

Abstract
The association of DNA with histones in chromatin impedes DNA repair enzymes from accessing DNA lesions. Nucleosomes exist in a dynamic equilibrium in which portions of the DNA molecule spontaneously unwrap, transiently exposing buried DNA sites. Thus, nucleosome dynamics in certain regions of chromatin may provide the exposure time and space needed for efficient repair of buried DNA lesions. We have used FRET and restriction enzyme accessibility to study nucleosome dynamics following DNA damage by UV radiation. We find that FRET efficiency is reduced in a dose-dependent manner, showing that the presence of UV photoproducts enhances spontaneous unwrapping of DNA from histones. Furthermore, this UV-induced shift in unwrapping dynamics is associated with increased restriction enzyme accessibility of histone-bound DNA after UV treatment. Surprisingly, the increased unwrapping dynamics is even observed in nucleosome core particles containing a single UV lesion at a specific site. These results highlight the potential for increased “intrinsic exposure” of nucleosome-associated DNA lesions in chromatin to repair proteins.
Keywords: Chromatin, DNA Nucleotide Excision Repair, Fluorescence Resonance Energy Transfer (FRET), Histones, Nucleic Acid Structure
Introduction
In eukaryotic cells, genomic DNA is organized in a fundamental repeating unit, the nucleosome, which is composed of the nucleosome core particle (NCP)2 and a linker region (1). Nucleosome core particles contain 147 bp of DNA wrapped around the histone octamer with two copies each of histones H2A, H2B, H3, and H4 (2). The neighboring core particles are connected by “linker DNA,” which is typically 30–50 bp in length, and linker histones (3).
Cellular DNA is being modified constantly by endogenous and exogenous DNA-damaging agents (4). Among these, UV light is one of the most prevalent environmental stresses that produce lesions in DNA. UV lesions inhibit DNA replication as well as RNA transcription, thus affecting genome stability (5). Therefore, failure to repair UV damage in humans has been associated with hypersensitivity to sunlight and a high risk of skin cancer (6, 7).
UV radiation mainly generates two types of lesions that covalently link adjacent DNA bases: cyclobutane pyrimidine dimers (CPDs) and pyrimidine(6-4)pyrimidone photoproducts (6-4PPs). CPDs bend DNA ∼30 ° toward the major groove (8), inducing helical unwinding of ∼9 °, whereas 6-4PPs bend DNA ∼44 ° (9). Although UV lesions distort the helical structure of DNA, both in vitro and in vivo experiments have shown that nucleosome structure persists after UV irradiation. Indeed, UV-damaged nucleosomes can be isolated from UV-irradiated cells (10, 11), and nucleosome cores can be UV-irradiated without significant disruption or destabilization (12–14). Therefore, the structural distortion caused by UV damage to many sites in nucleosome DNA does not appear to induce the disruption or destabilization necessary to facilitate lesion recognition and repair (15).
Access to buried UV DNA lesions in chromatin can be achieved by ATP-dependent chromatin remodeling factors, which use ATP hydrolysis to slide or unwrap DNA (16, 17). These multisubunit complexes also can catalyze eviction of histone octamers or promote histone variant replacement (18). In addition, post-translational modifications of histones can alter histone-DNA or histone-histone interactions (19). These two mechanisms have been demonstrated previously to facilitate repair of at least a subset of UV lesions (20–22). However, like DNA repair enzymes, both chromatin remodeling proteins and histone modification factors require initial localization to damaged sites. The mechanism by which these factors recognize UV-damaged DNA in nucleosomes is unclear, and the intrinsic features that distinguish damaged from undamaged nucleosomes remain a mystery.
Nucleosomes exist in a dynamic equilibrium where portions of the DNA molecule unwrap spontaneously, transiently exposing buried DNA in nucleosomes (23–25). Certain intrinsic properties, such as DNA sequence variation, may change the nucleosome structure and stability (26–28). The fraction of time that transient exposure renders DNA accessible can be as high as 10% (depending on sequence) for sites near the ends of the DNA-histone interface and as low as 0.001% for sites near the middle of nucleosomal DNA (29). Thus, nucleosome dynamics in certain regions of chromatin may provide the exposure time needed for efficient repair of buried DNA lesions. Indeed, it was reported that UV photolyase in yeast cells recognizes and repairs CPDs on a timescale of seconds (30), which is on par with that of spontaneous nucleosome unwrapping (29), as opposed to the longer times needed for histone dissociation or chromatin remodeling (30).
Understanding the effect of DNA damage on nucleosome dynamics is essential for understanding whether nucleosome dynamic variations can serve as a signal for damage recognition. In this study, we have used FRET and restriction enzyme accessibility (REA) to characterize the dynamics of NCPs containing different amounts of UV damage. We find that UV damage to NCPs enhances the spontaneous unwrapping of DNA, driving NCPs toward a more accessible conformation (on average) for REA at recognition sites located near the termini of NCPs. Furthermore, this change in FRET efficiency correlates with the formation of 6-4PPs in nucleosome DNA. Thus, increased dynamics of UV-damaged nucleosomes is an intrinsic property that drives nucleosomes toward the unwrapped states.
EXPERIMENTAL PROCEDURES
DNA Preparation
The 147-bp nucleosome positioning sequence 601 was used for NCP reconstitution (31). The 147-mer oligonucleotides were initially prepared from two separate fragments of 52 nts and 95 nts. These oligonucleotides were synthesized separately for fluorescent labeling (52 nts) and UV treatments (95 nts). The 52-mer oligonucleotides, containing internally labeled Cy3 or Cy5, were purified by HPLC (Integrated DNA Technologies). Cy3 was incorporated at position 33, and Cy5 was incorporated at position 113 on opposing strands (Fig. 1A). The 95-nt fragments were dissolved in water and irradiated on ice with (predominantly) 254 nm UV light under two low pressure Hg lamps (Sylvania, Model G30T8). UV doses were measured with a Spectroline DM-254N UV meter (Spectronics Corp., Westbury, NY). (We note that the 95-mer fragments were irradiated separately to avoid photobleaching of the Cy3 and Cy5 chromophores.) The 52- and 95-mer oligonucleotides were mixed at equal molar concentrations and annealed. The annealed products were then treated with ligase to seal the nicks. After ligation, the DNA was gel-purified to remove additional oligonucleotides and unligated products. For DNA containing a single lesion, the structure of the substrate is shown schematically in Fig. 6A. The fragments F1–F4 are 52, 83, 95, and 52 nt in length, respectively. The damage fragment (DF) is a 12-mer oligonucleotide, and the sequence is shown in Fig. 6A. DF1 is the sequence containing a single CPD, and DF2 is the sequence containing a single 6-4PP. These oligonucleotides were the generous gift of Dr. Nicholas Geacintov (Chemistry Department, New York University). The F3 sequence was changed according to the corresponding DF sequence. The DNA was prepared the same as UV-irradiated substrates described above. The 147-bp full-length DNA with both UV damage and internal fluorescent labels was then used for NCP reconstitution.
FIGURE 1.

FRET system to analyze the dynamic changes of nucleosomal DNA. A, locations of donor (Cy3) and acceptor (Cy5) on a 147-bp DNA. The DNA is the strongly positioned sequence 601. The upper panel shows the locations of Cy3 (green) and Cy5 (red) on the naked DNA. The two dyes are 80 bp apart, and the distance between the two dyes is ∼27 nm, which is far beyond the R0 (6 nm) for the Cy3-Cy5 pair. The lower panel is a schematic illustration of the locations of the dyes on the NCP structure. The distance between the two dyes is ∼3 nm, which falls into the R0, so that robust energy transfer could happen upon the excitation of the donor. The NCP structure was generated from the crystal structure with Protein Data Bank code 1KX5 (39). B, emission spectra of naked 601 DNA (pink) and 601 NCPs (blue) with excitation at 515 nm. C, salt-induced unwrapping of NCPs monitored by FRET. An appropriate volume of 5 m NaCl was added to the NCP sample, and the solution was allowed to equilibrate for 30 min at room temperature. Emission spectra were taken at different salt concentrations as shown. The data were normalized by the Cy5 signals excited at 615 nm.
FIGURE 6.

Incorporation of single UV photoproducts increases nucleosome dynamics. A, schematic illustration of the incorporation of a single CPD or 6-4PP into the 601 sequence. The DFs are 12-mer oligonucleotides with the sequences shown. Solid triangles indicate the positions of the photoproduct. The dark circle represents Cy5, and the open circle represents Cy3. B, FRET changes for the single CPD containing NCPs. The 601 sequence was changed according to the DF and denoted 601.DF1. The left panel shows emission spectra for the CPD containing NCPs compared with undamaged NCPs. The right panel shows the FRET efficiency values determined in each case, where the value for 601.DF1 NCPs was set to 1. The values shown are the mean ± 1 S.D. of three independent experiments. C, the same as B except the NCPs contained a single 6-4PP and the sequence denoted 601.DF2. D, relative FRET efficiency of salt dependence of NCPs containing a single CPD. The FRET efficiency at the lowest salt concentration (50 mm) was set to 1. Values are the mean ± 1 S.D. of three independent experiments. E, salt titration profile of NCPs containing a single 6-4PP. Values were plotted the same as in D.
Nucleosome Reconstitution
The four core histones of Xenopus laevis were prepared individually from Escherichia coli as described (32) with some modification. Histones were expressed as inclusion bodies, the cells were disrupted by sonication, and the pellets were collected by centrifugation. The pellets were washed and resuspended in unfolding buffer (7 m guanidinium hydrochloride, 20 mm Tris-HCl, pH 7.5, and 10 mm DTT), and the supernatant was dialyzed against freshly made urea dialysis buffer (7 m urea, 10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 0.1 m NaCl, 5 mm 2-mercaptoethanol, and 0.2 mm PMSF) with three changes. The samples were then separated on DEAE and CM Sepharose columns run in tandem. The DEAE column was removed, and samples were eluted from the CM Sepharose column by salt gradient (urea buffer A, 7 m urea, 10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1 mm DTT, and 0.2 mm PMSF; urea buffer B, 7 m urea, 10 mm Tris-HCl, pH 8.0, 1 mm EDTA, 1 m NaCl, 1 mm DTT, and 0.2 mm PMSF). Fractions were monitored by absorbance and 15% SDS-PAGE. The purified histones were dialyzed against deionized water containing 5 mm 2-mercaptoethanol and 0.2 mm PMSF and lyophilized to dryness.
Histone octamer was prepared as described (32) and mixed with DNA at different ratios to optimize reconstitution. The molar ratio of octamer:DNA at 1.1:1 was found to yield the highest reconstitution efficiency. Nucleosomes were reconstituted by salt gradient dialysis and verified on 5% native polyacrylamide gels run in 0.25×TBE. The final nucleosome concentration was 50 nm in all FRET measurements.
Measuring UV Photoproduct Yields
For qualitative analysis with specific antibodies, UV-irradiated DNA was denatured by boiling for 5 min in 0.1 m NaOH. At each UV dose, equal amounts of DNA (100 ng for 6-4PPs and 2 ng for CPDs) were immobilized on nitrocellulose membranes with a Bio-Dot SF microfiltration apparatus (Bio-Rad). To detect UV lesions, the membrane was probed with antibodies against either 6-4PPs or CPDs (Cosmo Bio Co., Ltd, Japan). The membranes were developed with ECL plus kits (GE Healthcare) and scanned on a STORM 840 FluorImager (GE Healthcare). To determine loading amounts, 601 DNA labeled with 32P (using random sequence primers and PCR) was used to reprobe the membranes.
For quantitative analysis, a T4 DNA polymerase-exonuclease blockage assay was performed as described previously (33). Briefly, samples were incubated with 2.5 units of T4 DNA polymerase-exonuclease (Fermentas) at 37 °C for 2 h. The reaction was stopped by heating at 65 °C for 10 min. The DNA was blotted onto a nylon membrane and probed with 32P-labeled 601 DNA. The average dimer yield was calculated as described previously (33).
Photoreactivation of CPDs
Photoreactivation of UV-damaged DNA was carried out as described (34). The UV-irradiated DNA (40 pmol) was mixed with 1 μl photolyase (70 μm) in repair buffer (50 mm Tris-HCl pH7.5, 100 mm NaCl, 1 mm EDTA, and 10 mm DTT). The mixture was incubated in the dark for 5 min to allow photolyase binding to the CPDs, and the samples were irradiated with 365 nm light (Spectroline, model ENF-240C, Spectronics Corp., Westbury, NY) through a Pyrex filter for 30 min.
Fluorescence measurements
Fluorescence experiments were carried out at room temperature (23 °C) on a Photon Technology International Quantamaster UV/Vis Steady State Fluorometer. A quartz microcuvette with cover (105.250 QS, Hellma, Germany) was used to measure the fluorescence intensity. The samples were excited at 515 nm, and emission spectra were recorded from 550 to 700 nm. The Cy5 acceptor was excited at 615 nm directly for recording the acceptor-only emission spectra.
Determination of FRET Efficiency
FRET efficiencies were measured from the sensitized emission of the acceptor (A) by the ratio (A) method (35). FRET efficiency (E) was calculated using the following equations (36),
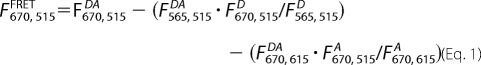 |
where F is the fluorescence signal, at the emission and excitation wavelengths indicated by subscripts, from the dyes indicated by superscripts (with D and A denoting Cy3 and Cy5, respectively); E is the FRET efficiency; ϵλ is the extinction coefficient at wavelength λ; and d is the fractional labeling of donor. These values are then used to determine the equilibrium constant (Keq) for the two states (wrapped and unwrapped), calculated from the relative FRET efficiency [p = (E/E0)] as Keq = (1 − P)/P (24).
Gel-based FRET
Naked DNA and nucleosomes were run on native 5% polyacrylamide gels in 0.25× TBE buffer. The gels were scanned on a Typhoon 9400 FluorImager (GE Healthcare), using the green laser (532 nm) and 580 BP 30 or 670 BP 30 bandpass emission filters. For acceptor-only fluorescence, gels were excited with a red laser (633 nm), and emission was captured through a 670 BP 30 bandpass filter. Images were displayed using FluorSepTM software (GE Healthcare).
Restriction Enzyme Digestion
The 601 sequence was prepared by EcoRV digestion of the plasmid pLMG601–23, which contains 23 tandem repeats of the 601 sequence (37). The released fragments (149 bp) were exposed to UV irradiation at different doses and used for nucleosome reconstitution, as described above. The reconstitution efficiency was verified on 5% native polyacrylamide gels, and only fully reconstituted NCPs were used for enzyme digestion. In each case, a 0.5-μg aliquot of NCPs was digested with 10 units of HaeIII or RsaI for 2 h at 37 °C, and the reaction was stopped by phenol-chloroform extraction. The digested samples were analyzed on 16% PAGE in TBE, and the gels were stained with SYBR gold and scanned on a STORM 840 FluorImager (GE Healthcare). Quantitative analysis of the digests was performed with ImageQuant software.
RESULTS
FRET System
To study nucleosome dynamics, we used purified recombinant histones, assembled into histone octamers, and the high affinity nucleosome-positioning sequence 601 (31), labeled with donor (D; Cy3) and acceptor (A; Cy5) fluorophores on opposite strands (Fig. 1A, upper panel). The 601 sequence, chosen to optimize the yield of homogeneously positioned DNA on the histone surface (31), was reconstituted into NCPs with histone octamers using stepwise salt dialysis (32). The FRET efficiency (E) relies on the inverse sixth power of the distance (R) between D and A [1 + (R/R0)6]−1 (38) and the Förster distance (R0), where E is 50%, is ∼6 nm for Cy3 and Cy5 (24). Therefore, in the naked 601 DNA, where R is ∼27 nm, little or no FRET is expected, whereas in NCPs, R is ∼3 nm (39), and efficient FRET is expected (Fig. 1A, lower panel). For these experiments, both naked DNA and NCPs were excited at 515 nm (donor excitation), and the emission spectra were measured from 550 nm to 700 nm. As expected, no FRET was observed with naked DNA (Fig. 1B), whereas robust FRET was observed with the NCP samples (Fig. 1B).
Nucleosomes show increased dissociation with increased salt concentration (24, 37, 40), and we used this property to further test the reliability of our FRET system. Widom and co-workers (24, 29) used 601-NCPs with dyes at the 5′-end of one of the DNA strands and at different positions on the core histones. In our case, both dyes are located in the internal region of the NCP DNA, analogous to the studies of Lohr and co-workers (26, 41) with different DNA sequences. To compare our data with these previous results, we examined the salt-induced dissociation of the 601 NCPs and found a clear decrease in FRET with increasing salt concentrations (Fig. 1C). At 0.75 m NaCl, Li and Widom (24) found that terminally labeled NCPs yield ∼40% FRET efficiency, whereas our internally labeled NCPs yield ∼60% FRET efficiency (Fig. 1C and see Fig. 4). These results highlight the increased dynamics associated with the terminal regions of NCP DNA (41).
FIGURE 4.

Salt-induced NCP dissociation profiles. The FRET efficiency at the lowest salt concentration (50 mm) was set as 1 for comparison of different UV-damaged NCPs. Values are means ± 1 S.D. for three independent experiments. Error bars for 0 kJ/m2 and 24 kJ/m2 are the only ones shown for clarity. The omitted error bars are all in the same range (≤4.5%) as those shown.
Nucleosomes Tolerate UV Photoproducts
We found that, in cells, DNA is damaged within the context of chromatin, and we tried to UV-irradiate the assembled NCPs for FRET measurement. However, a significant fraction of the Cy3- and Cy5-labeling dyes were photobleached following 254 nm UV irradiation in our experiments (see supplemental Fig. S1), confounding the FRET measurement. Therefore, we introduced photoproducts prior to nucleosome assembly, and the focus of this study is on the overall effect of UV lesions on nucleosome unwrapping, regardless of their nucleosome locations. In addition, we have shown that UV damage to the 5 S rDNA positioning sequence does not significantly change the rotational and translational settings in the nucleosome (12).
We synthesized two sets of oligonucleotides, one for fluorescent dye labeling and the other for UV treatment (see “Experimental Procedures”). Complete 147 bp 601 DNA containing both UV lesions and the fluorescent dyes was prepared by ligation and gel purification to ensure that the fragments contain exclusively DNA photoproducts (and essentially no DNA strand breaks). The UV-damaged DNA was reconstituted into NCPs with nearly the same efficiency as undamaged DNA (Fig. 2A), suggesting that incorporation of photolesions in the 601 sequence does not markedly affect the assembly of NCPs. This result is in agreement with our past studies using different sequences (42–44).
FIGURE 2.

UV damage does not affect NCP reconstitution with the 601 sequence. A, NCP reconstitution with UV-undamaged and -damaged DNA. The 147-bp 601 DNA containing both UV lesions and labeling were mixed with histone octamer at 2 m NaCl. The reconstitution was performed by stepwise salt dialysis, and the final NaCl concentration was 50 mm. The reconstituted products were resolved in 5% native polyacrylamide gel and stained with SYBR Gold. The 100-bp DNA markers are indicated on the left. B, presence of CPDs and 6-4PPs in UV-damaged DNA. The different UV-damaged DNA were blotted on the nitrocellulose and detected by lesion-specific antibodies. The same membranes were reprobed with 32P-labeled DNA to show equal loading. C, Southern blot of the photoproduct yield of the UV-irradiated DNA fragment. The DNA was treated with or without photolyase prior to the T4 DNA polymerase (pol) digestion. The digested samples were blotted on the nylon membrane and probed with with 32P-labeled DNA. D, quantification data of the photoproduct yield by Southern blots. The CPD signals were calculated by subtracting the total signals with the 6-4PPs signals. Three independent experiments were performed to show error bars.
Because the two DNA fragments used in these studies are short (95 bases each), we used UV doses in the kJ/m2 range to incorporate sufficient levels of UV lesions. Using specific antibodies to detect each photolesion, we observed that CPDs appear to saturate after ∼4 kJ/m2, whereas 6-4PPs appeared to increase over the UV dose range used (Fig. 2B). To quantify the level of these photoproducts, we used an exonuclease blockage assay developed previously (33). The 3′ → 5′ exonuclease activity of T4 DNA polymerase, in the absence of dNTPs, is blocked quantitatively by CPDs and 6-4PPs (45), and these phototproducts can be detected by slot blot after T4 DNA polymerase digestion (33). In some samples, CPDs were removed by treatment with UV photolyase, which allowed detection of either both photoproducts together (no photolyase) or just 6-4PPs (with photolyase) following T4 DNA polymerase digestion (Fig. 2C). The average numbers of CPD/fragment and 6-4PPs/fragment (33), show that the level of CPDs starts to saturate at 4 kJ/m2, and the maximum level was 0.25 CPDs/fragment for the highest dose tested (Fig. 2D). However, 6-4PPs increased almost linearly up to the highest UV dose used (24 kJ/m2; Fig. 2D). This data agrees well with the antibody data (Fig. 2B) and indicates that up to 40–45% of the fragments contain a UV lesion in these experiments.
UV Damage Drives NCPs to the Unwrapped State
To examine nucleosome unwrapping in damaged and undamaged NCPs, we initially used gel-based FRET, which enabled direct visualization of energy transfer on multiple samples (46). The labeled naked DNA and NCPs were resolved on native polyacrylamide gels and examined through different emission filters to visualize FRET in each sample. As shown in Fig. 3A (top panel), scanning gels with the green laser (532 nm; donor excitation) yields the unquenched Cy3 signals in both naked DNA and NCPs. In the second panel of Fig. 3A, excitation of the donor (green laser) results in Cy5 fluorescence (shown in red by using the 670 nm emission filter) only from the NCP bands. No FRET signal is detected in the naked DNA bands. Furthermore, the Cy5 signal from NCP bands becomes weaker with increased UV dose to the DNA when visualized either directly (Fig. 3A, second panel) or when the signals are merged (third panel). Quantifications of the fluorescence intensities show that the FRET efficiency decreases with increased UV irradiation (Fig. 3A, numbers between panels). Finally, scanning the gels by direct excitation of the acceptor with a red laser (633 nm) yields comparisons of labeling efficiency and loading, which are approximately the same for the gel shown (Fig. 3A, bottom panel). Therefore, the gel-based FRET results indicate that there is reduced energy transfer efficiency in the UV damaged NCPs.
FIGURE 3.

Incorporation of UV lesions drives nucleosome unwrapping. A, energy transfer shown by gel-based FRET. NCPs reconstituted with undamaged and damaged DNA were resolved on a 5% native polyacrylamide gel and then scanned on Typhoon 9400. The excitation lasers and emission filters are shown on the left. The green laser was excited at 532 nm, and the red laser was excited at 633 nm. 580 BP 30 and 670 BP 30 are the band pass emission filters that pass the band of lights centered at 580 nm or 670 nm. The FRET efficiency was calculated as E = IA/(IA + γID) (26), where γ is 1.0 in this case and is shown in the middle of the panels. B, emission spectra of damaged 601 NCPs following different UV doses. The final NaCl concentration was 50 mm. The samples were excited at 515 nm, and emission spectra from 550 nm to 700 nm were recorded. The data were normalized by Cy5 signals excited at 615 nm. C, relative FRET efficiency (E/E0) for the NCP irradiated at different UV doses. E is the FRET efficiency of UV-damaged NCPs, and E0 is the FRET efficiency of undamaged NCPs, which was set as 1. D, equilibrium constants (Keq) for partial DNA unwrapping as a function of UV damage. The constants were calculated from the data in B as described before (24).
To quantify the effect of UV dose on the FRET signal, we measured the FRET efficiency in solution. The emission spectra clearly demonstrate that the FRET efficiency decreases with increased UV dose (Fig. 3B). For comparison, the FRET efficiency from the sensitized emission of the acceptor was normalized to the FRET efficiency of the nondamaged, fully reconstituted NCPs. As seen in Fig. 3C, the efficiency decreased more over the first 8 kJ/m2 UV dose than at higher UV doses. At the highest UV dose used (24 kJ/m2), the efficiency was reduced by at least 30% compared with nondamaged NCPs (Fig. 3C).
At a low salt concentration, NCPs exhibited a dynamic conformational equilibrium between fully wrapped and partially unwrapped states, with equilibrium constant, Keq (24). We used this constant to describe the change in conformational equilibrium induced by UV DNA lesions. This analysis clearly shows that the equilibrium shifts from wrapped to unwrapped states with increased DNA damage (Fig. 3D), suggesting that a given UV-damaged nucleosome spends more time in an unwrapped state compared with that of an undamaged nucleosome. At the highest dose tested (24 kJ/m2), we calculate that the NCP population in our samples spends almost 50% more time in an unwrapped state. This is the case, even though <50% of the NCPs in our samples contain a UV-induced lesion (Fig. 2D). Thus, the change in equilibrium between wrapped and unwrapped states of just the UV-damaged NCPs in these samples is substantial.
Salt Dependence of UV-damaged NCPs
The electrostatic interaction between histone octamers and DNA is weakened by increased ionic strength of the solvent (47), and consequently, the dynamic properties of NCPs are affected (24). To determine whether UV damage changes the salt-induced dynamic properties of NCPs, we titrated NCPs with increasing salt and compared the FRET efficiency with different levels of UV lesions. For NCPs damaged at a given UV dose, the FRET efficiency (E) was normalized to the efficiency at the lowest salt concentration (50 mm NaCl). At low salt concentrations (below 250 mm NaCl), where changes are associated with DNA “breathing” (37, 40), there was little change in FRET (Fig. 4). However, we observed a small, yet significant, variation in FRET between 0.25 m and 1 m NaCl salt (Fig. 4). The undamaged NCPs yield a higher FRET in this salt concentration range compared with UV-damaged NCPs. This indicates that, on average, the distance between the two dyes is larger in the NCPs containing UV lesions, most likely due to increased unwrapping of the DNA in damaged NCPs. At a 1.5 m NaCl concentration, the FRET efficiency decreases to <10% of the low salt value, indicating the DNA is dissociated from histone octamers.
Restriction Enzyme Accessibility of Damaged NCPs
The FRET data indicate that UV-damaged NCPs exist in open structures more often than nondamaged NCPs. Therefore, UV-damaged nucleosomes may be more accessible to site-specific DNA-binding proteins. REA is a method that has been successfully used to measure spontaneous nucleosomal DNA exposure (23, 25, 48). We tested the REA of nucleosome DNA using two restriction sites in the 601 sequence to examine digestion at different locations from the DNA ends in NCPs. A HaeIII site is located near an end of DNA within the NCP, whereas an RsaI site is close to the dyad center (Fig. 5A). For these experiments, we decreased the UV dose to adjust for the exposure of the full-length 601 fragment (149 bp) versus the shorter single-strand fragment (95-mer) used in the FRET experiments. As a control, naked DNA (± UV damage) was digested by the enzymes and showed no differences in activity (supplemental Fig. S2), highlighting the fact that any UV-associated changes in REA should reflect nucleosome dynamic variations rather than DNA structural changes.
FIGURE 5.

Accessibility of restriction enzyme to UV-damaged NCPs. A, schematic illustration of the restriction enzyme digestion sites on the 601 sequence. The DNA was prepared from an EcoRV digestion of the plasmid pLMG601-23, which contained 23 tandem repeats of the 149-bp 601 sequence (37). B, RsaI digestion of NCPs. The UV doses were reduced corresponding to the increase of the DNA length (from 95 bp to 149 bp). S denotes substrates and P1 and P2 denote digestion products. The digestion products were separated on a 16% native polyacrylamide gel and stained with SYBR Gold. The gels were scanned on a STORM 840 FluorImager. C, HaeIII digestion of NCPs. The experiments were performed the same way as in B. D, quantitative analysis of the digestion of UV-damaged NCPs. The fraction undigested was defined as (counts in S)/(counts in S + P1 + P2).
As shown in Fig. 5, these two restriction sites are only slightly accessible in NCP DNA (Fig. 5, B and C, P1 and P2 fragments in lane 0). Furthermore, there are no obvious differences in RsaI REA among UV-treated NCPs at different UV doses (Fig. 5B, lanes 2.5–15, kJ/m2), where only ∼10% of the DNA is cleaved. This result suggests that UV damage does not lead to long range opening events of nucleosome DNA, near the dyad center of NCPs. However, NCPs bearing UV lesions show enhanced REA to HaeIII (Fig. 5C), reflected by enhanced yields of long (P1) and short (P2) restriction fragments with increased UV dose (Fig. 5C, lanes 2.5–15 kJ/m2). Quantification of these results shows that up to ∼30% more HaeIII sites in NCPs are rendered accessible over this UV dose range (Fig. 5D). (We note that a minor fraction of NCPs can dissociate during mixing with REA buffer (23, 49), giving rise to a small drop in the fraction of undigested DNA (Fig. 5D, lane 0.)) These results indicate that UV lesions promote greater NCP unwrapping within at least the first ∼30 bp from the ends of NCP DNA. Moreover, the UV-dose dependence suggests that the magnitude and rate of the unwrapping-rewrapping fluctuations are influenced by the extent of UV damage. These data support the “site-exposure” model, where NCP associated DNA peels off from the end, facilitating invasion of nucleosomes by DNA binding proteins (24).
A Single UV Lesion Drives NCPs Unwrapping
Our analysis with UV-irradiated and nonirradiated NCPs demonstrates that UV lesions enhance nucleosome dynamics (Fig. 3). However, as “global” UV irradiation incorporates both 6-4PPs and CPDs at variable nucleosome positions and in only a fraction of the NCPs (Fig. 2), we generated site-specific UV-damaged 601 fragments by incorporating a single CPD (601.DF1 CPD) or 6-4PP (601.DF2 6-4PP) into the same position of 601 NCPs (58 nt from the 5′-end; Fig. 6A and supplemental Fig. S3). As observed with other DNA sequences (43, 44, 50), these UV-damaged 601 fragments can be reconstituted successfully with histone octamers (supplemental Fig. S4). In addition, we observed significant retardation of the naked 601.DF2 6-4PP fragments on gels, possibly the result of enhanced DNA bending of these fragments, although this did not prevent NCP formation (Fig. S4B). Finally, the presence of a single CPD in the 601.DF1 CPD fragments was confirmed by complete digestion with T4 endoV (data not shown).
We then examined the effect of each of these lesions on nucleosome dynamics. As can be seen in Fig. 6, B and C, the FRET efficiency was reduced by ∼16% in CPD-containing NCPs, and ∼9% in 6-4PP-containing NCPs compared with undamaged NCPs. Given the fact that the donor-acceptor distance (R) for undamaged NCPs is 3 nm (39), R for NCPs containing a single CPD or 6-4PP lesion is ∼4.6 nm and ∼4.2 nm, respectively. Furthermore, in each case, the FRET efficiency is reduced, indicating that the introduction of a single UV lesion in nucleosome DNA can increase NCP unwrapping. This supports the UV dose-dependent FRET change we observed with UV-irradiated NCP DNA (Fig. 3). Moreover, NCPs with either a single CPD or 6-4PP are more sensitive to salt (Fig. 6, D and E). In the range of 0.25–1 m salt concentrations, the FRET efficiency in NCPs with single UV lesions is significantly lower than that of undamaged NCPs, indicating that NCPs containing just one DNA photoproduct are significantly less stable with increasing salt.
DISCUSSION
The interactions between histones and DNA create a hindrance to recognition and access to DNA lesions by DNA repair proteins. This barrier may be overcome, however, if DNA lesions change the structural and/or dynamic properties of nucleosomes to promote the accessibility of repair factors, including ancillary proteins such as chromatin remodeling complexes. In this study, the unwrapping dynamics of nucleosomes containing UV lesions, either by UV irradiation or incorporation of a single UV photoproduct, was monitored in reconstituted NCPs consisting of the model 601 DNA sequence and recombinant histone octamers. The presence of UV lesions did not hinder the reconstitution of NCPs, allowing for measurement of their dynamic properties. Results from two experimental strategies, FRET and REA, indicate that the equilibrium of dynamic unwrapping-rewrapping fluctuations shifts toward the unwrapped states with increased DNA damage. Furthermore, we found that a single UV photoproduct (either CPD or 6-4PP) in NCPs is sufficient to drive nucleosomes toward the more open state. Thus, UV damaged nucleosomes spend more time in unwrapped states.
The fact that UV lesions do not prevent bulk nucleosome assembly (Fig. 2) but do affect salt stability (Fig. 4) may seem conflicting. However, as UV lesions cause local distortion in DNA structure that is not necessarily propagated along the DNA (e.g. blocked by the strong positioning ability of the 601 sequence), the local distortion may not be able to disrupt the overall interaction between histones and DNA, translating into successful nucleosome reconstitution with different UV damaged DNA fragments (Fig. 2A). In addition, UV lesions may change the local “sequence adjustment” of the DNA, which may affect the salt stability and intrinsic site exposure of individual bases, yielding an increased sensitivity to salt (Fig. 4). Finally, UV photoproducts may destabilize histone-DNA interactions at specific damaged sites, which could promote DNA peeling off the histone core and partial unwrapping from the end (Fig. 5).
Interestingly, a CPD at position 58 (15 bp from the dyad center; or SHL1.5 (2)) appears to be more efficient in driving unwrapping of NCPs than a 6-4PP at this position (Fig. 6, B and C). The histone DNA binding surface at this location causes an outward bulge in the DNA and shows significant bending (2). The solution structure of duplexes containing a 6-4PP shows a greater extent of helix distortion than identical duplexes containing a CPD (supplemental Fig. S4, naked DNA). The ∼44° kinking angle (9) around the 6-4PP at site 58 is expected to alter the DNA curvature at this position, which could moderate the increase in rate of nucleosome unwrapping and/or change the average distance (R) between the dyes. At the same time, CPDs should cause less structural deformity at this site, and the enhanced nucleosome unwrapping dynamics caused by this lesion may be less affected by changes in DNA curvature. Clearly, a definitive answer only may be obtained with crystal structures of NCPs containing either of these two DNA lesions at this specific location. Furthermore, it will be interesting to examine the effects of these DNA photoproducts at other locations in NCPs on nucleosome-unwrapping dynamics.
The REA results indicate that unwrapped states of the UV-damaged NCPs occur primarily in the terminal regions of the DNA (Fig. 5). Previous work on undamaged nucleosomes by FRET and REA has shown that DNA unwraps as far into NCPs as the dyad axis (25, 41). Our results suggest that the presence of UV lesions mainly drive the unwrapping (or breathing) of DNA near the edges of nucleosomes, as UV lesions did not enhance the REA of RsaI near the dyad center of NCPs. As the change in conformational equilibrium and enhanced accessibility of damaged nucleosomes does not require the involvement of other factors, the UV damage-induced unwrapping is an intrinsic property of NCPs.
In the context of damaged nucleosomes, lesions must be exposed and accessed by the repair machinery to remove the damaged bases. The breathing of nucleosomal DNA ends, which exist in equilibrium between associated and dissociated from the histone octamer, may serve to allow this accession to take place. Partial dissociation of the DNA ends from histone octamers has been demonstrated by monitoring the salt-dependent changes in FRET (40). Furthermore, NCPs are assembled with DNA that binds tightly to the (H3-H4)2 tetramer and position about the dyad center. The (H2A-H2B) dimers can exchange dynamically in vivo (51) and this may play an important role in ATP-dependent nucleosome remodeling (52, 53). Moreover, dissociation of (H2A-H2B) dimers has been detected in vitro by FRET over increased salt concentrations (37). Using this technique, we found that both UV irradiated NCPs and NCPs containing a single UV lesion are significantly less stable to salt-induced dissociation (Figs. 4, 6, D and E). It is possible that, UV-damaged NCPs undergo conformational changes more easily, and/or preferentially release (H2A-H2B) dimers, with increased salt, both of which would facilitate DNA lesion access by repair proteins.
Enhanced unwrapping of damaged nucleosomes may provide sufficient time for passive binding of repair proteins. Indeed, Suter and Thoma have shown that the single-subunit protein, UV photolyase, is strongly inhibited by nucleosomes in vitro, yet is capable of gaining rapid access to CPDs in nucleosomes of intact yeast cells (54). Importantly, these authors found that repair rates are slower in the central regions of nucleosome DNA and faster near the terminal ends (54). These observations are consistent with the intrinsic site-exposure model and the dynamic enhancement of damaged nucleosomes yielding more time for UV photolyase proteins to recognize CPDs in chromatin. Access and binding to nucleosomes by UV photolyase may be possible through the spontaneous “trapping” of partially unwrapped nucleosomes, in which the terminal DNA is transiently released from the histone surface. Thus, compared with nucleosome sliding or histone eviction, the site-exposure mechanism is a rapid, energy-efficient way for repair proteins to gain access to DNA lesions, at least near the ends of nucleosome DNA.
Nucleotide excision repair is the main pathway used in most organisms (including human) to repair UV lesions (5), and nucleosome rearrangements occur during nucleotide excision repair in chromatin (55, 56). Furthermore, it has been shown that ATP-dependent chromatin remodeling factors and histone modification are involved in the DNA repair process (14, 20, 21). However, it is unclear how these remodeling and modification factors are recruited to sites of damaged DNA associated with nucleosomes. Our data implies that intrinsic nucleosome dynamics, especially increased unwrapping of UV-damaged NCPs, facilitate the invasion of factors involved in repair and/or those involved in remodeling or histone modifications. The binding of damage specific factors should further shift the equilibrium toward the unwrapped states. Thus, once repair recognition factors and/or remodeling factors are recruited to the damaged nucleosomes, disruption of local chromatin structure could initiate the “cascade” of recruitment of nucleotide excision repair proteins (5).
Supplementary Material
Acknowledgments
We thank Drs. William Davis and Lisa Gloss (School of Molecular Biosciences, Washington State University) for help with FRET measurements and for providing the pLMG601–23 plasmid, respectively. We also thank Dr. Aziz Sancar (Department of Biochemistry and Biophysics, University of North Carolina) for providing purified Caulobacter crescentus UV photolyase, and Drs. Nicholas Geacintov and Aleksandr Kolbanovskiy (Chemistry Department, New York University) for providing purified single CPD and 6-4PP containing oligonucleotides. Finally, we thank Drs. John Hinz and Lisa Gloss for critical evaluation of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant ES004106 from the NIEHS.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- NCP
- nucleosome core particle
- DF
- damage fragment
- REA
- restriction enzyme accessibility
- CPD
- cyclobutane pyrimidine dimer
- 6-4PP
- pyrimidine-6-4-pyrimidone photoproduct
- nt
- nucleotide
- TBE
- Tris borate-EDTA.
REFERENCES
- 1.Kornberg R. D. (1974) Science 184, 868–871 [DOI] [PubMed] [Google Scholar]
- 2.Luger K., Mäder A. W., Richmond R. K., Sargent D. F., Richmond T. J. (1997) Nature 389, 251–260 [DOI] [PubMed] [Google Scholar]
- 3.Kornberg R. D., Lorch Y. (1999) Cell 98, 285–294 [DOI] [PubMed] [Google Scholar]
- 4.Hanawalt P. C. (1998) Mutat Res. 400, 117–125 [DOI] [PubMed] [Google Scholar]
- 5.Friedberg E. C., Walker G. C., Siede W. (2006) DNA Repair and Mutagenesis, 2nd Ed., pp. 617–620, ASM Press, Washington, DC [Google Scholar]
- 6.Cleaver J. E. (2005) Nat. Rev. Cancer 5, 564–573 [DOI] [PubMed] [Google Scholar]
- 7.Hoeijmakers J. H. (2001) Nature 411, 366–374 [DOI] [PubMed] [Google Scholar]
- 8.Park H., Zhang K., Ren Y., Nadji S., Sinha N., Taylor J. S., Kang C. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 15965–15970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim J. K., Choi B. S. (1995) Eur. J. Biochem. 228, 849–854 [DOI] [PubMed] [Google Scholar]
- 10.Gale J. M., Nissen K. A., Smerdon M. J. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 6644–6648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gale J. M., Smerdon M. J. (1990) Photochem. Photobiol. 51, 411–417 [DOI] [PubMed] [Google Scholar]
- 12.Liu X., Mann D. B., Suquet C., Springer D. L., Smerdon M. J. (2000) Biochemistry 39, 557–566 [DOI] [PubMed] [Google Scholar]
- 13.Schieferstein U., Thoma F. (1998) EMBO J. 17, 306–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaillard H., Fitzgerald D. J., Smith C. L., Peterson C. L., Richmond T. J., Thoma F. (2003) J. Biol. Chem. 278, 17655–17663 [DOI] [PubMed] [Google Scholar]
- 15.Thoma F. (2005) DNA Repair 4, 855–869 [DOI] [PubMed] [Google Scholar]
- 16.Smith C. L., Peterson C. L. (2005) Curr. Top Dev. Biol. 65, 115–148 [DOI] [PubMed] [Google Scholar]
- 17.Zhang L., Jones K., Gong F. (2009) Biochem. Cell Biol. 87, 265–272 [DOI] [PubMed] [Google Scholar]
- 18.Li B., Carey M., Workman J. L. (2007) Cell 128, 707–719 [DOI] [PubMed] [Google Scholar]
- 19.Allis C. D., Jenuwein T., Reinberg D. (2007) Epigenetics, pp. 193–206, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York [Google Scholar]
- 20.Gong F., Fahy D., Smerdon M. J. (2006) Nat. Struct. Mol. Biol. 13, 902–907 [DOI] [PubMed] [Google Scholar]
- 21.Yu Y., Teng Y., Liu H., Reed S. H., Waters R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 8650–8655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Teng Y., Yu Y., Ferreiro J. A., Waters R. (2005) DNA Repair 4, 870–883 [DOI] [PubMed] [Google Scholar]
- 23.Polach K. J., Widom J. (1995) J. Mol. Biol. 254, 130–149 [DOI] [PubMed] [Google Scholar]
- 24.Li G., Widom J. (2004) Nat. Struct. Mol. Biol. 11, 763–769 [DOI] [PubMed] [Google Scholar]
- 25.Anderson J. D., Thåström A., Widom J. (2002) Mol. Cell. Biol. 22, 7147–7157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kelbauskas L., Chan N., Bash R., Yodh J., Woodbury N., Lohr D. (2007) Biochemistry 46, 2239–2248 [DOI] [PubMed] [Google Scholar]
- 27.Widom J. (2001) Q. Rev. Biophys. 34, 269–324 [DOI] [PubMed] [Google Scholar]
- 28.Kaplan N., Moore I. K., Fondufe-Mittendorf Y., Gossett A. J., Tillo D., Field Y., LeProust E. M., Hughes T. R., Lieb J. D., Widom J., Segal E. (2009) Nature 458, 362–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li G., Levitus M., Bustamante C., Widom J. (2005) Nat. Struct. Mol. Biol. 12, 46–53 [DOI] [PubMed] [Google Scholar]
- 30.Bucceri A., Kapitza K., Thoma F. (2006) EMBO J. 25, 3123–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lowary P. T., Widom J. (1998) J. Mol. Biol. 276, 19–42 [DOI] [PubMed] [Google Scholar]
- 32.Luger K., Rechsteiner T. J., Richmond T. J. (1999) Methods Enzymol. 304, 3–19 [DOI] [PubMed] [Google Scholar]
- 33.Liu X., Smerdon M. J. (1995) Anal. Biochem. 229, 323–328 [DOI] [PubMed] [Google Scholar]
- 34.Oztürk N., Kao Y. T., Selby C. P., Kavakli I. H., Partch C. L., Zhong D., Sancar A. (2008) Biochemistry 47, 10255–10261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clegg R. M. (1992) Methods Enzymol. 211, 353–388 [DOI] [PubMed] [Google Scholar]
- 36.Yang J. G., Madrid T. S., Sevastopoulos E., Narlikar G. J. (2006) Nat. Struct. Mol. Biol. 13, 1078–1083 [DOI] [PubMed] [Google Scholar]
- 37.Hoch D. A., Stratton J. J., Gloss L. M. (2007) J. Mol. Biol. 371, 971–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lakowicz J. R. (2006) Principles of Fluorescence Spectroscopy, 3rd Ed., pp. 13–15, Springer, New York [Google Scholar]
- 39.Davey C. A., Sargent D. F., Luger K., Maeder A. W., Richmond T. J. (2002) J. Mol. Biol. 319, 1097–1113 [DOI] [PubMed] [Google Scholar]
- 40.Park Y. J., Dyer P. N., Tremethick D. J., Luger K. (2004) J. Biol. Chem. 279, 24274–24282 [DOI] [PubMed] [Google Scholar]
- 41.Kelbauskas L., Sun J., Woodbury N., Lohr D. (2008) Biochemistry 47, 9627–9635 [DOI] [PubMed] [Google Scholar]
- 42.Mann D. B., Springer D. L., Smerdon M. J. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 2215–2220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kosmoski J. V., Smerdon M. J. (1999) Biochemistry 38, 9485–9494 [DOI] [PubMed] [Google Scholar]
- 44.Svedruzić Z. M., Wang C., Kosmoski J. V., Smerdon M. J. (2005) J. Biol. Chem. 280, 40051–40057 [DOI] [PubMed] [Google Scholar]
- 45.Doetsch P. W., Chan G. L., Haseltine W. A. (1985) Nucleic Acids Res. 13, 3285–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramirez-Carrozzi V., Kerppola T. (2001) Methods 25, 31–43 [DOI] [PubMed] [Google Scholar]
- 47.Mihardja S., Spakowitz A. J., Zhang Y., Bustamante C. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 15871–15876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poirier M. G., Bussiek M., Langowski J., Widom J. (2008) J. Mol. Biol. 379, 772–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Polach K. J., Widom J. (1999) Methods Enzymol. 304, 278–298 [DOI] [PubMed] [Google Scholar]
- 50.Hara R., Sancar A. (2003) Mol. Cell. Biol. 23, 4121–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kimura H., Cook P. R. (2001) J. Cell Biol. 153, 1341–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bruno M., Flaus A., Stockdale C., Rencurel C., Ferreira H., Owen-Hughes T. (2003) Mol. Cell 12, 1599–1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vicent G. P., Nacht A. S., Smith C. L., Peterson C. L., Dimitrov S., Beato M. (2004) Mol. Cell 16, 439–452 [DOI] [PubMed] [Google Scholar]
- 54.Suter B., Thoma F. (2002) J. Mol. Biol. 319, 395–406 [DOI] [PubMed] [Google Scholar]
- 55.Smerdon M. J., Lieberman M. W. (1978) Proc. Natl. Acad. Sci. U.S.A. 75, 4238–4241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Smerdon M. J. (1991) Curr. Opin Cell Biol. 3, 422–428 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.