leppert, et al., 1990; Miyahara, Mizunuma, Hirata, Tsuchiya, & Miyakawa, 1996; Molin, Norbeck, & Blomberg, 2003; Wehner, Rao, & Brendel, 1993). In addition, although many core processes are conserved between yeast and human, several “metazoan-specific” processes are not. Nonetheless, a number of labs have designed clever screens to study processes such as neurodegeneration (Miyano, 2005), diabetes (Kohlwein, 2010), and angiogenesis (McGary, et al., 2010) in yeast models.
2. Drug-induced HaploInsufficiency Profiling (HIP)
The Yeast KnockOut (YKO) collection consists of a complete set of deletion strains, including haploid strains of both yeast mating types and heterozygous and homozygous diploid deletions. Each strain carries a precise start to stop deletion of a single gene (Giaever, et al., 2002; Winzeler, et al., 1999). A key feature of these collections is that each deletion strain is tagged or “barcoded” with two unique 20 base pair sequences that serve as strain identifiers. These collections can be pooled and grown competitively in any condition of choice which allows the identification of genes most important for growth in a given condition (e.g. compound/drug treatment) because strains carrying deletions of these genes will become depleted from the pool over time. The relative abundance of each strain is measured by the abundance of the barcodes. Specifically, following pooled cell growth, genomic DNA is extracted from cells, barcodes are PCR amplified using the primers common to every strain, and relative strain abundance quantified based on hybridization signal from a DNA barcode microarray (TAG4 microarray; Affymetrix part no. 511331) containing the barcode complements (Giaever, et al., 2004; Pierce, et al., 2006; Winzeler, et al., 1999) (Figure 1A). Alternatively, barcodes can be detected by next-generation sequencing (Smith, et al., 2009). Barcodes that decrease in abundance over the time course of the experiment versus the control identify strains deleted for genes required for survival in the tested condition.
Figure 1.
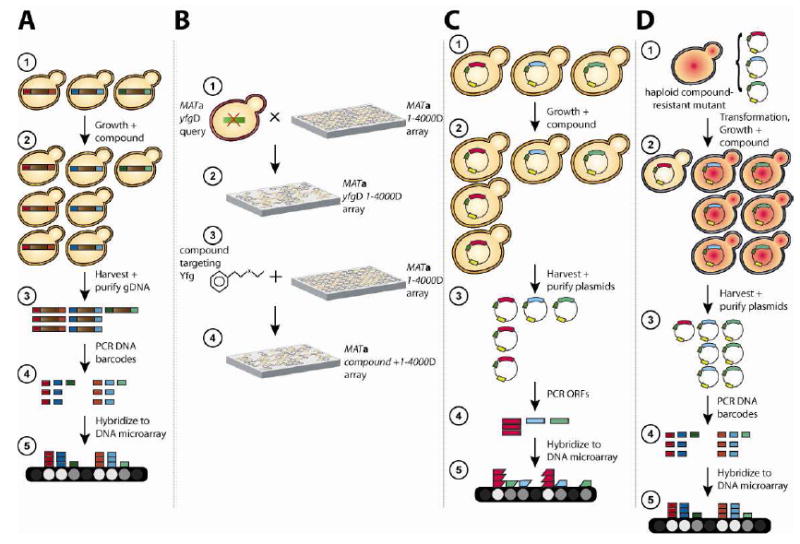
A) Diagram of the HIPHOP assay
(1) The yeast deletion collection is pooled and each strain is included at approximately equal abundance. (2) The pool is grown competitively in a compound of choice. (3) Genomic DNA is isolated from the pooled compound treated sample. (4) Up and down barcodes are PCR amplified in 2 separate reactions. (5) The PCR product is hybridized to a TAG4 barcode microarray to assess relative abundance of each strain by hybridization intensity. The intensity on the microarray serves as a proxy for strain abundance, intensities that are significantly reduced compared with the control identify strains sensitive to compound.
B) Comparison of genetic interactions and compound-gene interactions
(1) A query strain consisting of a mutation in Your Favourite Gene (yfgΔ) is crossed into an ordered array of ~4,000 non-essential deletion strains (designated as 1-4000Δ) of the opposite mating type using the Synthetic Genetic Array (SGA) protocol and (2) the resulting double mutant haploid progeny are selected on plates containing the appropriate media (Tong & Boone, 2006). Colony size is used to identify those strains that are reduced in sized and represent genetic interactions. (3) The same ordered array of ~4,000 non-essential deletion strains, as in (1), is pinned onto plates containing drug targeting Yfg. (4) Colony size is used to identify those colonies that are reduced in sized and therefore identify deletion mutants sensitive to compound.
C) Multi-copy suppression profiling
(1) An ORFeome library constructed by one of several methods (See Table 1) is transformed en masse, into a wildtype yeast strain. (2) The resulting pool is grown in a compound of choice. (3) Plasmid DNA is isolated. (4) Inserts are amplified using plasmid primers that flank each insert. (5) Amplicons are then labeled using a biotin labeling mix and a Klenow fragment, to generate short strands of labeled DNA molecules (denoted as coloured trapezoids), that are hybridized to a TAG4 microarray carrying the complementary ORF-specific probes. The trapezoid generated from the ORFs are used to distinguish them as ORF probes from the rectangular barcode amplicons in Figure 1A. In this scenario, intensities that are significantly increased on the array compared to the control identify ORFs that confer drug resistance.
D) Complementation of compound resistant mutants
(1) A haploid drug resistant (resistance designated as the red-hue in the yeast) strain is isolated and confirmed that the resistant phenotype is recessive by crossing to a wildtype haploid strain to verify that drug sensitivity is restored. (1) The original resistant strain is transformed with the MoBY-ORF library. (2) The resulting pool is grown in a high concentration of drug; strains that are sensitive due to complementation by plasmid are depleted from the pool. (3) Plasmid DNA is isolated. (4) Barcodes are amplified using universal primers that flank each barcode. (5) Amplicons are hybridized to a TAG4 microarray carrying the complementary barcode probes. Intensities that are significantly reduced compared with the control identify strains that harbor the ORF carried by plasmid responsible for drug resistance. In a complementary approach, dominant compound-sensitive strains can be transformed with the MoBY-ORF library and those ORFs that render the mutant compound-sensitive can be identified.
Drug-induced HaploInsufficiency Profiling or HIP was one of the first assays to take advantage of this parallelized growth strategy. HIP is based on the observation that a heterozygous deletion strain is specifically sensitized to a drug that targets the product of the heterozygous locus (as measured by a decrease in growth rate or fitness) (Giaever, et al., 1999). When all possible heterozygous deletion strains are screened in parallel the heterozygous deletion strain most sensitive to a particular drug often identifies the drug target(s) (Giaever, et al., 2004; Giaever, et al., 1999; Lum, et al., 2004). A key advantage of this assay is that it simultaneously identifies both the inhibitory compound and its candidate targets without prior knowledge of either. These candidate targets represent genes most important for growth and are therefore relevant for identification of antiproliferative targets that may have potential in antifungal or oncology indications. The feasibility and robustness of this assay has been demonstrated by screening well-characterized and novel compounds (Giaever, et al., 2004; Giaever, et al., 1999; Hillenmeyer, et al., 2008; Lum, et al., 2004; Pierce, Davis, Nislow, & Giaever, 2007; Smith, et al., 2009; St Onge, et al., 2007; Yan, et al., 2008). In addition, screens in which the direct target does not exist in yeast (e.g. neuropleptic agents), provide insight into the off-target mechanism of action (Ericson, et al., 2008).
An alternative to the competitive pooling approach is to pin the heterozygous (or other) yeast strain collections onto agar plates that contain a compound of interest and to monitor fitness based on colony size (Baetz, et al., 2004; Carroll, et al., 2009). A drawback of plate-based assays is that they require significantly more compound (1 to 2 orders of magnitude) versus miniaturized, pooled liquid assays. Although it is still a matter of debate how well growth data obtained from liquid or solid media correlate, recent data show that genetic interactions monitored by growth in liquid media correlate well with interactions identified on solid media when using robotic plating combined with sophisticated data analysis (Costanzo, et al., 2010).
Despite the successes of HIP, in some cases, reducing gene copy by half (in heterozygotes) may be insufficient to identify drug targets and gene dose must be further decreased. To address this, Yan et al. (2008) barcoded a yeast allele collection of haploid essential gene mutants, the DAmP (Decreased Abundance by mRNA Perturbation) where a drug resistance marker is placed upstream of the 3’ UTR (UnTranslated Region) of each gene. These DAmP truncations or strains have been shown to express, on average, about 10% of the wild type protein levels (Schuldiner, et al., 2005). This collection of hypomorphic alleles allowed detection of drug-induced haploinsufficiency not observed in the heterozygote case (Yan, et al., 2008), thereby broadening the ability of the assay to identify compound target candidates. Because the collections of heterozygote and DAmP essential alleles carry non-overlapping barcode sequences, both assays can be performed in parallel (and hybridized to the same microarray), resulting in an increase in the dynamic range and sensitivity of the pooled assays. If, however, the target of a drug requires a dosage decrease beyond that of the heterozygous case this suggests that the target that is well-characterized does not represent the primary mechanisms of drug action. Alternatively, inhibiting the primary target may induce sensitivity in a large number of additional strains, thereby confounding interpretation of the screen.
Haploinsufficiency Profiling is a timely and powerful approach, particularly in light of recent studies in which it has become apparent that few drugs target single gene products (e.g. imatinib (Gleevec) (Buchdunger, et al., 1996; Druker, et al., 1996)), therefore, an in vivo view of the relative sensitivity of all targets in the cell is invaluable to understand the complete mechanism of drug action. Yeast cells obviously differ from human cells, accordingly any human targets that lack a yeast homolog will not be identified. Moreover, the HIP assay relies on a growth phenotype resulting from drug/target binding; targets whose inhibition does not affect growth will not be identified. Despite these caveats, based on our screening of >2000 compounds, we have not yet failed to identify a target in yeast when that target is 1) well-characterized and 2) target inhibition impairs cell growth. We do observe off-target effects that likely reflect actual in vivo interactions. As mentioned, decreasing gene dosage by a single copy may not be sufficient to reveal drug-induced haploinsufficiency for a particular target. In principle, further lowering gene dose may reveal the true target when simply raising the compound concentration would not, due to general cellular toxicity which could obscure the results. However, the failure to detect a target as a heterozygote (and only in a more severe DAmP or temperature-sensitive allele) may imply that the suspected/known target is actually not the major mechanism of action of a particular compound. For example, 5-FU is thought to act by inhibiting Cdc21. However, yeast lacks thymidine kinase and therefore the inhibition of Cdc21 can only occur indirectly through a series of metabolic interactions (Goodman, Hardman, Limbird, & Gilman, 2001). Indeed, genome-wide yeast assays reveal primary mechanism of action is via misincorporation of fluorinated nucleotides into RNA(Giaever, et al., 2004; Goodman, et al., 2001; Lum, et al., 2004; Scherf, et al., 2000). Finally, targets that are either not essential and/or are highly redundant are unlikely to be detected in the loss of function assays because inhibition of all homologs would be required.
3. Homozyous Profiling (HOP)/Haploid deletion chemical-genetic profiling
Homozygous profiling (HOP) is analogous to the HIP assay, except that the strains are completely deleted for non-essential genes in either haploid or diploid strains. Relative growth rate, in the condition of choice (e.g. drug treatment), is measured by microarray signal intensity as described above.
In the HOP or haploid assays (Hillenmeyer, et al., 2008; Lee, et al., 2005; Parsons, et al., 2004; Parsons, et al., 2006), strains most sensitive to a drug become depleted from a pool over time, as in the HIP assay. However, because these strains carry complete deletions of non-essential genes they do not identify the target directly because the target is absent. Rather, these assays identify genes that act to buffer the drug target pathway and are therefore required for growth in the presence of compound. This assay can be particularly informative for compounds that lack a direct protein target. For example, genes involved in the DNA damage response, while non-essential under standard growth conditions, are required for survival when challenged with DNA damaging agents (Birrell, Giaever, Chu, Davis, & Brown, 2001; Chang, Bellaoui, Boone, & Brown, 2002; Lee, et al., 2005; Workman, et al., 2006; L. Yu, et al., 2008). For example, one study (Lee et al., 2005) defined the relative importance of different DNA-repair modules for resistance to 12 DNA damaging agents and revealed functional interactions that comprise the DNA-damage response. While many of these compounds share similar mechanisms of actions (e.g. a subset were alkylating agents), each compound produced a unique genome-wide profile, or “signature”. By screening a collection of compounds across non-essential genes, these genome-wide profiles can be clustered which allow one to infer the mechanism of action (Parsons, et al., 2004; Parsons, et al., 2006) when compared to those profiles obtained from drugs with well-characterized mechanisms. Like HIP, this assay can be performed either competitively in pools using barcode-based assays (Giaever, et al., 2004; Giaever, et al., 1999; Hillenmeyer, et al., 2008; Parsons, et al., 2006; Pierce, et al., 2007; Smith, et al., 2009; St Onge, et al., 2007; Xu, et al., 2009; Yan, et al., 2008) or on agar plates where drug sensitivity is measured by colony size (Parsons, et al., 2004).
A related approach for identifying drug-target interactions involves correlating HOP profiles with Synthetic Genetic Analysis (SGA) profiles (Tong, et al., 2001; Tong, et al., 2004) where a conditionally essential gene is used as a query gene to create comprehensive double mutant collections (Costanzo, et al., 2010). In this case, genetic interactions identified often correlate with non-essential deletion strains detected by HOP in the presence of drug, and the essential gene used as a query can be inferred to be the drug target (Figure 1B). A recent example of the power of this approach indentified Ero1 as the target of a novel small molecule (Costanzo et al., 2010). In a variation of this approach, Carroll et al. (2009) screened a yeast mutant collection to probe the mechanism of action of the yeast K28 toxin. In this screen, the inhibition of growth by secreted K28 toxin was monitored using a traditional halo assay to identify novel genes involved in cellular pathways essential for the response to this toxin (Carroll, et al., 2009).
Because HIP and HOP assays are complementary, combining the results of both heterozygous and homozygous/haploid loss-of-function chemical genomic screens can be particularly powerful for understanding the mode-of-action (MOA) of compounds. A caveat of all HIP and HOP-based screens (and other cell-based screens) is that while these assays screen compound against all potential targets simultaneously, definitive demonstration of a drug-target interaction requires independent confirmatory approaches such as in vitro binding or activity assays (Chan, et al., 2009).
4. Multi-copy Suppression Profiling (MSP)
One approach to identify or confirm a drug-target interaction is to demonstrate that overexpression of the target in vivo confers resistance to drug (R. A. Butcher, et al., 2006; Hoon, et al., 2008; X. Li, et al., 2004; Rine, Hansen, Hardeman, & Davis, 1983). In a feasibility study demonstrating that drug targets can be identified de novo, Rine et al. (1983) used a high copy plasmid carrying randomly generated yeast genomic inserts to identify genes that, when overexpressed, conferred resistance to tunicamycin when plated on solid media containing this compound. Plasmids were then isolated from resistant colonies and sequenced to identify ALG7, which encodes the known target of tunicamycin. This approach has been miniaturized to use pools of strains in liquid culture screened in parallel in a manner analogous to the HIP assay (Hoon, et al., 2008). Specifically, a high copy plasmid collection containing yeast genomic DNA fragments (with genes expressed from native promoters) is screened in yeast at high inhibitory concentrations of compounds (e.g. doses that inhibit wild type yeast by ~90%). Strains are grown competitively in compound, such that only one or a few strains that confer resistance are selected from the population. Plasmids are then isolated from resistant cells, and inserts are amplified by PCR and hybridized to a DNA TAG4 microarray carrying probes complementary to each yeast open reading frame (ORF). Microarray signal intensities are determined (Hoon, et al., 2008) (see Figure 1C) and resistance scored by comparing strain abundance between drug treatment pools and untreated reference pool. This approach correctly identified Dfr1, Erg11 and Tor1 as the targets of methotrexate, fluconazole and rapamycin, respectively. A caveat when using this approach as currently described is that drug pumps or other “indirect” targets may dominate the set of strains resistant to compound. Creating similar overexpression libraries in diverse drug pump resistant mutants can alleviate this challenge (Paulsen, Sliwinski, Nelissen, Goffeau, & Saier, 1998).
Several recently constructed libraries offer advantages over the traditional genomic DNA library used by Hoon et al. (2008). The Yeast Genome Tiling collection (Jones, et al., 2008) contains overlapping fragments of the yeast genome (~10Kb in size) cloned into high-copy 2μ vectors. The ends of each insert of this library have been sequenced, and the plasmids organized in a tiling fashion across the yeast genome, ensuring near-saturation (97.2%) coverage of the yeast genome. However, because each insert contains multiple genes, once a resistant fragment is identified, the exact gene target must be subcloned and confirmed. Another library consists of 3,900 yeast strains, each carrying a plasmid containing a single yeast ORF under expression of the GAL1 promoter (available from http://www.hip.harvard.edu/). Butcher et al. (2006) performed a proof-of-principle experiment with this library using the immunosuppressant rapamycin and found that plasmids carrying the Target Of Rapamycin (TOR) genes were correctly identified as conferring the greatest level of resistance (R. A. Butcher, et al., 2006; Chiu, Katz, & Berlin, 1994). Sopko et al. (2006) created a yeast library consisting of over 80% of all yeast ORFs, with gene expression controlled by a galactose-inducible promoter (Zhu, et al., 2001), such that high-levels of expression can be obtained (Johnston, 1987). A benefit of using an inducible system is that gene expression can be induced at specific times during the course of an experiment. On the other hand, galactose induction often does not accurately reflect endogenous gene expression levels, and overexpression can cause toxicity to the host cell (Sopko, et al., 2006). Because PCR was used to create this library, the ORFs may contain PCR-induced mutations, a concern addressed with a new, fully sequenced library (Hu, et al., 2007). Arguably, the optimal ORF library for MSP is the Molecular-Barcoded Yeast Open Reading Frame (MoBY-ORF) collection where each plasmid includes individual yeast ORFs flanked by their endogenous 5’ and 3’ UTRs, representing 90% of all yeast ORFs (Ho, et al., 2009). Because these plasmids are CEN-based, copy number is low (1-3 copies/cell) and predictable (Apostol & Greer, 1988), minimizing instances of overexpression toxicity. Each ORF in the MoBY collection is linked to the same two DNA barcodes associated with the corresponding deletion strain from the YKO permitting abundance measurements by microarray hybridization or sequencing (Ho, et al., 2009) (see Table 1 for a summary of yeast ORF libraries).
Table 1.
List of yeast plasmid libraries
| MoBY-ORF library | Yeast Tiling Collection | Random genomic fragment libraries | Galactose inducible libraries | |
|---|---|---|---|---|
| Sequence verified | √ŧ | √+ | - | √* |
| PCR free | - | √ | √ | - |
| Native promoter and terminator | √ | √ | √ | - |
| High Copy plasmid | - | √ | √ | √ |
| Low Copy plasmid | √ | - | - | - |
| Inducible promoter | - | - | - | √ |
| Barcoded | √ | - | - | - |
| Percent genome coverage | 90% | 97.2% | ~85% | 80% |
| Average fragment size | 2Kb | 10Kb | 5Kb | 1.5Kb |
| Number of genes per fragment | 1 | 4-6 | 2-3 | 1 |
| References | (Ho, et al., 2009) | (Jones, et al, 2008) | (Hoon, et al., 2008) | (Hu, et al., 2007; Zhu, et al., 2001) |
barcodes/partial ORFs sequenced
clones end-sequenced
library of Hu et al. (2007) has been fully sequenced, but other libraries are only partially sequenced including Zhu et al. (2001) and Ho et al. (2009).
A caveat (and occasional advantage) of these yeast clone banks is that a subset of genes, which when overexpressed, are toxic to yeast. 15% of yeast genes are toxic to the cell when overexpressed (Sopko, et al., 2006). This cohort of toxic genes can be informative regarding how regulation of gene products can alter cell physiology and they can be used as a starting point for chemical suppressor screens to identify compounds that suppress the toxicity and which, by extension, may interact with that toxic gene product. Chemical suppression has been successfully employed to find inhibitors of the bacteria Pseudomonas aeruginosa by overexpressing Pseudomonas genes in yeast (Arnoldo, et al., 2008). Tugenreich et al. (2001), similarly identified human genes that, when expressed in yeast, result in a growth defect (Tugendreich, et al., 2001). The authors selected a p38 overexpressing strain from a collection of “toxic” human genes to screen commercial libraries to identify chemical suppressors of the fitness defect which represent potential p38 inhibitors.
An additional application of the MoBY-ORF library is the identification of recessive genes responsible for drug resistance (Ho, et al., 2009). After a recessive drug resistant mutant strain is identified, it is transformed with the MoBY-ORF collection (Ho, et al., 2009) and complementation by one or more wildtype alleles from the collection that restores drug-sensitivity identifies the recessive gene(s) conferring drug resistance (see figure 1D). The feasibility of the assay was demonstrated by identifying fpr1 as a mutant resistant to the drug rapamycin (Heitman, Movva, & Hall, 1991; Sabatini, Erdjument-Bromage, Lui, Tempst, & Snyder, 1994). This assay complements the MSP assay in that it identifies drug targets in cases where the compound must interact with another protein to become toxic. In this case, rapamycin binds to Fpr1 to form a toxic complex, which, in turn, inhibits the Tor1 protein (Heitman, et al., 1991; Sabatini, et al., 1994). Complementation of recessive drug resistant alleles can also be used to systematically uncover general and specific resistance mechanisms. For example, Ho et al. (2009) used MoBY-ORF complementation to identify an essential enzyme in the ergosterol biosynthesis pathway as resistant to the natural product theopalauamide. Subsequent confirmations indicated that theopalauamide binds to ergosterol, defining a novel class of sterol-binding compounds.
MSP is flexible in that it can be used with diverse genomes. For example, ORF libraries exist for several organisms, all of which can be cloned into yeast expression vectors and expressed in yeast. As a test case, we used a genomic DNA library from Candida albicans expressed in Saccharomyces cerevisiae to identify a Candida ortholog of Saccharomyces cerevisiae Glc7 (a type 1 protein phosphatase) as resistant to the phosphatase inhibitor calyculin A (Hoon, et al., 2008). Indeed, yeast mutants have been rescued using human genes by several groups to identify human gene function by complementation (Mushegian, Bassett, Boguski, Bork, & Koonin, 1997; Osborn & Miller, 2007; Tugendreich, et al., 2001; Zhang, et al., 2003).
5. Comparative expression profiling
The transcriptional response of yeast cells to drug can correlate with the transcriptional response of strains deleted for the drug’s target. In a proof-of-principle study, Marton et al. (1998) demonstrated that the expression profile of cells treated with 3-amino triazole (an inhibitor of the His3 protein) correlates with that obtained from a his3Δ mutant (Marton, et al., 1998). This correlation does not, however, always hold, for example the authors found that FK506 treatment manifested a gene expression profile that correlated poorly (r = -0.23) with that observed for a fpr1 mutant, the known protein target of FK506 (Marton, et al., 1998). In a subsequent study, this transcriptional comparison approach was performed systematically using 300 mutant expression profiles and several dozen drug treatments, to create a compendium of gene expression profiles (Hughes, et al., 2000). The authors demonstrated that transcription profiles obtained from mutants correlated with the expression profiles of several well-characterized drugs. An advantage of this assay is that the drug does not necessarily need to inhibit yeast growth to cause a transcriptional response. However, the environmental stress response can often overwhelm an expression profile, particularly when samples are collected shortly after the drug is applied. To be comprehensive, this method would require transcription profiles for all deletion alleles and drug profiles under a variety of concentrations to be robust and predictive. Moreover, the biological differences between a knock-down (characteristic of drug inhibition or RNA interference) versus a knockout are significant.
Recently, Golub and colleagues completed a comprehensive study that analyzed the transcriptional response of diverse human cell lines to a library of small molecule inhibitors, to produce “The connectivity map” (C-map) (Lamb, et al., 2006). The authors used pattern matching to examine these transcription profiles for commonalities and demonstrated they could identify transcriptional signatures shared by drugs with common MOAs. This original C-map suggested novel connections between human genes and therapeutic compounds. The success of the C-map (and its on-going efforts) demonstrates another example of how concepts and methods are originally developed in yeast and more importantly, are transferable to assays in other organisms. For a complete summary of advantages of different yeast chemogenomic approaches see Table 2.
Table 2.
List of yeast chemogenomic applications
| HIP | DAmP | MSP | Complementation of drug resident mutant | Transcriptional profiling | Chemical suppression | Chemical Genetic interactions HOP | |
|---|---|---|---|---|---|---|---|
| Genome wide screen | √ | - | √ | √ | √ | √ | √ |
| Identification of drug target | √ | √ | √ | √ | - | √ | - |
| Identification of buffering pathways | - | - | - | - | √ | - | √ |
| Minimal drug consumption | √ | √ | √ | - | - | √ | √ |
| Performed in vivo | √ | √ | √ | √ | √ | √ | √ |
| Compound must inhibit cell growth | √ | √ | √ | √ | - | √ | √ |
| References | (Giaever, et al., 2004; Giaever, et al., 1999; Hillenmeyer,et al., 2008; Lum, et al.,2004; Pierce, et al., 2007; Smith, et al.,2009; St Onge, et al., 2007) | (Breslow, et al., 2008; Yan, et al., 2008) | (R. A. Butcher, et al., 2006; Hoon, et al., 2008; X. Li, et al.,2004; Rine, et al., 1983) | (Ho, et al.,2009) | (Hughes, et al., 2000; Marton, et al., 1998) | (Arnoldo, et al., 2008;Tugendreich,et al., 2001) | (Hillenmeyer,et al., 2008; Lee, et al.,2005; Parsons,et al., 2004; Parsons, et al.,2006) |
6. Looking ahead: Yeast chemical genomics and its translation to other model systems
Chemical genomic tools developed in yeast have contributed to our understanding of compound and drug mechanisms. Given current progress, chemogenomic methods, yeast-based and otherwise, are advancing the field of drug discovery. Table 3 describes several examples where yeast chemogenomic approaches were used to identify different compound targets. There are numerous additional examples of these assays identifying novel chemicals that are effective tools to probe biological function (e.g. Dorer, et al., 2005). A more difficult question to answer with certainty, is how many compounds, identified as yeast inhibitors have been developed into approved drugs? A complete answer to this question would require access to privileged Pharmaceutical and Biotech Company programs. Several small and large drug discovery companies licensed key yeast assay patents (Davis, Giaever and Shoemaker, USPTO 6,046,002 and Roemer et al., USPTO 6,783,985) so one can infer that these assays have been utilized for discovery purposes. An often-cited example of yeast contributing to a novel drug, is rapamycin, which targets yeast Tor1/Tor2 (Heitman, et al., 1991) and more recently, a yeast assay was essential in establishing the target of AN260, an antifungal agent that acts against tRNA synthetase and currently in phase 2 clinical trials (Rock, et al., 2007).
Table 3.
Known drug-target pairs confirmed using yeast chemical genetic methods
proposed target
novel compound and target
The past two decades have been enormously fruitful for yeast functional genomics, due in large part, to the rich genetic history of this model organism. We speculate that one feature of the next decade of yeast “omics” especially as it relates to our understanding of human disease will come from the integration of the diverse genomic datasets that have been generated. Indeed the discipline of “interactomics” (H. Yu, et al., 2008) and the concept of the “diseaseome” (Goh, et al., 2007) are combining to help predict the global effects of cellular perturbations and remedies at a systems level.
The primary advantage of yeast in contributing to drug discovery is the ability to identify the mechanism of action of compounds where they are not known. An additional advantage is the ability to assess all targets in the cell simultaneously in vivo; yeast is currently the only system where this is possible. This is of particular importance in repurposing already approved drugs and identifying novel, specific compounds. These compounds would not identify drug candidates themselves, but certainly have potential to be developed into drug candidates by medicinal chemistry and pharmacokinetic optimization. A second strength of yeast with respect to drug discovery is the unbiased identification of novel targets that are presumed “undruggable” by the pharmaceutical industry and are therefore not actively perused in other venues.
The identification of novel chemical probes in yeast assays will provide tools to probe novel biology. Accordingly, one of the key contributions from yeast may not necessarily be drugs in the clinic, but the starting point for development efforts. Specifically, such chemical probes that exhibit a high degree of specificity (but which require optimization by medicinal chemistry beyond the scope of many academic labs (Frye, 2010)) will, 1) identify novel druggable targets, 2) identify novel chemical scaffolds for medicinal chemical consideration 3) encourage efforts to investigate conserved targets and target pathways in human cells and 4) allow for modeling of such processes in human cells. Indeed, the movement away from therapeutic “magic bullets” which may be too specific and result in on-target toxicities (Liebler & Guengerich, 2005) towards magic shotguns where multiple targets are simultaneously targeted (Liebler & Guengerich, 2005), has led to a renewed interest in both multi-target drugs and combination drug therapies (Cirstea, Vallet, & Raje, 2009; Potti, et al., 2006). Given the enormous number of combinatorial possibilities to test, yeast is poised to make significant contributions in the area as thousands of potential combinations can systematically be tested in diverse sensitized backgrounds quite rapidly (Lehar, Stockwell, Giaever, & Nislow, 2008).
Recently, methods originally developed in yeast have been applied to other cellular systems. As described above, MSP has been used successfully with libraries of yeast ORFs to detect resistance to compounds. MSP can be expanded to other organisms, for example the human ORFeome collection (Lamesch, et al., 2007; Rual, Hirozane-Kishikawa, et al., 2004) can be overexpressed in yeast to identify human genes that confer resistance to compound (unpublished data). Recently, a collection of Escherichia coli overexpressing essential genes has become available and used in an MSP assay in E. coli by Pathania et al. (2009) to identify targets of growth inhibitory compounds. This method is advantageous because it overexpresses genes in the native host, allowing for proper post-translational modifications.
To understand human biology by direct observation in the relevant cellular context, loss-of-function assays, analogous to the HIP assay, have been developed for mammalian cells. Various RNAi (RNA interference) assays have been employed to knock down gene expression to understand gene function (Moffat, et al., 2006; Silva, et al., 2005). Because the number of siRNA (small interfering RNAs) is quite large (50,000-100,000+), several laboratories have adopted various pooled screening strategies (Schlabach, et al., 2008; Silva, et al., 2008). These screens have proven useful for identifying and assigning gene function. For example, a screen of diverse human cell lines using a pooled RNAi library identified many genes that, when knocked-down, showed anti-proliferative effects, some of which were cell line specific and others that were universal across many cell lines (Schlabach, et al., 2008; Silva, et al., 2008). While these screens are, not surprisingly, more labor-intensive than those in yeast (they require long culture times and analyzing the effects of siRNA knockdowns is more complex than that required for complete knockout alleles) the success of yeast chemogenomic screens inform the design and encourage the development of these mammalian screens. Using this approach, McManus et al. (2009), demonstrated that yeast synthetic lethal interactions could be used to prioritize human genetic interactions by testing for interactions amongst homologous genes in human cell lines (McManus, Barrett, Nouhi, & Hieter, 2009).
A recent application of pooled RNAi technology was demonstrated in a series of chemical synthetic lethality screens in human cancer cell lines (Luo, Emanuele, et al., 2009; Scholl, et al., 2009). Luo et al. (2009) screened a cell line whose oncogenic phenotype depended on the k-Ras mutation with a pool of interfering shRNAs to identify potential synthetic lethal gene pairs (Luo, Emanuele, et al., 2009). The genes identified represent potential cancer-specific vulnerabilities, which can be mimicked with drugs that specifically inhibit these proteins. The authors demonstrated that a PLK1 inhibitor had selectivity toxicity in this cell line, in both in vitro and in vivo models (Luo, Emanuele, et al., 2009). Synthetic lethality screens, pioneered in yeast, have recently been harnessed for developing novel therapeutic interventions in the treatment of cancer. Fong et al. (2009) exploited tumor-specific mutations in BRCA1 or BRCA2 genes, both of which are involved in DNA repair. The PARP family of enzymes (also involved in DNA repair) has is synthetically lethal with BRCA mutations (Fong, et al., 2009). The benefits of these types of studies to conventional chemotherapy are encouraging and will have a great impact on our understanding of cancer specific vulnerabilities (Luo, Solimini, & Elledge, 2009).
In summary, yeast assays have provided an unprecedented amount of information regarding gene-gene and gene-drug interactions and we are now witnessing the transfer of these technologies to mammalian cell assays. Yeast will continue, however, to be invaluable in the contribution of these studies with respect to confirmation of results, importantly, continuing to provide an important test-bed for feasibility of unique approaches to understanding the biology of the cell.
Abbreviations
- YKO
Yeast KnockOut collection
- HIP
HaploInsufficiency Profiling
- MSP
Multi-copy Suppression Profiling
- SGA
Synthetic Genetic Array
- ORF
Open Reading Frame
- MoBY-ORF
Molecular-Barcoded Yeast Open Reading Frame
- MOA
Mode-Of-Action
- C-MAP
Connectivity Map
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alberts AW, Chen J, Kuron G, Hunt V, Huff J, Hoffman C, et al. Mevinolin: a highly potent competitive inhibitor of hydroxymethylglutaryl-coenzyme A reductase and a cholesterol-lowering agent. Proc Natl Acad Sci U S A. 1980;77(7):3957–3961. doi: 10.1073/pnas.77.7.3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostol B, Greer CL. Copy number and stability of yeast 2 mu-based plasmids carrying a transcription-conditional centromere. Gene. 1988;67(1):59–68. doi: 10.1016/0378-1119(88)90008-x. [DOI] [PubMed] [Google Scholar]
- Arnoldo A, Curak J, Kittanakom S, Chevelev I, Lee VT, Sahebol-Amri M, et al. Identification of small molecule inhibitors of Pseudomonas aeruginosa exoenzyme S using a yeast phenotypic screen. PLoS Genet. 2008;4(2):e1000005. doi: 10.1371/journal.pgen.1000005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader GD, Heilbut A, Andrews B, Tyers M, Hughes T, Boone C. Functional genomics and proteomics: charting a multidimensional map of the yeast cell. Trends in Cell Biology. 2003;13(7):344–356. doi: 10.1016/s0962-8924(03)00127-2. [DOI] [PubMed] [Google Scholar]
- Baetz K, McHardy L, Gable K, Tarling T, Reberioux D, Bryan J, et al. Yeast genome-wide drug-induced haploinsufficiency screen to determine drug mode of action. Proc Natl Acad Sci U S A. 2004;101(13):4525–4530. doi: 10.1073/pnas.0307122101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birrell GW, Giaever G, Chu AM, Davis RW, Brown JM. A genome-wide screen in Saccharomyces cerevisiae for genes affecting UV radiation sensitivity. Proc Natl Acad Sci U S A. 2001;98(22):12608–12613. doi: 10.1073/pnas.231366398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff H, Angerbauer R, Boberg M, Petzinna D, Schmidt D, Steinke W, et al. Preclinical review of cerivastatin sodium--a step forward in HMG-CoA reductase inhibition. Atherosclerosis. 1998;139(Suppl 1):S7–13. doi: 10.1016/s0021-9150(98)00188-9. [DOI] [PubMed] [Google Scholar]
- Breslow DK, Cameron DM, Collins SR, Schuldiner M, Stewart-Ornstein J, Newman HW, et al. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5(8):711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56(1):100–104. [PubMed] [Google Scholar]
- Butcher EC, Berg EL, Kunkel EJ. Systems biology in drug discovery. Nat Biotechnol. 2004;22(10):1253–1259. doi: 10.1038/nbt1017. [DOI] [PubMed] [Google Scholar]
- Butcher RA, Bhullar BS, Perlstein EO, Marsischky G, LaBaer J, Schreiber SL. Microarray-based method for monitoring yeast overexpression strains reveals small-molecular targets in the TOR pathway. Nature Chemical Biology. 2006;2:103–109. doi: 10.1038/nchembio762. [DOI] [PubMed] [Google Scholar]
- Carroll SY, Stirling PC, Stimpson HE, Giesselmann E, Schmitt MJ, Drubin DG. A yeast killer toxin screen provides insights into a/b toxin entry, trafficking, and killing mechanisms. Dev Cell. 2009;17(4):552–560. doi: 10.1016/j.devcel.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cayley PJ, Dunn SM, King RW. Kinetics of substrate, coenzyme, and inhibitor binding to Escherichia coli dihydrofolate reductase. Biochemistry. 1981;20(4):874–879. doi: 10.1021/bi00507a034. [DOI] [PubMed] [Google Scholar]
- Chan JN, Nislow C, Emili A. Recent advances and method development for drug target identification. Trends Pharmacol Sci. 2009 doi: 10.1016/j.tips.2009.11.002. [DOI] [PubMed] [Google Scholar]
- Chang M, Bellaoui M, Boone C, Brown GW. A genome-wide screen for methyl methanesulfonate-sensitive mutants reveals genes required for S phase progression in the presence of DNA damage. Proc Natl Acad Sci U S A. 2002;99(26):16934–16939. doi: 10.1073/pnas.262669299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, et al. Comparison of the complete protein sets of worm and yeast: orthology and divergence. Science. 1998;282(5396):2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu MI, Katz H, Berlin V. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc Natl Acad Sci U S A. 1994;91(26):12574–12578. doi: 10.1073/pnas.91.26.12574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirstea D, Vallet S, Raje N. Future novel single agent and combination therapies. Cancer J. 2009;15(6):511–518. doi: 10.1097/PPO.0b013e3181c51c8e. [DOI] [PubMed] [Google Scholar]
- Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, et al. The genetic landscape of a cell. Science. 2010;327(5964):425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo M, Giaever G, Nislow C, Andrews B. Experimental approaches to identify genetic networks. Curr Opin Biotechnol. 2006;17(5):472–480. doi: 10.1016/j.copbio.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Cowen LE, Steinbach WJ. Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot Cell. 2008;7(5):747–764. doi: 10.1128/EC.00041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouzet M, Perrot M, Nogueira M, Begueret J. Genetic and biochemical analysis of cycloheximide resistance in the fungus Podospora anserina. Biochem Genet. 1978;16(3-4):271–286. doi: 10.1007/BF00484084. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. Systematic mapping of genetic interaction networks. Annu Rev Genet. 2009;43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR. Identifying druggable disease-modifying gene products. Curr Opin Chem Biol. 2009;13(5-6):549–555. doi: 10.1016/j.cbpa.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorer RK, Zhong S, Tallarico JA, Wong WH, Mitchison TJ, Murray AW. A small-molecule inhibitor of Mps1 blocks the spindle-checkpoint response to a lack of tension on mitotic chromosomes. Curr Biol. 2005;15(11):1070–1076. doi: 10.1016/j.cub.2005.05.020. [DOI] [PubMed] [Google Scholar]
- Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2(5):561–566. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- Ericson E, Gebbia M, Heisler LE, Wildenhain J, Tyers M, Giaever G, et al. Off-target effects of psychoactive drugs revealed by genome-wide assays in yeast. PLoS Genet. 2008;4(8):e1000151. doi: 10.1371/journal.pgen.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361(2):123–134. doi: 10.1056/NEJMoa0900212. [DOI] [PubMed] [Google Scholar]
- Frye SV. The art of the chemical probe. Nat Chem Biol. 2010;6(3):159–161. doi: 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- Giaever G, Chu AM, Ni L, Connelly C, Riles L, Veronneau S, et al. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418(6896):387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Giaever G, Flaherty P, Kumm J, Proctor M, Nislow C, Jaramillo DF, et al. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proc Natl Acad Sci U S A. 2004;101(3):793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G, Shoemaker DD, Jones TW, Liang H, Winzeler EA, Astromoff A, et al. Genomic profiling of drug sensitivities via induced haploinsufficiency. Nat Genet. 1999;21(3):278–283. doi: 10.1038/6791. [DOI] [PubMed] [Google Scholar]
- Goh KI, Cusick ME, Valle D, Childs B, Vidal M, Barabasi AL. The human disease network. Proc Natl Acad Sci U S A. 2007;104(21):8685–8690. doi: 10.1073/pnas.0701361104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman LS, Hardman JG, Limbird LE, Gilman AG. Goodman and Gilman’s the pharmacological basis of therapeutics. 10. New York: McGraw-Hill; 2001. [Google Scholar]
- Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science. 1991;253(5022):905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- Higgins MJ, Graham SJ. Intellectual property. Balancing innovation and access: patent challenges tip the scales. Science. 2009;326(5951):370–371. doi: 10.1126/science.1176116. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME, Fung E, Wildenhain J, Pierce SE, Hoon S, Lee W, et al. The chemical genomic potrait of yeast: uncovering a phenotype for all genes. Science. 2008;320(5874):362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CH, Magtanong L, Barker SL, Gresham D, Nishimura S, Natarajan P, et al. A molecular barcoded yeast ORF library enables mode-of-action analysis of bioactive compounds. Nat Biotechnol. 2009;27(4):369–377. doi: 10.1038/nbt.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoon S, Smith AM, Wallace IM, Suresh S, Miranda M, Fung E, et al. An integrated platform of genomic assays reveals small-molecule bioactivities. Nat Chem Biol. 2008;4(8):498–506. doi: 10.1038/nchembio.100. [DOI] [PubMed] [Google Scholar]
- Hopkins AL, Groom CR. The druggable genome. Nat Rev Drug Discov. 2002;1(9):727–730. doi: 10.1038/nrd892. [DOI] [PubMed] [Google Scholar]
- Hu Y, Rolfs A, Bhullar B, Murthy TV, Zhu C, Berger MF, et al. Approaching a complete repository of sequence-verified protein-encoding clones for Saccharomyces cerevisiae. Genome Res. 2007;17(4):536–543. doi: 10.1101/gr.6037607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes TR. Yeast and drug discovery. Funct Integr Genomics. 2002;2(4-5):199–211. doi: 10.1007/s10142-002-0059-1. [DOI] [PubMed] [Google Scholar]
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, et al. Functional discovery via a compendium of expression profiles. Cell. 2000;102(1):109–126. doi: 10.1016/s0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- Johnston M. A model fungal gene regulatory mechanism: the GAL genes of Saccharomyces cerevisiae. Microbiological Reviews. 1987;51(4):458–476. doi: 10.1128/mr.51.4.458-476.1987. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones GM, Stalker J, Humphray S, West A, Cox T, Rogers J, et al. A systematic library for comprehensive overexpression screens in Saccharomyces cerevisiae. Nat Methods. 2008;5(3):239–241. doi: 10.1038/nmeth.1181. [DOI] [PubMed] [Google Scholar]
- Keiser MJ, Setola V, Irwin JJ, Laggner C, Abbas AI, Hufeisen SJ, et al. Predicting new molecular targets for known drugs. Nature. 2009;462(7270):175–181. doi: 10.1038/nature08506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlwein SD. Obese and anorexic yeasts: experimental models to understand the metabolic syndrome and lipotoxicity. Biochim Biophys Acta. 2010;1801(3):222–229. doi: 10.1016/j.bbalip.2009.12.016. [DOI] [PubMed] [Google Scholar]
- Kuo SC, Lampen JO. Tunicamycin--an inhibitor of yeast glycoprotein synthesis. Biochem Biophys Res Commun. 1974;58(1):287–295. doi: 10.1016/0006-291x(74)90925-5. [DOI] [PubMed] [Google Scholar]
- Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science. 2006;313(5795):1929–1935. doi: 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- Lamesch P, Li N, Milstein S, Fan C, Hao T, Szabo G, et al. hORFeome v3.1: a resource of human open reading frames representing over 10,000 human genes. Genomics. 2007;89(3):307–315. doi: 10.1016/j.ygeno.2006.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar JD, Wilner KD. Drug interactions with fluconazole. Rev Infect Dis. 1990;12(Suppl 3):S327–333. doi: 10.1093/clinids/12.supplement_3.s327. [DOI] [PubMed] [Google Scholar]
- Lee W, St Onge RP, Proctor M, Flaherty P, Jordan MI, Arkin AP, et al. Genome-Wide Requirements for Resistance to Functionally Distinct DNA-Damaging Agents. PLoS Genet. 2005;1(2):e24. doi: 10.1371/journal.pgen.0010024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehar J, Stockwell BR, Giaever G, Nislow C. Combination chemical genetics. Nat Chem Biol. 2008;4(11):674–681. doi: 10.1038/nchembio.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppert G, McDevitt R, Falco SC, Van Dyk TK, Ficke MB, Golin J. Cloning by gene amplification of two loci conferring multiple drug resistance in Saccharomyces. Genetics. 1990;125(1):13–20. doi: 10.1093/genetics/125.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JW, Vederas JC. Drug discovery and natural products: end of an era or an endless frontier? Science. 2009;325(5937):161–165. doi: 10.1126/science.1168243. [DOI] [PubMed] [Google Scholar]
- Li X, Zolli-Juran M, Cechetto JD, Daigle DM, Wright GD, Brown ED. Multicopy suppressors for novel antibacterial compounds reveal targets and drug efflux susceptibility. Chem Biol. 2004;11(10):1423–1430. doi: 10.1016/j.chembiol.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Liebler DC, Guengerich FP. Elucidating mechanisms of drug-induced toxicity. Nat Rev Drug Discov. 2005;4(5):410–420. doi: 10.1038/nrd1720. [DOI] [PubMed] [Google Scholar]
- Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell. 1991;66(4):807–815. doi: 10.1016/0092-8674(91)90124-h. [DOI] [PubMed] [Google Scholar]
- Lum PY, Armour CD, Stepaniants SB, Cavet G, Wolf MK, Butler JS, et al. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116(1):121–137. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, et al. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137(5):835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell. 2009;136(5):823–837. doi: 10.1016/j.cell.2009.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal S, Moudgil M, Mandal SK. Rational drug design. Eur J Pharmacol. 2009;625(1-3):90–100. doi: 10.1016/j.ejphar.2009.06.065. [DOI] [PubMed] [Google Scholar]
- Marton MJ, DeRisi JL, Bennett HA, Iyer VR, Meyer MR, Roberts CJ, et al. Drug target validation and identification of secondary drug target effects using DNA microarrays. Nat Med. 1998;4(11):1293–1301. doi: 10.1038/3282. [DOI] [PubMed] [Google Scholar]
- McGary KL, Park TJ, Woods JO, Cha HJ, Wallingford JB, Marcotte EM. Systematic discovery of nonobvious human disease models through orthologous phenotypes. Proc Natl Acad Sci U S A. 2010;107(14):6544–6549. doi: 10.1073/pnas.0910200107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus KJ, Barrett IJ, Nouhi Y, Hieter P. Specific synthetic lethal killing of RAD54B-deficient human colorectal cancer cells by FEN1 silencing. Proc Natl Acad Sci U S A. 2009;106(9):3276–3281. doi: 10.1073/pnas.0813414106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara K, Mizunuma M, Hirata D, Tsuchiya E, Miyakawa T. The involvement of the Saccharomyces cerevisiae multidrug resistance transporters Pdr5p and Snq2p in cation resistance. FEBS Lett. 1996;399(3):317–320. doi: 10.1016/s0014-5793(96)01353-1. [DOI] [PubMed] [Google Scholar]
- Miyano S. proceedings; Research in computational molecular biology : 9th annual International Conference, RECOMB 2005; Cambridge, MA, USA. May 14-18, 2005; Berlin: Springer; 2005. [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124(6):1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Molin M, Norbeck J, Blomberg A. Dihydroxyacetone kinases in Saccharomyces cerevisiae are involved in detoxification of dihydroxyacetone. J Biol Chem. 2003;278(3):1415–1423. doi: 10.1074/jbc.M203030200. [DOI] [PubMed] [Google Scholar]
- Mushegian AR, Bassett DE, Jr, Boguski MS, Bork P, Koonin EV. Positionally cloned human disease genes: patterns of evolutionary conservation and functional motifs. Proc Natl Acad Sci U S A. 1997;94(11):5831–5836. doi: 10.1073/pnas.94.11.5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, Miller JR. Rescuing yeast mutants with human genes. Brief Funct Genomic Proteomic. 2007;6(2):104–111. doi: 10.1093/bfgp/elm017. [DOI] [PubMed] [Google Scholar]
- Parsons AB, Brost RL, Ding H, Li Z, Zhang C, Sheikh B, et al. Integration of chemical-genetic and genetic interaction data links bioactive compounds to cellular target pathways. Nat Biotechnol. 2004;22(1):62–69. doi: 10.1038/nbt919. [DOI] [PubMed] [Google Scholar]
- Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, et al. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell. 2006;126(3):611–625. doi: 10.1016/j.cell.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Paulsen IT, Sliwinski MK, Nelissen B, Goffeau A, Saier MH., Jr Unified inventory of established and putative transporters encoded within the complete genome of Saccharomyces cerevisiae. FEBS Lett. 1998;430(1-2):116–125. doi: 10.1016/s0014-5793(98)00629-2. [DOI] [PubMed] [Google Scholar]
- Pena-Castillo L, Hughes TR. Why are there still over 1000 uncharacterized yeast genes? Genetics. 2007;176(1):7–14. doi: 10.1534/genetics.107.074468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce SE, Davis RW, Nislow C, Giaever G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nat Protoc. 2007;2(11):2958–2974. doi: 10.1038/nprot.2007.427. [DOI] [PubMed] [Google Scholar]
- Pierce SE, Fung EL, Jaramillo DF, Chu AM, Davis RW, Nislow C, et al. A unique and universal molecular barcode array. Nat Methods. 2006;3(8):601–603. doi: 10.1038/nmeth905. [DOI] [PubMed] [Google Scholar]
- Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2006;12(11):1294–1300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- Provart NJ, McCourt P. Systems approaches to understanding cell signaling and gene regulation. Curr Opin Plant Biol. 2004;7(5):605–609. doi: 10.1016/j.pbi.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Quon K, Kassner PD. RNA interference screening for the discovery of oncology targets. Expert Opin Ther Targets. 2009;13(9):1027–1035. doi: 10.1517/14728220903179338. [DOI] [PubMed] [Google Scholar]
- Richardson K, Cooper K, Marriott MS, Tarbit MH, Troke PF, Whittle PJ. Discovery of fluconazole, a novel antifungal agent. Rev Infect Dis. 1990;12(Suppl 3):S267–271. doi: 10.1093/clinids/12.supplement_3.s267. [DOI] [PubMed] [Google Scholar]
- Rine J, Hansen W, Hardeman E, Davis RW. Targeted selection of recombinant clones through gene dosage effects. Proceedings of the National Academy of Sciences USA. 1983;80:6750–6754. doi: 10.1073/pnas.80.22.6750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rix U, Superti-Furga G. Target profiling of small molecules by chemical proteomics. Nat Chem Biol. 2009;5(9):616–624. doi: 10.1038/nchembio.216. [DOI] [PubMed] [Google Scholar]
- Rock FL, Mao W, Yaremchuk A, Tukalo M, Crepin T, Zhou H, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316(5832):1759–1761. doi: 10.1126/science.1142189. [DOI] [PubMed] [Google Scholar]
- Rual JF, Hill DE, Vidal M. ORFeome projects: gateway between genomics and omics. Curr Opin Chem Biol. 2004;8(1):20–25. doi: 10.1016/j.cbpa.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Rual JF, Hirozane-Kishikawa T, Hao T, Bertin N, Li S, Dricot A, et al. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res. 2004;14(10B):2128–2135. doi: 10.1101/gr.2973604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH. RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell. 1994;78(1):35–43. doi: 10.1016/0092-8674(94)90570-3. [DOI] [PubMed] [Google Scholar]
- Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, et al. A gene expression database for the molecular pharmacology of cancer. Nat Genet. 2000;24(3):236–244. doi: 10.1038/73439. [DOI] [PubMed] [Google Scholar]
- Schlabach MR, Luo J, Solimini NL, Hu G, Xu Q, Li MZ, et al. Cancer proliferation gene discovery through functional genomics. Science. 2008;319(5863):620–624. doi: 10.1126/science.1149200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137(5):821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- Schreiber SL. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science. 2000;287(5460):1964–1969. doi: 10.1126/science.287.5460.1964. [DOI] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, et al. Exploration of the Function and Organization of the Yeast Early Secretory Pathway through an Epistatic Miniarray Profile. Cell. 2005;123(3):507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Sidhu SS, Bader GD, Boone C. Functional genomics of intracellular peptide recognition domains with combinatorial biology methods. Curr Opin Chem Biol. 2003;7(1):97–102. doi: 10.1016/s1367-5931(02)00011-x. [DOI] [PubMed] [Google Scholar]
- Silva JM, Li MZ, Chang K, Ge W, Golding MC, Rickles RJ, et al. Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet. 2005;37(11):1281–1288. doi: 10.1038/ng1650. [DOI] [PubMed] [Google Scholar]
- Silva JM, Marran K, Parker JS, Silva J, Golding M, Schlabach MR, et al. Profiling essential genes in human mammary cells by multiplex RNAi screening. Science. 2008;319(5863):617–620. doi: 10.1126/science.1149185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AM, Heisler LE, Mellor J, Kaper F, Thompson MJ, Chee M, et al. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009;19(10):1836–1842. doi: 10.1101/gr.093955.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder M, Gallagher JE. Systems biology from a yeast omics perspective. FEBS Lett. 2009;583(24):3895–3899. doi: 10.1016/j.febslet.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CM, Lim SJ, Tong JC. Recent advances in computer-aided drug design. Brief Bioinform. 2009;10(5):579–591. doi: 10.1093/bib/bbp023. [DOI] [PubMed] [Google Scholar]
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, et al. Mapping pathways and phenotypes by systematic gene overexpression. Mol Cell. 2006;21(3):319–330. doi: 10.1016/j.molcel.2005.12.011. [DOI] [PubMed] [Google Scholar]
- St Onge RP, Mani R, Oh J, Proctor M, Fung E, Davis RW, et al. Systematic pathway analysis using high-resolution fitness profiling of combinatorial gene deletions. Nat Genet. 2007;39(2):199–206. doi: 10.1038/ng1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockwell BR. Exploring biology with small organic molecules. Nature. 2004;432(7019):846–854. doi: 10.1038/nature03196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suganuma M, Fujiki H, Furuya-Suguri H, Yoshizawa S, Yasumoto S, Kato Y, et al. Calyculin A, an inhibitor of protein phosphatases, a potent tumor promoter on CD-1 mouse skin. Cancer Res. 1990;50(12):3521–3525. [PubMed] [Google Scholar]
- Teotico DG, Babaoglu K, Rocklin GJ, Ferreira RS, Giannetti AM, Shoichet BK. Docking for fragment inhibitors of AmpC beta-lactamase. Proc Natl Acad Sci U S A. 2009;106(18):7455–7460. doi: 10.1073/pnas.0813029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH, Boone C. Synthetic genetic array analysis in Saccharomyces cerevisiae. Methods Mol Biol. 2006;313:171–192. doi: 10.1385/1-59259-958-3:171. [DOI] [PubMed] [Google Scholar]
- Tong AH, Evangelista M, Parsons AB, Xu H, Bader GD, Page N, et al. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294(5550):2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Tong AH, Lesage G, Bader GD, Ding H, Xu H, Xin X, et al. Global mapping of the yeast genetic interaction network. Science. 2004;303(5659):808–813. doi: 10.1126/science.1091317. [DOI] [PubMed] [Google Scholar]
- Tugendreich S, Perkins E, Couto J, Barthmaier P, Sun D, Tang S, et al. A streamlined process to phenotypically profile heterologous cDNAs in parallel using yeast cell-based assays. Genome Res. 2001;11(11):1899–1912. doi: 10.1101/gr.191601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner BK, Clemons PA. Connecting synthetic chemistry decisions to cell and genome biology using small-molecule phenotypic profiling. Curr Opin Chem Biol. 2009;13(5-6):539–548. doi: 10.1016/j.cbpa.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehner EP, Rao E, Brendel M. Molecular structure and genetic regulation of SFA, a gene responsible for resistance to formaldehyde in Saccharomyces cerevisiae, and characterization of its protein product. Mol Gen Genet. 1993;237(3):351–358. doi: 10.1007/BF00279438. [DOI] [PubMed] [Google Scholar]
- Winzeler EA, Shoemaker DD, Astromoff A, Liang H, Anderson K, Andre B, et al. Functional Characterization of the S. cerevisiae genome by gene deletion and parallel analysis. Science. 1999;285:901–906. doi: 10.1126/science.285.5429.901. [DOI] [PubMed] [Google Scholar]
- Workman CT, Mak HC, McCuine S, Tagne JB, Agarwal M, Ozier O, et al. A systems approach to mapping DNA damage response pathways. Science. 2006;312(5776):1054–1059. doi: 10.1126/science.1122088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Sillaots S, Davison J, Hu W, Jiang B, Kauffman S, et al. Chemical genetic profiling and characterization of small-molecule compounds that affect the biosynthesis of unsaturated fatty acids in Candida albicans. J Biol Chem. 2009;284(29):19754–19764. doi: 10.1074/jbc.M109.019877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Z, Costanzo M, Heisler LE, Paw J, Kaper F, Andrews BJ, et al. Yeast Barcoders: a chemogenomic application of a universal donor-strain collection carrying bar-code identifiers. Nat Methods. 2008;5(8):719–725. doi: 10.1038/nmeth.1231. [DOI] [PubMed] [Google Scholar]
- Yu H, Braun P, Yildirim MA, Lemmens I, Venkatesan K, Sahalie J, et al. High-quality binary protein interaction map of the yeast interactome network. Science. 2008;322(5898):104–110. doi: 10.1126/science.1158684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Lopez A, Anaflous A, El Bali B, Hamal A, Ericson E, et al. Chemical-genetic profiling of imidazo[1,2-a]pyridines and -pyrimidines reveals target pathways conserved between yeast and human cells. PLoS Genet. 2008;4(11):e1000284. doi: 10.1371/journal.pgen.1000284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Osborn M, Gitsham P, Yen K, Miller JR, Oliver SG. Using yeast to place human genes in functional categories. Gene. 2003;303:121–129. doi: 10.1016/s0378-1119(02)01142-3. [DOI] [PubMed] [Google Scholar]
- Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, et al. Global analysis of protein activities using proteome chips. Science. 2001;293:2101–2105. doi: 10.1126/science.1062191. [DOI] [PubMed] [Google Scholar]