

Abstract
Neuropeptide Y (NPY), a sympathetic cotransmitter, acts via G protein-coupled receptors to stimulate constriction and vascular smooth muscle cell (VSMC) proliferation through interactions with its Y1 receptors. However, VSMC proliferation appears bimodal, with high- and low-affinity peaks differentially blocked by antagonists of both Y1 and Y5 receptors. Here, we sought to determine the signaling mechanisms of NPY-mediated bimodal mitogenesis. In rat aortic VSMCs, NPY's mitogenic effect at all concentrations was blocked by pertussis toxin and was associated with decreased forskolin-stimulated cAMP levels. NPY also increased intracellular calcium levels; in contrast to mitogenesis, this effect was dose dependent. The rise in intracellular Ca2+ depended on extracellular Ca2+ and was mediated via activation of Y1 receptors, but not Y5 receptors. Despite differences in calcium, the signaling pathways activated at low and high NPY concentrations were similar. The mitogenic effect of the peptide at all doses was completely blocked by inhibitors of calcium/calmodulin-dependent kinase II (CaMKII), protein kinase C (PKC), and mitogen-activated protein kinase kinase, MEK1/2. Thus, in VSMCs, NPY-mediated mitogenesis signals primarily via Y1 receptors activating 2 Ca2+-dependent, growth-promoting pathways—PKC and CaMKII. At the high-affinity peak, these 2 pathways are amplified by Y5 receptor-mediated, calcium-independent inhibition of the adenylyl cyclase–protein kinase A (PKA) pathway. All 3 mechanisms converge to the extracellular signal-regulated kinases (ERK1/2) signaling cascade and lead to VSMC proliferation.
Keywords: neuropeptide Y, vascular smooth muscle, signaling
Introduction
Neuropeptide Y (NPY) is a sympathetic neurotransmitter widely expressed throughout the central and peripheral nervous system, where it exerts pleiotropic actions mediated by multiple receptors (Y1–Y5, as well as Y6 in mice) (Balasubramaniam 2002). The release of the peptide is increased during sympathetic nerve activation such as during a stress situation (Zukowska et al. 2003), when it is involved in the regulation of neurotransmitter release, cell growth, immune functions, and metabolism (Kitlinska et al. 2005; Kuo et al. 2007; Wheway et al. 2007; Zukowska-Grojec et al. 1998a, 1998b). These actions are both cell- and receptor-specific (Berglund et al. 2003). For example, NPY-induced neuroinhibition is Y2 receptor dependent, whereas its neuroproliferative actions are mediated by Y1 receptors (Hansel et al. 2001; Wahlestedt et al. 1990).
Owing to the high expression of NPY receptors in endothelium and vascular smooth muscle cells (VSMCs), the peptide is also involved in the regulation of cardiovascular functions. Originally, it was characterized as a vasoconstrictor acting at nanomolar concentrations through Y1 receptors present on VSMCs (Malmstrom 2000; Wahlestedt et al. 1990), yet we and others have shown that NPY is also a potent growth factor for vascular cells. It stimulates endothelial cell proliferation and migration via its Y2 receptors and, consequently, is involved in angiogenesis in both physiological and pathological conditions (Ekstrand et al. 2003; Koulu et al. 2004; Lee et al. 2003; Movafagh et al. 2006).
In VSMCs, the mitogenic effect of NPY is mediated by Y1 receptors; however, our previous in vitro and in vivo studies suggest that this action is potentiated by Y5 receptors (Li et al. 2003; Pons et al. 2003; Zukowska-Grojec et al. 1998a). Another unusual property of the NPY proliferative effect in VSMCs that we noted is that it begins at subpicomolar concentrations—significantly lower than the Kds of known NPY receptors (approximately 0.1–1 nmol·L−1) (Michel et al. 1998)—and has a bimodal pattern (Pons et al. 2003; Zukowska-Grojec et al. 1998b). We observed similar bimodality in NPY's effects in endothelial cells and neuroblastomas (Kitlinska et al. 2005; Kitlinska et al. 2002; Movafagh et al. 2006), which also express multiple NPY receptors, suggesting a more generalized phenomenon of interactions of multiple peptide receptors in the regulation of cell growth.
In support of this concept of multireceptor action, NPY's high- and low-affinity mitogenic responses are differentially blocked by NPY-receptor antagonists (Pons et al. 2003). The high-affinity peak is not affected by Y1- or Y5-receptor antagonists alone, but a combination of both completely blocks the proliferative response to low concentrations of NPY. The low-affinity peak is partially inhibited by the Y1-receptor antagonist alone; however, only a combination of Y1- and Y5-receptor antagonists blocks it completely. Therefore, the goal of the current study was to determine whether these interactions between Y1 and Y5 receptors result in differences in cell signaling between the high- and low-affinity peaks. To date, many pathways of NPY-mediated signaling have been identified, but rarely have they been directly related to the peptide's cellular activities, and never have they been assessed at concentrations below its receptor Kds.
Materials and methods
Cell culture
Primary rat aortic VSMCs (Cambrex, Walkersville, Md.) at passages 4–8 were grown in a 5% CO2 humidified incubator at 37 °C in smooth muscle basal medium (SmBM)-2 bullet kit (Cambrex) and subcultured at 90%–95% confluency, as indicated in Pons et al. (2003).
Mitogenic assays
VSMCs (1.0 × 103) were plated onto 96-well plates, grown for 2 days, serum-starved for 24 h, and treated with NPY in the presence of [3H]thymidine to measure DNA synthesis levels, as previously described (Pons et al. 2003). VSMCs were incubated with 100 ng·mL−1 pertussis toxin (PTX) in serum-free SmBM for 6 h (concentration and time according to Pellieux et al. (2000)) before treatment in 0.25% FBS SmBM with the following: PD98059 (2.5 × 10−6 mol·L−1, 30 min) (Tsuda et al. 2002); GF109203X or chelerythrine chloride (10−7 mol·L−1, 10 min); and KN-93 (0.5 × 10−6 mol·L−1, 60 min). Later, NPY (in 0.5% BSA) was added to 0.25% FBS SmBM for 24 h (5 wells per treatment group per experiment).
cAMP ELISA
VSMCs were plated, grown, and serum-starved as indicated previously. After incubation with 10−4 mol·L−1 3-isobutyl-1-methylxanthine (IBMX) (according to Cabrele et al. (2000)) for 5 min, treatment with NPY and 10−6 mol·L−1 forskolin (concentration from Pons et al. (2003)) in 0.25% FBS SmBM for 60 min at 37 °C (Cabrele et al. 2000) and cell lysis, intracellular cAMP levels were measured using the Biotrak enzyme immunoassay system (Amersham Biosciences, Piscataway, N.J.).
Fluorescent confocal microscopy
Cells were loaded with the Ca2+ fluorescence dye Fluo-4/AM at a final concentration of 13 μmol·L−1 in Tyrode–BSA, as previously described (Bkaily et al. 1997, 1999; Claing et al. 2002). In brief, VSMCs cultured on 25-millimetre coverslips were incubated with Fluo-4/AM for 1 h at room temperature, washed, and then left for 15 min to ensure complete hydrolysis of the acetoxymethyl ester groups before starting the experiments. Cells were examined with a Multiprobe 2001 confocal argon laser scanning system (CLSM) (Molecular Dynamics, Sunnyvale, Calif.) equipped with a Nikon Diaphot epifluorescence inverted microscope and a 60× (1.4 NA) Nikon Plan Apochromat oil objective (Bkaily et al. 1997, 1999). The argon laser line (488 nm, 9.0 mV) was directed toward the sample via a primary dichroic filter (510 nm) and attenuated with a 3% neutral-density filter to reduce photobleaching. Pinhole size was set at 100 μm. The image size was 512 pixels × 512 pixels with a pixel size of 0.34 μm. Laser line intensity, photometric gain, PMT settings, and filter attenuations were kept constant throughout the experimental procedures (Bkaily et al. 1997, 1999). Measurements of sustained (steady-state) cytosolic and nuclear Ca2+ fluorescence were performed from 3-dimensional (3D) reconstructions of the cells (including the nucleus) and expressed in fluorescence intensity relative to the volume (μm3) of the cell, as described previously (Bkaily et al. 1999). In all experiments, the cells were superfused with a balanced Tyrode salt solution containing 5 mmol·L−1 HEPES, 136 mmol·L−1 NaCl, 2.7 mmol·L−1 KCl, 1 mmol·L−1 MgCl2, 1.9 mmol·L−1 CaCl2, and 5.6 mmol·L−1 glucose, buffered to pH 7.4 with Tris base. The osmolarity of the buffer solution was adjusted with sucrose to 310 mosmol·L−1 using a Digimatic osmometer (Advanced Instruments Inc., Needham Heights, Mass.). For the dose–response curve of the effect of NPY on sustained cytosolic ([Ca]c) and nucleoplasmic ([Ca]n) Ca2+ levels, the concentration of the peptide was increased by sequential addition of NPY until a steady-state effect for each concentration was reached. The sustained [Ca]c and [Ca]n levels were expressed as mean absolute Ca2+–Fluo-4 fluorescence intensity levels (scale from 0 to 255 AU (arbitrary units)) or as the percentage increase of sustained Ca2+–Fluo-4 fluorescence intensity from control levels (Bkaily et al. 2000). At the end of each experiment, the nucleus was stained with Syto 11 (Bkaily et al. 1997, 1999). The 3D images were presented as viewed from the top (horizontal) using Image Space software (Molecular Dynamics). The top view 3D image permits a look through the whole cell.
Fluorescent Fura-2 microfluorometric measurements
The loading procedure with Fura-2 has been described elsewhere (Bkaily et al. 1998). In brief, for calcium measurements using a two-dimensional (2D) imaging system, cultured cells were washed with 2 mL of Tyrode-equilibrated solution (osmolarity adjusted to 310 mosmol·L−1) containing 1.8 mmol·L−1 CaCl2 and supplemented with 0.1% BSA. The cells were loaded with 1 μmol·L−1 of the calcium probe Fura-2/AM for 60 min in the dark at room temperature and then washed twice with Tyrode–BSA solution followed by 2 washes with normal Tyrode solution. A hydrolysis period of 20 min was necessary to ensure the complete hydrolysis of the acetoxymethyl ester groups. Finally, the cells were washed again in Tyrode solution, placed in a chamber, and visualized by a fluorescent microscope (Photon Technology International Inc. (PTI), Princeton, N.J.).
To monitor intracellular calcium changes in rat vascular smooth muscle cells, fluorescent measurements were made by using the double excitatory wavelength method (340/380 ratio) using a Deltascan and Imagescan microfluorometer (PTI) equipped with an NEC Power Mate 386/20 and accompanying PTI software-enabling instrument control, data acquisition, and analysis (Bkaily et al. 1998). Calculation of intracellular calcium was performed using the standard Grynkiewicz equation (Grynkiewicz et al. 1985; Poenie and Tsien 1986) included in the software, in which [Ca]i = Kd[(F – Rmin)/Rmax – F], where the Kd for Fura-2 is 224 nmol·L–1, F is the experimental fluorescence value, Rmax is the maximal fluorescence of Fura-2 in the presence of saturating calcium, and Rmin is the minimal fluorescence in the presence of minimal calcium. Rmax and Rmin were determined at the end of each experiment using the divalent cation ionophore ionomycin (2 × 10−5 mol·L−1) to permeabilize the cell (Rmax) and 30 mmol·L−1 of the calcium chelator EGTA (Rmin).
Western blot analysis
VSMCs were plated (2.5 × 105 cells, 100 mm dishes), grown until 60%–70% confluent, and serum-starved for 24 h. The cells were primed with PTX, as above, and treated with NPY. VSMCs were washed and harvested with the following lysis buffers: 50 mmol·L−1 Tris–HCl (pH 8.0), 150 mmol·L−1 NaCl, 100 μg·mL−1 phenylmethylsulfonyl fluoride (PMSF), 1 μg·mL−1 aprotinin, and 1% NP-40. The samples were resolved on 4%–20% Tris–glycine SDS-PAGE gels, transferred to Protran nitrocellulose membranes (Schleicher & Schuell, Germany), and immunoblotted with anti-phospho-p44/42-MAPK (Thr202/Tyr204) or anti-p44/42-MAPK antibodies (Cell Signaling Technology, Danvers, Mass.), according to the manufacturer's recommendations.
Statistical analysis of data
Data are presented as means ± SE, percentage of control. Analysis of data was by two-way ANOVA followed by Tukey's test using SAS (SAS Institute Inc. Cary, N.C.); one-way repeated-measures (RM) ANOVA followed by either Dunnett's t test or Student–Newman–Keuls method using SigmaStat 3.5 (SPSS Science, Chicago, Ill.); or Student's t test using Prism 3.02 (GraphPad Software, San Diego, Calif.), as noted. A p level < 0.05 was considered statistically significant for the indicated n per group. Non-significant results are indicated as p = NS.
Materials
Porcine NPY1–36 was from Peninsula Laboratories (San Carlos, Calif.). GF109203X and chelerythrine chloride were from Calbiochem (San Diego, Calif.). KN-93 was from Seikagaku America (East Falmouth, Mass.). PTX, forskolin, IBMX, and all other chemicals were from Sigma-Aldrich (St. Louis, Mo.).
Results
NPY-mediated bimodal proliferation of primary rat aortic VSMCs
To determine the pattern of the mitogenic response to NPY, primary rat aortic VSMCs were growth-arrested for 24 h and then stimulated with NPY at concentrations ranging from 10−14–10−7 mol·L−1 in the presence of [3H]thymidine. The peptide stimulated proliferation of VSMCs at all tested concentrations, with 2 distinct peaks of activity—a high-affinity growth peak at NPY 10−12 mol·L−1 (137 ± 7%, p < 0.05) and a second, low-affinity peak at NPY 10−8 mol·L−1 (162% ± 12%, p < 0.05), as measured by increases in [3H]thymidine uptake over control (media containing 0.25% FBS). After the high-affinity peak of mitogenic activity, there was a corresponding decrease in DNA synthesis levels at NPY 10−11–10−10 mol·L−1 (114% ± 6% and 123% ± 7%, respectively), forming a “valley” between the 2 growth peaks, and at NPY 10−7 mol·L−1 (132% ± 4%), forming a decline after the second growth peak (Fig. 1). On the basis of these results, the 3 representative doses of NPY corresponding to the high-affinity peak (10−12 mol·L−1), the “valley” (10−10 mol·L−1), and the low-affinity peak (10−8 mol·L−1) were selected for further studies designed to compare cell-signaling pathways at different NPY concentrations.
Fig. 1.

NPY-induced bimodal VSMC proliferation. Rat aortic VSMCs were serum-starved and treated with NPY for 24 h. NPY stimulated proliferation, measured as [3H]thymidine uptake, in a bimodal fashion with 2 growth peaks at 10−12 and 10−8 mol·L−1. Significant at *, p < 0.05 compared with control by one-way RM ANOVA followed by Dunnett's t test, n = 3 separate experiments. NPY, neuropeptide Y; VSMC, vascular smooth muscle cell.
NPY's mitogenic effect in VSMCs is mediated by Gi/o proteins
Since NPY is known to act via Gi/o proteins in other cells, we sought to determine if its proliferative effects in VSMCs are also mediated by this G protein at all concentrations of the peptide. To this end, rat aortic VSMCs were pretreated for 6 h with 100 ng·mL−1 PTX, a selective Gi/o protein inhibitor, before NPY stimulation. PTX pretreatment blocked NPY-induced [3H]thymidine uptake at all 3 concentrations investigated—from 127% ± 3% (p < 0.05) to 82% ± 7% at NPY 10−12 mol·L−1, from 113% ± 3% (p < 0.05) to 100% ± 5% at 10−10 mol·L−1, and from 125% ± 3% (p < 0.05) to 85% ± 7% at NPY 10−8 mol·L−1 (Fig. 2A).
Fig. 2.

NPY-induced VSMC proliferation is mediated by Gi/o proteins. (A) Pertussis toxin (PTX) (100 ng·mL−1, pretreatment for 6 h) blocked the proliferative effect of NPY in rat aortic VSMCs, measured as an increase in [3H]thymidine uptake, at both high- and low-affinity growth peaks. Significant at *, p < 0.05 compared with control using two-way ANOVA followed by Tukey's test. #, p< 0.05 compared with samples treated with NPY only at the same concentration. n = 3 separate experiments. (B) NPY, at all concentrations, inhibited forskolin-stimulated increases in cAMP levels in VSMCs. VSMCs were incubated with IBMX (10−4 mol·L−1) for 5 min, then treated with NPY and forskolin (10−6 mol·L−1) in 0.25% FBS SmBM for 60 min; intracellular cAMP concentrations were measured by ELISA. Significant at *, p < 0.05 compared with forskolin and IBMX alone by one-way RM ANOVA followed by Dunnett's t test. n = 3 separate experiments, 5 wells per experiment.
NPY inhibits forskolin-stimulated intracellular cAMP production in VSMCs
Since our results indicated that NPY's mitogenic response in VSMCs involves Gi/o protein activation and cAMP is one of the signaling molecules connecting G proteins to downstream signaling pathways, we next examined whether the growth peaks are linked to the inhibition of adenylyl cyclase and a decrease in intracellular cAMP levels. Thus, VSMCs were treated with NPY in the presence of 10−6 mol·L−1 forskolin, an adenylyl cyclase activator, and 10−4 mol·L−1 IBMX, a phosphodiesterase inhibitor. Cellular cAMP levels were measured 1 h after NPY stimulation. NPY inhibited forskolin-induced intracellular cAMP accumulation at all 3 concentrations—by 44% ± 7% at NPY 10−12 mol·L−1, 38% ± 7% at NPY 10−10 mol·L−1, and 58% ± 6% at NPY 10−8 mol·L−1. There was no statistically significant difference between the percentages of inhibition at the 3 NPY doses (Fig. 2B).
NPY increases intracellular calcium concentration in VSMCs via activation of Y1 receptors
An increase in intracellular calcium is another signaling mechanism associated with the activation of Gi/o proteins, independent of cAMP. We again stimulated VSMCs with NPY (10−16–10−6 mol·L−1) and followed intracellular calcium levels in the cytoplasm ([Ca]c) and nucleus ([Ca]n) using 3D Fluo-4 confocal fluorescent microscopy. The representative 3D reconstruction showed that NPY caused a sustained increase in calcium fluorescence intensity in the cytoplasm and nucleus in a single VSMC (Fig. 3A). NPY dose-dependently increased the steady state basal free-calcium levels in both the cytoplasm and nucleus with EC50 values of 9.3 × 10−12 and 3.2 × 10−12 mol·L−1, respectively (Fig. 3B).
Fig. 3.
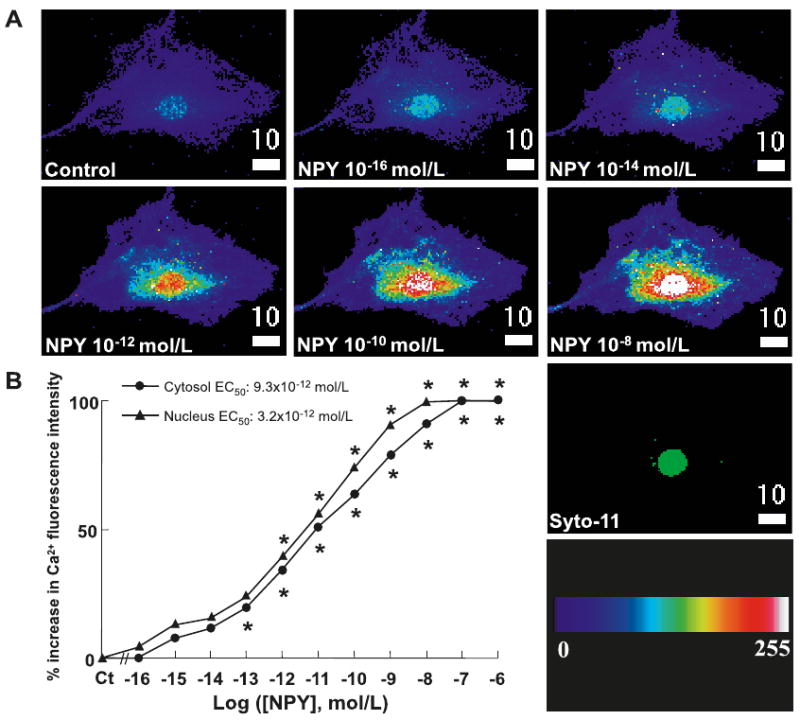
NPY's effect on intracellular calcium mobilization in primary rat aortic VSMCs. (A) Representative 3D upper-view reconstruction of a VSMC, before and after administration of NPY (10−16–10−8 mol·L−1), showing a dose-dependent increase in steady-state, resting, cytosolic ([Ca]c) and nuclear ([Ca]n) calcium concentrations. Syto-11 shows nuclear localization (green color has no measurable meaning). The color scale represents the pseudocolor-Fluo-4/Ca2+ complex intensity levels from 0 to 255 AU. The white scale bar is in micrometres (μm). (B) Dose-dependent effect of NPY (10−16–10−6 mol·L−1) on the steady-state, resting, 3D reconstruction of [Ca]c and [Ca]n of VSMCs using Fluo-4 confocal microscopy technique. n = 3–7 separate experiments. p = NS between the 2 curves. Significant at *, p < 0.05 compared with control (Ct) for cytosol and nucleus alone by Student's t test.
In another series of experiments, we verified whether the effect of NPY on intracellular calcium is mediated via an increase of calcium entry. Our results obtained from confocal microscopy and microfluorometry showed that in the absence of extracellular calcium, NPY failed to induce an effect on the calcium level at all concentrations tested (control = 9.9 ± 0.6 nmol·L−1; NPY (at 10−12 mol·L−1) = 10.2 ± 0.8 nmol·L−1 (microfluorometry measurement); control = 63.3 ± 5.2 nmol·L−1; NPY (at 10−10 mol·L−1) = 66.3 ± 5.9 nmol·L−1 (microfluorometry measurement); control = 9.5 ± 0.9 AU; NPY (at 10−8 mol·L−1) = 9.1 ± 0.8 AU) (confocal measurement). Results not shown.
To determine which receptors mediate NPY's effect on cellular Ca2+ levels, we measured NPY-induced increases in [Ca]i in the presence of Y1- (BIBP3226, 10−6 mol·L−1) or Y5- (CGP71683, 10−6 mol·L−1) receptor antagonists. In these series of experiments, using confocal microscopy and microfluorometry techniques, we first induced increases of [Ca]i by using 10−12, 10−10, or 10−8 mol·L−1 NPY (Fig. 4). Then, NPY was washed-out from the medium, and in some cells, the peptide was re-applied at all concentrations to test whether the second application induced changes in [Ca]i, similar to the first, which it did (not shown). In other cells, which only received the first NPY application, the Y1- or Y5-receptor antagonists (10−6 mol·L−1) were added for 15 min. In the presence of the antagonist, NPY was then added at 10−12, 10−10, or 10−8 mol·L−1. At all concentrations, the NPY-induced increase in [Ca]i was completely prevented by the Y1-receptor antagonist, BIBP3226 at a concentration of 10−6 mol·L−1 (Fig. 4A). However, blockade of the Y5 receptor (CGP71683, 10−6 mol·L−1) did not prevent NPY from inducing an increase of [Ca]i at any of the concentrations of NPY used (Fig. 4B).
Fig. 4.

Effect of the Y1-receptor antagonist BIBP3226 (A) and the Y5-receptor antagonist CGP71683 (B) on calcium response induced by 10−12 mol·L−1, 10−10 mol·L−1, and 10−8 mol·L−1 NPY. (A) Pretreatment with BIBP3226 (10−6 mol·L−1) prevents the NPY-induced calcium rise at the 3 concentrations used (black bars). (B) In the presence of CGP71683 (10−6 mol·L−1), NPY, at the 3 different concentrations, induces a significant increase in intracellular free Ca2+ (black bars). In both graphs, white bars represent the Ca2+ increase in the presence of NPY alone. The data are means ± SE, and n is the number of different cell culture experiments. Significant at *, p < 0.05, **, p < 0.01, and ***, p < 0.001 vs. respective control. #, p < 0.05, ##, p < 0.01, and ###, p < 0.001 vs. respective NPY response.
NPY's mitogenic effect in VSMCs is PKC and CaMKII dependent
Since NPY stimulated an increase in intracellular calcium in VSMCs, we next examined the role of downstream targets of calcium-stimulated signaling, protein kinase C (PKC) and calcium/calmodulin-dependent kinase II (CaMKII). The cells were pretreated with 2 different PKC inhibitors — an inhibitor of PKC's ATP-binding site, GF109203X (10−7 mol·L−1), or an inhibitor of its catalytic domain, chelerythrine chloride (10−7 mol·L−1)—as well as with the selective CaMKII inhibitor KN-93 (5 × 10−5 mol·L−1), then stimulated with the selected doses of NPY. Both PKC and CaMKII inhibitors completely blocked NPY's mitogenic effect at both growth peaks (10−12 and 10−8 mol·L−1), as well as at 10−10 mol·L−1, corresponding to the “valley” (Fig. 5).
Fig. 5.

The proliferative effect of NPY in VSMCs is PKC- and CaMKII-dependent. Two different PKC inhibitors, GF109203X and chelerythrine chloride (10−7 mol·L−1, pretreatment for 10 min), as well as a specific CaMKII inhibitor, KN-93 (0.5 × 10−6 mol·L−1, pretreatment for 60 min), abolished NPY-induced [3H]thymidine uptake at both growth peaks. Significant at *, p< 0.05 compared with 0.25% FBS SmBM alone (control) by two-way ANOVA followed by Tukey's test. #, p < 0.05 compared with the samples treated with NPY only at the same concentration. n = 3 separate experiments.
NPY-induced VSMC proliferation is associated with ERK1/2 activation
Although G protein-coupled receptor (GPCR)-mediated proliferation in VSMCs can be stimulated by various signaling molecules, most of them seem to converge to a common downstream pathway, namely Ras–Raf–MEK–ERK. Therefore, we sought to determine the role of this pathway in NPY-mediated VSMC growth. As shown in Fig. 6A, treatment of the cells with PD98059, a MAPK kinase (MEK1/2) inhibitor, completely blocked NPY-mediated increases in [3H]thymidine uptake levels at both 10−12 and 10−8 mol·L−1 growth peaks from 134% ± 3% (p < 0.05) to 104% ± 4% (NPY 10−12 mol·L−1) and from 145% ± 3% (p < 0.05) to 102% ± 3% (NPY 10−8 mol·L−1), as well as at 10−10 mol·L−1, corresponding to the “valley”, from 122% ± 3% (p < 0.05) to 104% ± 4%. Moreover, NPY stimulated phosphorylation of extracellular signal-regulated kinase (ERK)1/2 with a peak between 2 and 10 min for all 3 concentrations (Fig. 6B). This NPY-induced ERK1/2 activation was blocked by PTX (Fig. 6C). We did not observe any activation of other MAPKs—p38 or SAPK/JNK1, 2, 3—at any of the investigated NPY concentrations (data not shown).
Fig. 6.
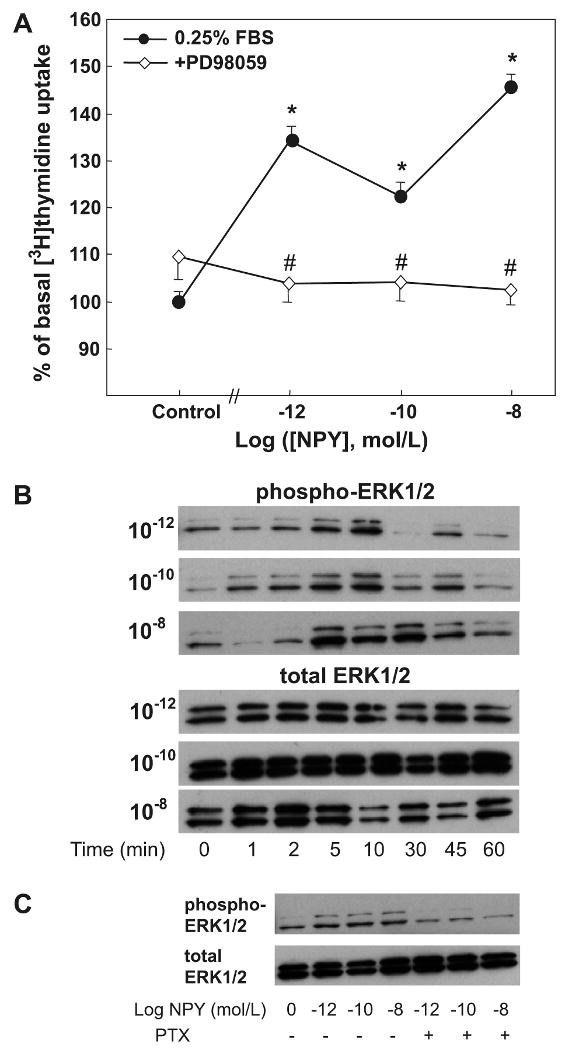
NPY-induced VSMC proliferation is mediated by ERK1/2 pathway. (A) A specific inhibitor of ERK1/2 MAPK, PD98059 (2.5 × 10−6 mol·L−1, pretreatment for 30 min), blocked NPY's mitogenic effect, measured as [3H]thymidine uptake, at all concentrations of the peptide. Significant at *, p< 0.05 compared with 0.25% FBS SmBM alone (control) by two-way ANOVA followed by Tukey's test. #, p< 0.05 compared with the samples treated with NPY only at the same concentration. n = 3 separate experiments. (B) Representative Western blot showing the effect of NPY on ERK1/2 phosphorylation. NPY activated ERK1/2 at all 3 selected doses with a peak between 2 and 10 min. (C) NPY-induced ERK1/2 phosphorylation was blocked by PTX (100 ng·mL−1, pretreatment for 6 h).
Discussion
Previously, we (Zukowska-Grojec et al. 1993) and others (Nilsson and Edvinsson 2000; Shigeri and Fujimoto 1993) have shown that NPY stimulates VSMC proliferation by activating Y1 receptors at concentrations similar to those causing vasoconstriction, at or above the Kds of known NPY receptors (Dumont and Quirion 2006). In doing so, NPY's vascular mitogenic activity resembled that of other vasoconstrictive peptides activating GPCRs, such as angiotensin II or endothelin (Schieffer et al. 1997). However, more recent studies in which NPY concentrations were extended below receptor Kds revealed that the peptide exerts a bimodal mitogenic action in VSMCs, with a high-affinity peak at 10−12 mol·L−1 and a low-affinity peak at 10−8 mol·L−1, separated by a “valley” at 10−10 mol·L−1 (Pons et al. 2003) (although the maxima of these peaks vary between cells and passages). Understanding how NPY elicits these 2 growth response peaks — whether or not such bimodality involves differential activation of multiple signaling pathways—has not been previously addressed. In the present study, we have identified signaling molecules involved in NPY-induced VSMC proliferation for the first time, starting at ligand concentrations below the traditionally accepted Kds of NPY receptors.
Here, we showed that in VSMCs, PTX blocked NPY's mitogenic effect at all concentrations, indicating that Gi/o proteins mediated the proliferative activity of NPY at both growth peaks. Similarly, NPY also inhibited forskolin-stimulated cAMP levels in these cells. cAMP, a secondary messenger commonly involved in GPCR signaling, activates PKA, which, depending on the cell type, either stimulates or inhibits cell growth (Liebmann 2001). In VSMCs, PKA is known to inhibit proliferation by phosphorylation of Raf-1, interfering with MAPK signaling (Bornfeldt and Krebs 1999; Hafner et al. 1994). Thus, by decreasing cAMP levels, NPY can block PKA growth-inhibitory activity and, as a result, augment VSMC proliferation.
Another secondary messenger linked to Gi/o protein activation is intracellular calcium (Noda et al. 2004). Here, we have shown that NPY stimulated a dose-dependent rise in intracellular calcium levels starting at sub-picomolar concentrations and did not display a bimodal pattern. Such an elevation of calcium levels at picomolar concentrations of NPY has been also reported in both human VSMCs (Jacques et al. 2000) and endocardial endothelial cells (Jacques et al. 2003). Our results also showed that the effect of NPY on [Ca]i was dependent on extracellular calcium, again similar to results reported in human VSMCs (Jacques et al. 2000). Thus, the increased [Ca]i by NPY seems to be mainly due to stimulation of calcium influx through the R-type calcium channels (Bkaily et al. 1997, 2006; Jacques et al. 2000). Previously, however, NPY-induced increases in calcium levels in VSMCs have been shown to be dependent on phospholipase C (PLC) (Shigeri et al. 1995), which can contribute to both influx of extracellular calcium and IP3-induced release from sarcoplasmic reticulum (Putney 2002). Thus, it is likely that mobilization of calcium from intracellular stores can also contribute to NPY-induced increases in calcium levels (Fig. 7); however, this would require further investigation.
Fig. 7.

Putative model for the intracellular signaling pathways involved in NPY-induced VSMC proliferation. NPY stimulates VSMC proliferation at concentrations ranging from 10−14 to 10−7 mol·L−1, in a characteristic bimodal fashion with 2 growth peaks — high-affinity (below known NPY receptor Kds) and low-affinity (10−9–10−7 mol·L−1). At all concentrations, NPY activates Gi/o proteins, which stimulate 2 major signaling pathways: Y1 receptor-mediated PLC activation and Y1 and Y5 receptor-mediated inhibition of adenylyl cyclase. PLC activation leads to the increase in intracellular calcium, mainly by extracellular calcium influx, but possibly also by its mobilization from intracellular stores. Increase in calcium activates PKC, which stimulates the Ras–Raf–MEK–ERK1/2 cascade, and CaMKII, which facilitates this by direct interactions with Raf-1. Decrease in adenylyl cyclase activity and cAMP level inhibits PKA and, as a consequence, blocks its inhibitory effects on the Ras and Raf proteins. This pathway is particularly important at low concentrations of NPY, where Y5-dependent PKA inhibition provides additional amplification of Y1 receptor-mediated mitogenic signal.
Increases in intracellular calcium observed after NPY stimulation can result in activation of several calcium-dependent kinases, such as PKC. Indeed, our studies indicated that NPY-induced VSMC proliferation was blocked by selective PKC inhibitors at both high- and low-affinity peaks. Thus, PKC activation appears to be necessary for NPY's proliferative actions in VSMCs, as shown for other GPCR ligands (Liebmann 2001). Interestingly, however, the same mitogenic response was also completely abolished by inhibitors of another calcium-activated kinase, CaMKII.
CaMKII has been implicated in the proliferation of a variety of cells, including VSMCs (Ginnan et al. 2004; Xiao et al. 2005). The proposed mechanisms of its growth-promoting actions include multiple signaling pathways. In myeloblastic leukemia cells, CaMKII activates cytoplasmic phospholipase A2 (cPLA2) and stimulates proliferation in a MAPK-independent manner (Muthalif et al. 2001). On the other hand, in VSMCs and thyroid cells, its actions seem to activate ERK1/2, either by direct interactions with Raf-1 or transactivation of the epidermal growth factor (EGF) receptor (Ginnan et al. 2004; Illario et al. 2003). CaMKII has also been reported as an inhibitor of adenylyl cyclase 3, preventing its growth-inhibitory effect in arterial smooth muscle cells (Wong et al. 2001). Our data suggest that in VSMCs, NPY-activated CaMKII signals through the ERK1/2 pathway, as NPY-induced growth peaks in VSMCs are completely blocked by inhibitors of MAPK kinase (i.e., MEK1/2) and NPY stimulates phosphorylation of ERK1/2 at all concentrations. Moreover, the NPY-mediated activation of MAPK is abolished by the Gi/o protein inhibitor PTX. These results strongly indicate that even though NPY seems to activate multiple signaling pathways in VSMCs, all the pathways converge to the common ERK1/2 pathway.
Surprisingly, both PKC and CaMKII inhibitors blocked the proliferative effect of NPY completely, which indicates that activation of both pathways is essential for this process to occur and suggests cross-talk between these pathways. Examples of such interactions have already been reported for other cells. In thyroid cells, fibronectin simultaneously activates 2 signaling pathways, Ras–Raf–MEK–ERK and CaMKII. However, ERK1/2 cannot be activated without stimulation of CaMKII, which forms a complex with Raf-1 and facilitates its binding to Ras (Illario et al. 2003; Illario et al. 2005). Therefore, activation of both pathways is necessary for fibronectin-induced thyroid cell proliferation. Moreover, in neuronal cells, a direct cross-talk between PKC and CaMKII is necessary for N-methyl d-aspartate (NMDA) receptor clustering and signaling (Gardoni et al. 2001; Wang and Kelly 1995).
Similar activation of the calcium-dependent kinases PKC and CaMKII at the high- and low-affinity growth peaks occurred in the presence of markedly different levels of [Ca2+]i, which increased in a dose-dependent manner. This indicated that even low levels of calcium released by picomolar concentrations of NPY were sufficient to activate those kinases, suggesting the existence of an amplifying mechanism. Interestingly, NPY's calcium response was fully dependent on Y1 receptors, and Y5 receptors were not involved. This is similar to what was reported for Y5 receptor signaling in HEC-1B cells, where it was limited to a decrease in cAMP levels (Bischoff et al. 2001). However, for the high-affinity mitogenic response, both Y1 and Y5 receptors were required. This may indicate that the Y5 receptor acts as an “amplifier” of Y1 receptor's proliferative signal by inhibition of adenylyl cyclase and the PKA pathway, particularly at low NPY concentrations when Y1 receptor-dependent increases in calcium levels are less pronounced. Such crosstalk between calcium-dependent kinases and PKA pathway may explain the presence of the high-affinity peak exclusively in cells expressing multiple NPY receptors.
Based on the data presented here, we propose the following model for the NPY-signaling cascade leading to VSMC proliferation (Fig. 7). At both growth peaks NPY acts through Gi/o proteins, which trigger 2 events: PLC activation and inhibition of adenylyl cyclase. PLC activation is a major proliferative pathway mediated by Y1 receptors. It leads to the increase in intracellular calcium, mainly by extracellular calcium influx, but possibly also by mobilization of intracellular calcium stores. This activates PKC, which stimulates the Ras–Raf–MEK–MAPK cascade, and CaMKII, which facilitates this process by its direct interactions with Raf-1. Moreover, the possibility that CaMKII also acts via inhibition of adenylyl cyclase, as previously shown in VSMCs (Wong et al. 2001), cannot be excluded. NPY-activated pathways converge to the MAPK signaling cascade, which directly leads to VSMC proliferation. On the other hand, a decrease in adenylyl cyclase activity and cAMP level inhibits PKA and, as a consequence, blocks its inhibitory effects on the Ras–Raf proteins. This pathway can be activated by both Y1 and Y5 receptors, but it is particularly important at low concentrations of NPY, where Y5 receptor-dependent PKA inhibition provides an additional amplification signal, compensating for a lower Y1 receptor-induced increase in calcium.
Taken together, our study shows that the bimodal NPY growth-promoting effect in VSMCs is mediated through 2 receptors, Y1 and Y5, which differ in their intracellular calcium response but not other signaling pathways studied so far. Since the presence of the high-affinity mitogenic peak is observed only in the cells expressing multiple types of NPY receptors (Kitlinska et al. 2005; Movafagh et al. 2006; Pons et al. 2003), this phenomenon is most likely associated with interactions between the receptors and (or) their signaling pathways. This may be due to receptor heterodimerization, since increases in receptor affinity were shown in other systems (AbdAlla et al. 2001) and Y1 and Y5 receptors have been found to form heterodimers (Gehlert et al. 2007). Alternatively, or in addition to the direct receptor interactions, multiple receptor induced amplification may be due to crosstalk between a calcium-dependent PKC–CaMKII–ERK1/2 pathway and a calcium-independent cAMP–PKA–ERK pathway, which can enhance the response to low NPY concentrations.
The high-affinity proliferative NPY actions may be relevant to conditions when levels of NPY are below those responsible for the low-affinity peak, that is, less than nanomolar concentrations, as shown by us in the rat model of carotid artery angioplasty (Li et al. 2003). In this model, doubling of vascular NPY concentrations in the injured artery either by a placement of an NPY slow-release pellet or by cold stress dramatically augmented neointima formation and led to the development of an occlusive lesion; both completely blocked by a Y1-receptor antagonist and partially inhibited by a Y5-receptor antagonist (Li et al. 2005). Thus, elucidating the intracellular signaling pathways and receptor interactions involved in this process may therefore lead to developing new therapeutic strategies for treatment of cardiovascular diseases, such as restenosis, atherosclerosis, and hypertension.
Acknowledgments
This work was supported by a grant to Zofia Zukowska from the National Institutes of Health (HL55310).
Contributor Information
Jennifer Pons, Department of Physiology and Biophysics, Georgetown University Medical Center, Box 571460, Washington, DC 20057-1460, USA.
Joanna Kitlinska, Department of Physiology and Biophysics, Georgetown University Medical Center, Box 571460, Washington, DC 20057-1460, USA.
Danielle Jacques, Department of Anatomy and Cell Biology, Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC J1H 5N4, Canada.
Claudine Perreault, Department of Anatomy and Cell Biology, Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC J1H 5N4, Canada.
Moni Nader, Department of Anatomy and Cell Biology, Faculty of Medicine, University of Sherbrooke, Sherbrooke, QC J1H 5N4, Canada.
Lindsay Everhart, Department of Physiology and Biophysics, Georgetown University Medical Center, Box 571460, Washington, DC 20057-1460, USA.
Ying Zhang, Lombardi Cancer Center, Georgetown University Medical Center, Washington, DC 20057, USA.
Zofia Zukowska, Department of Physiology and Biophysics, Georgetown University Medical Center, Box 571460, Washington, DC 20057-1460, USA.
References
- AbdAlla S, Lother H, el Massiery A, Quitterer U. Increased AT(1) receptor heterodimers in preeclampsia mediate enhanced angiotensin II responsiveness. Nat Med. 2001;7:1003–1009. doi: 10.1038/nm0901-1003. [DOI] [PubMed] [Google Scholar]
- Balasubramaniam A. Clinical potentials of neuropeptide Y family of hormones. Am J Surg. 2002;183:430–434. doi: 10.1016/S0002-9610(02)00803-6. [DOI] [PubMed] [Google Scholar]
- Berglund MM, Hipskind PA, Gehlert DR. Recent developments in our understanding of the physiological role of PP-fold peptide receptor subtypes. Exp Biol Med (Maywood) 2003;228:217–244. doi: 10.1177/153537020322800301. [DOI] [PubMed] [Google Scholar]
- Bischoff A, Puttmann K, Kotting A, Moser C, Buschauer A, Michel MC. Limited signal transduction repertoire of human Y(5) neuropeptide Y receptors expressed in HEC-1B cells. Peptides. 2001;22:387–394. doi: 10.1016/S0196-9781(01)00346-1. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Pothier P, D'Orléans-Juste P, Simaan M, Jacques D, Jaalouk D, et al. The use of confocal microscopy in the investigation of cell structure and function in the heart, vascular endothelium and smooth muscle cells. Mol Cell Biochem. 1997;172:171–194. doi: 10.1023/A:1006840228104. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Naik R, Jaalouk D, Jacques D, Economos D, D'Orléans-Juste P, Pothier P. Endothelin-1 and insulin activate the steady-state voltage dependent R-type Ca2+ channel in aortic smooth muscle cells via a pertussis toxin and cholera toxin sensitive G-protein. Mol Cell Biochem. 1998;183:39–47. doi: 10.1023/A:1006887714302. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Jacques D, Pothier P. Use of confocal microscopy to investigate cell structure and function. Methods Enzymol. 1999;307:119–135. doi: 10.1016/s0076-6879(99)07010-x. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Choufani S, Hassan G, El-Bizri N, Jacques D, D'Orléans-Juste P. Presence of functional endothelin-1 receptors in nuclear membranes of human aortic vascular smooth muscle cells. J Cardiovasc Pharmacol. 2000;36(Suppl 1):S414–417. doi: 10.1097/00005344-200036051-00121. [DOI] [PubMed] [Google Scholar]
- Bkaily G, Nader M, Avedanian L, Choufani S, Jacques D, D'Orléans-Juste P, et al. G-protein-coupled receptors, channels, and Na+–H+ exchanger in nuclear membranes of heart, hepatic, vascular endothelial, and smooth muscle cells. Can J Physiol Pharmacol. 2006;84:431–441. doi: 10.1139/Y06-002. [DOI] [PubMed] [Google Scholar]
- Bornfeldt KE, Krebs EG. Crosstalk between protein kinase A and growth factor receptor signaling pathways in arterial smooth muscle. Cell Signal. 1999;11:465–477. doi: 10.1016/S0898-6568(99)00020-0. [DOI] [PubMed] [Google Scholar]
- Cabrele C, Langer M, Bader R, Wieland HA, Doods HN, Zerbe O, et al. The first selective agonist for the neuropeptide YY5 receptor increases food intake in rats. J Biol Chem. 2000;275:36043–36048. doi: 10.1074/jbc.M000626200. [DOI] [PubMed] [Google Scholar]
- Claing A, Shbaklo H, Plante M, Bkaily G, D'Orléans-Juste P. Comparison of the contractile and calcium-increasing properties of platelet-activating factor and endothelin-1 in the rat mesenteric artery and vein. Br J Pharmacol. 2002;135:433–443. doi: 10.1038/sj.bjp.0704441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont Y, Quirion R. An overview of neuropeptide Y: pharmacology to molecular biology and receptor localization. In: Zukowska Z, Feuerstein G, editors. NPY family of peptides in neurobiology, cardiovascular and metabolic disorders: from genes to therapeutics. Bikrhauser Verlag; Basel, Switzerland: 2006. pp. 7–33. [DOI] [PubMed] [Google Scholar]
- Ekstrand AJ, Cao R, Bjorndahl M, Nystrom S, Jonsson-Rylander AC, Hassani H, et al. Deletion of neuropeptide Y (NPY) 2 receptor in mice results in blockage of NPY-induced angiogenesis and delayed wound healing. Proc Natl Acad Sci USA. 2003;100:6033–6038. doi: 10.1073/pnas.1135965100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardoni F, Bellone C, Cattabeni F, Di Luca M. Protein kinase C activation modulates alpha-calmodulin kinase II binding to NR2A subunit of N-methyl-d-aspartate receptor complex. J Biol Chem. 2001;276:7609–7613. doi: 10.1074/jbc.M009922200. [DOI] [PubMed] [Google Scholar]
- Gehlert DR, Schober DA, Morin M, Berglund MM. Co-expression of neuropeptide Y Y1 and Y5 receptors results in heterodimerization and altered functional properties. Biochem Pharmacol. 2007;74:1652–1664. doi: 10.1016/j.bcp.2007.08.017. [DOI] [PubMed] [Google Scholar]
- Ginnan R, Pfleiderer PJ, Pumiglia K, Singer HA. PKC-delta and CaMKII-delta 2 mediate ATP-dependent activation of ERK1/2 in vascular smooth muscle. Am J Physiol Cell Physiol. 2004;286:C1281–1289. doi: 10.1152/ajpcell.00202.2003. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz GE, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hafner S, Adler HS, Mischak H, Janosch P, Heidecker G, Wolfman A, et al. Mechanism of inhibition of Raf-1 by protein kinase A. Mol Cell Biol. 1994;14:6696–6703. doi: 10.1128/mcb.14.10.6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansel DE, Eipper BA, Ronnett GV. Neuropeptide Y functions as a neuroproliferative factor. Nature. 2001;410:940–944. doi: 10.1038/35073601. [DOI] [PubMed] [Google Scholar]
- Illario M, Cavallo AL, Bayer KU, Di Matola T, Fenzi G, Rossi G, et al. Calcium/calmodulin-dependent protein kinase II binds to Raf-1 and modulates integrin-stimulated ERK activation. J Biol Chem. 2003;278:45101–45108. doi: 10.1074/jbc.M305355200. [DOI] [PubMed] [Google Scholar]
- Illario M, Cavallo AL, Monaco S, Di Vito E, Mueller F, Marzano LA, et al. Fibronectin-induced proliferation in thyroid cells is mediated by alphavbeta3 integrin through Ras/Raf-1/MEK/ERK and calcium/CaMKII signals. J Clin Endocrinol Metab. 2005;90:2865–2873. doi: 10.1210/jc.2004-1520. [DOI] [PubMed] [Google Scholar]
- Jacques D, Sader S, El-Bizri N, Chouffani S, Hassan G, Shbaklo H. Neuropeptide Y induced increase of cytosolic and nuclear Ca2+ in heart and vascular smooth muscle cells. Can J Physiol Pharmacol. 2000;78:162–172. doi: 10.1139/cjpp-78-2-162. [DOI] [PubMed] [Google Scholar]
- Jacques D, Sader S, Perreault C, Fournier A, Pelletier G, Beck-Sickinger AG, et al. Presence of neuropeptide Y and the Y1 receptor in the plasma membrane and nuclear envelope of human endocardial endothelial cells: modulation of intracellular calcium. Can J Physiol Pharmacol. 2003;81:288–300. doi: 10.1139/y02-165. [DOI] [PubMed] [Google Scholar]
- Kitlinska J, Lee EW, Movafagh S, Pons J, Zukowska Z. Neuropeptide Y-induced angiogenesis in aging. Peptides. 2002;23:71–77. doi: 10.1016/S0196-9781(01)00581-2. [DOI] [PubMed] [Google Scholar]
- Kitlinska J, Abe K, Kuo L, Pons J, Yu M, Li L, et al. Differential effects of neuropeptide Y on the growth and vascularization of neural crest-derived tumors. Cancer Res. 2005;65:1719–1728. doi: 10.1158/0008-5472.CAN-04-2192. [DOI] [PubMed] [Google Scholar]
- Koulu M, Movafagh S, Tuohimaa J, Jaakkola U, Kallio J, Pesonen U, et al. Neuropeptide Y and Y2-receptor are involved in development of diabetic retinopathy and retinal neovascularization. Ann Med. 2004;36:232–240. doi: 10.1080/07853890410031236. [DOI] [PubMed] [Google Scholar]
- Kuo LE, Kitlinska JB, Tilan JU, Li L, Baker SB, Johnson MD, et al. Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat Med. 2007;13:803–811. doi: 10.1038/nm1611. [DOI] [PubMed] [Google Scholar]
- Lee EW, Michalkiewicz M, Kitlinska J, Kalezic I, Switalska H, Yoo P, et al. Neuropeptide Y induces ischemic angiogenesis and restores function of ischemic skeletal muscles. J Clin Invest. 2003;111:1853–1862. doi: 10.1172/JCI16929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lee EW, Ji H, Zukowska Z. Neuropeptide Y-induced acceleration of postangioplasty occlusion of rat carotid artery. Arterioscler Thromb Vasc Biol. 2003;23:1204–1210. doi: 10.1161/01.ATV.0000071349.30914.25. [DOI] [PubMed] [Google Scholar]
- Li L, Jonsson-Rylander AC, Abe K, Zukowska Z. Chronic stress induces rapid occlusion of angioplasty-injured rat carotid artery by activating neuropeptide Y and its Y1 receptors. Arterioscler Thromb Vasc Biol. 2005;25:2075–2080. doi: 10.1161/01.ATV.0000179601.1988.8.19. [DOI] [PubMed] [Google Scholar]
- Liebmann C. Regulation of MAP kinase activity by peptide receptor signalling pathway: paradigms of multiplicity. Cell Signal. 2001;13:777–785. doi: 10.1016/S0898-6568(01)00192-9. [DOI] [PubMed] [Google Scholar]
- Malmstrom RE. Neuropeptide Y Y1 receptor mediated mesenteric vasoconstriction in the pig in vivo. Regul Pept. 2000;95:59–63. doi: 10.1016/S0167-0115(00)00128-2. [DOI] [PubMed] [Google Scholar]
- Michel MC, Beck-Sickinger A, Cox H, Doods HN, Herzog H, Larhammar D, et al. XVI. International Union of Pharmacology recommendations for the nomenclature of neuropeptide Y, peptide YY, and pancreatic polypeptide receptors. Pharmacol Rev. 1998;50:143–150. [PubMed] [Google Scholar]
- Movafagh S, Hobson JP, Spiegel S, Kleinman HK, Zukowska Z. Neuropeptide Y induces migration, proliferation, and tube formation of endothelial cells bimodally via Y1, Y2, and Y5 receptors. FASEB J. 2006;20:1924–1926. doi: 10.1096/fj.05-4770fje. [DOI] [PubMed] [Google Scholar]
- Muthalif MM, Ljuca F, Roaten JB, Pentapaty N, Uddin MR, Malik KU. Ca2+/calmodulin-dependent protein kinase II and cytosolic phospholipase A2 contribute to mitogenic signaling in myeloblastic leukemia U-937 cells. J Pharmacol Exp Ther. 2001;298:272–278. [PubMed] [Google Scholar]
- Nilsson T, Edvinsson L. Neuropeptide Y stimulates DNA synthesis in human vascular smooth muscle cells through neuropeptide Y Y1 receptors. Can J Physiol Pharmacol. 2000;78:256–259. doi: 10.1139/cjpp-78-3-256. [DOI] [PubMed] [Google Scholar]
- Noda M, Higashida H, Aoki S, Wada K. Multiple signal transduction pathways mediated by 5-HT receptors. Mol Neurobiol. 2004;29:31–39. doi: 10.1385/MN:29:1:31. [DOI] [PubMed] [Google Scholar]
- Pellieux C, Sauthier T, Domenighetti A, Marsh DJ, Palmiter RD, Brunner HR, et al. Neuropeptide Y (NPY) potentiates phenylephrine-induced mitogen-activated protein kinase activation in primary cardiomyocytes via NPY Y5 receptors. Proc Natl Acad Sci USA. 2000;97:1595–1600. doi: 10.1073/pnas.030533197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poenie M, Tsien R. Fura-2: a powerful new tool for measuring and imaging [Ca2+]i in single cells. Prog Clin Biol Res. 1986;210:53–56. [PubMed] [Google Scholar]
- Pons J, Kitlinska J, Ji H, Lee EW, Zukowska Z. Mitogenic actions of neuropeptide Y in vascular smooth muscle cells: synergetic interactions with the beta-adrenergic system. Can J Physiol Pharmacol. 2003;81:177–185. doi: 10.1139/y02-166. [DOI] [PubMed] [Google Scholar]
- Putney JW. PLC-gamma: an old player has a new role. Nat Cell Biol. 2002;4:E280–281. doi: 10.1038/ncb1202-e280. [DOI] [PubMed] [Google Scholar]
- Schieffer B, Drexler H, Ling BN, Marrero MB. G protein-coupled receptors control vascular smooth muscle cell proliferation via pp60c-src and p21ras. Am J Physiol. 1997;272:C2019–2030. doi: 10.1152/ajpcell.1997.272.6.C2019. [DOI] [PubMed] [Google Scholar]
- Shigeri Y, Fujimoto M. Neuropeptide Y stimulates DNA synthesis in vascular smooth muscle cells. Neurosci Lett. 1993;149:19–22. doi: 10.1016/0304-3940(93)90337-K. [DOI] [PubMed] [Google Scholar]
- Shigeri Y, Nakajima S, Fujimoto M. Neuropeptide YY1 receptors-mediated increase in intracellular Ca2+ concentration via phospholipase C-dependent pathway in porcine aortic smooth muscle cells. J Biochem. 1995;118:515–520. doi: 10.1093/oxfordjournals.jbchem.a124938. [DOI] [PubMed] [Google Scholar]
- Tsuda Y, Okazaki M, Uezono Y, Osajima A, Kato H, Okuda H, et al. Activation of extracellular signal-regulated kinases is essential for pressure-induced proliferation of vascular smooth muscle cells. Eur J Pharmacol. 2002;446:15–24. doi: 10.1016/S0014-2999(02)01811-3. [DOI] [PubMed] [Google Scholar]
- Wahlestedt C, Hakanson R, Vaz CA, Zukowska-Grojec Z. Norepinephrine and neuropeptide Y: vasoconstrictor cooperation in vivo and in vitro. Am J Physiol. 1990;258:R736–742. doi: 10.1152/ajpregu.1990.258.3.R736. [DOI] [PubMed] [Google Scholar]
- Wang JH, Kelly PT. Postsynaptic injection of CA2+/CaM induces synaptic potentiation requiring CaMKII and PKC activity. Neuron. 1995;15:443–452. doi: 10.1016/0896-6273(95)90048-9. [DOI] [PubMed] [Google Scholar]
- Wheway J, Herzog H, Mackay F. The Y1 receptor for NPY: a key modulator of the adaptive immune system. Peptides. 2007;28:453–458. doi: 10.1016/j.peptides.2006.09.030. [DOI] [PubMed] [Google Scholar]
- Wong ST, Baker LP, Trinh K, Hetman M, Suzuki LA, Storm DR, et al. Adenylyl cyclase 3 mediates prostaglandin E(2)-induced growth inhibition in arterial smooth muscle cells. J Biol Chem. 2001;276:34206–34212. doi: 10.1074/jbc.M103923200. [DOI] [PubMed] [Google Scholar]
- Xiao W, Liu Y, Templeton DM. Ca2+/calmodulin-dependent protein kinase II inhibition by heparin in mesangial cells. Am J Physiol Renal Physiol. 2005;288:F142–149. doi: 10.1152/ajprenal.00145.2004. [DOI] [PubMed] [Google Scholar]
- Zukowska Z, Pons J, Lee EW, Li L. Neuropeptide Y: a new mediator linking sympathetic nerves, blood vessels and immune system. Can J Physiol Pharmacol. 2003;81:89–94. doi: 10.1139/y03-006. [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Pruszczyk P, Colton C, Yao J, Shen GH, Myers AK, et al. Mitogenic effect of neuropeptide Y in rat vascular smooth muscle cells. Peptides. 1993;14:263–268. doi: 10.1016/0196-9781(93)90040-N. [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Fisher TA, Ji H. Mechanisms of vascular growth-promoting effects of neuropeptide Y: role of its inducible receptors. Regul Pept. 1998a;75–76:231–238. doi: 10.1016/S0167-0115(98)00073-1. [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, et al. Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res. 1998b;83:187–195. doi: 10.1161/01.res.83.2.187. [DOI] [PubMed] [Google Scholar]