

Abstract
Mammalian airways normally regulate the volume of a thin liquid layer, the periciliary liquid (PCL), to facilitate the mucus clearance component of lung defense. Studies under standard (static) culture conditions revealed that normal airway epithelia possess an adenosine-regulated pathway that blends Na+ absorption and Cl− secretion to optimize PCL volume. In cystic fibrosis (CF), the absence of CF transmembrane conductance regulator results in a failure of adenosine regulation of PCL volume, which is predicted to initiate mucus stasis and infection. However, under conditions that mimic the phasic motion of the lung in vivo, ATP release into PCL was increased, CF ion transport was rebalanced, and PCL volume was restored to levels adequate for lung defense. This ATP signaling system was vulnerable, however, to insults that trigger CF bacterial infections, such as viral (respiratory syncitial virus) infections, which up-regulated extracellular ATPase activity and abolished motion-dependent ATP regulation of CF PCL height. These studies demonstrate (i) how the normal coordination of opposing ion transport pathways to maintain PCL volume is disrupted in CF, (ii) the hitherto unknown role of phasic motion in regulating key aspects of normal and CF innate airways defense, and (iii) that maneuvers directed at increasing motion-induced nucleotide release may be therapeutic in CF patients.
The lung must continually defend itself against bacteria that deposit on airway surfaces during normal tidal breathing. It appears that mechanical clearance of bacteria mediated by mucus transport is the principal innate defense mechanism of mammalian airways (1–4). Recent data have shown that a critical component of this defense system is the thin (~7) μm liquid layer lining airway surfaces, the periciliary liquid (PCL),2 that provides a low viscosity solution for ciliary beating and acts a lubricant layer for mucus transport (5, 6). In cystic fibrosis (CF) lung disease, it appears that the primary pathophysiologic defect is the depletion of PCL volume, resulting in a failure of mucus clearance of bacteria and persistent airways infection (7, 8).
However, questions have been raised as to the relevance of PCL depletion to CF pathogenesis in vivo (9). For example, whereas in vitro data from standard (static) culture systems describe rapid depletion of PCL height and a complete failure of mucus transport (7), young CF patients exhibit reduced but measurable rates of mucus clearance in vivo (10). This inconsistency suggests that mechanisms for PCL height regulation operating in vivo are absent from standard static culture systems. In addition, clinical observations suggest that CF lung disease exacerbates intermittently and is heterogeneous. Often, viral infections trigger these disease exacerbations (11, 12), but no links between viral infection and PCL regulation have been reported.
To investigate these questions, we used a well differentiated airway epithelial culture system that exhibits PCL volume regulation and mucus transport (7). Based on the observations that (i) tidal volume expansion and airflow impart shear to airway surfaces (13), (ii) shear releases nucleotides from many cell types into the extracellular environment (14, 15), and (iii) extracellular nucleotides interact with P2Y2 and P2X purinoceptors to regulate airway ion transport (16, 17), we hypothesized that the failure to recapitulate in vitro the phasic motion associated with tidal breathing has hindered the identification of the pathways for PCL volume autoregulation. Accordingly, we developed a novel culture system that mimics the best characterized shear stress in the lung, i.e. the phasic motion-induced shear that is associated with normal tidal breathing. Finally, we used a respiratory syncitial virus (RSV) encoding the GFP protein (18) to study the relationship between RSV infection and the capacity of CF cells to maintain a PCL under phasic motion culture conditions.
EXPERIMENTAL PROCEDURES
Human Airway Epithelial Cultures
Cells were obtained from freshly excised bronchial specimens from normal (NL; n = 15; 4–55 years; 9 males, 6 females) and CF subjects (n = 21; 13–49 years; 12 males; 9 females; 13 ΔF/ΔF, 4 ΔF/other, 4 unknown genotype) by protease digestion (7), seeded directly as primary cultures on 12-mm Transwell Col membranes (T-Col; Costar) in modified BEGM (62) media under air-liquid interface (ALI) conditions, and studied when fully differentiated (2–5 weeks). Cultures with transepithelial resistances (Rt) of >200 ohms-cm2 were studied and were kept in a highly humidified incubator between readings (7).
Confocal Microscopy Measurement of PCL
PBS (20 μl) containing 2 mg/ml Texas Red-dextran (10 kDa; Molecular Probes) was added to cultures. To measure the average height of the PCL, 5 predetermined points, 1 central and four 2 mm from the edge of the culture, were XZ scanned (19). For all studies, perfluorocarbon (PFC; FC-77) was added mucosally to prevent evaporation of the PCL (20), and the culture was placed on the stage of the confocal microscope over a serosal reservoir (80 μl TES-buffered Ringer).
Measurement of Transepithelial Potential Difference (Vt)
A macro-electrode (polyethylene tubing with 3 M KCl, 4% agar) was placed in the serosal bath, and a 3 M KCl-filled glass microelectrode was positioned by a micromanipulator into the PCL to stably record Vt. PFC was added to the mucosal surface during the period of recording to avoid PCL evaporation.
Morphologic Studies
We utilized a non-aqueous fixative that preserves the topology of the PCL in situ (7). Cultures were fixed using 1% osmium tetraoxide in perfluorocarbon (OsO4/PFC; FC-72). After fixation, samples were directly immersed in 100% ethanol, processed for epon/araldite resin embedding using conventional procedures, and sections prepared for electron microscopy.
Measurement of Mucus Transport Rates
PBS (20 μl), containing a 0.02% v/v fluorescent microsphere suspension (1 μm, green fluorescence), was added to cultures screened for rotational mucus and Na+ transport. Measurements of mucus transport were made at t = 0 h and 48 h later for cultures under static versus phasic motion conditions from 5-s exposure images acquired with an inverted epifluorescence microscope (Leica, Germany) and a CCD camera (Hamamatsu C5985, Japan), and the linear velocity of bead transport was analyzed as previously reported (7).
Phasic Motion Culture System and Generation of Shear Forces
We elected to accelerate/decelerate the cultures inside a highly humidified incubator to deliver phasic shear stress to avoid airflow-induced evaporation of the PCL. The change in velocity caused by this phasic motion generated shear stress over the apical cell membrane with profiles similar to airflow-induced shear stress (see the supplemental material). We built a series of devices to control the magnitude and frequency of the phasic motion/shear stress exerted on human bronchial airway epithelial cultures. The prototype device was built from motorized Lego (Billund, Denmark) coupled to a signal generator, which provided the voltage to power the motor. Subsequent, computer controlled rotational devices were machined in-house with identical dimensions. Four cultures housed in a 6-well plate (4.5 cm equidistant from the center of rotation) were placed in this motorized device, and a square wave was generated at 0.5–1 Hz to drive the DC motor, rotating the cultures in a stop/go fashion. Because of the inertia of the device, this resulted in an acceleration period of 100 ms rather than an instantaneous increase in velocity as implied by the square wave signal. Consequently, the device produced a maximal velocity of 16.6 cm/s at the center of each culture for 550 ms, with acceleration/deceleration periods of 100 ms and 7 stop/starts per 360°. This change in velocity was similar to changes seen during in vivo inspiration during normal tidal breathing as a recorded by a pneumotachometer (see the supplemental material).
For the dose-response to phasic motion, the rates of acceleration/deceleration were altered from 1 s (0.06 dynes/cm2) to 10 ms (6 dynes/cm2) while maintaining 28 cycles/min of phasic motion (note that to generate 0.06 dynes/cm2 of phasic motion while maintaining 28 cycles/min, it was also necessary to decrease the maximum angular velocity to generate the reduced rate of shear stress). Note, normal tidal breathing has both inspiratory and expiratory airflows, so for 14 breaths/min there are 28 cycles/min of shear stress.
Measurement of Intracellular Ca2+ (Ca2+i) and Vt
Ca2+i and Vt were measured in CF epithelial cultures on T-Col membranes interfaced to a microfluorimeter (21). Luminal perfusion (Na+-free KBr ± apyrase) was initiated and stopped at 5-min intervals, whereas KBr perfusion was continuous in the basolateral compartment. Calculations based on the geometry (volume) of the chamber and flow rate to predict shear force were performed as follows.
The inlet flow rate of Ringers solution (ρ ~ 1 g/cm3) into the luminal chamber was 4 cm3/min (Q) through 20-gauge tubing (r = 0.0292 cm inner radius, V = Q/πr2), which directed flow downward to the luminal surface of the culture at a 30° angle. Thus, the force exerted on the surface of the culture was F = ρQV sinθ = 0.83 dynes. This inlet jet exited from tubing 0.6 cm away from the culture surface, directed toward the center of the culture surface, a circular disk of ~0.15 cm in radius. Jet flow upon leaving tubing entrains the surrounding fluid and expands, and the momentum of the flow is transferred to the surrounding fluid. For a water system the jet entrainment angle for a circular jet is about 14°. In this case this would distribute force over almost the entire disk culture surface of 0.1 cm2. Therefore, the expected shear stress applied in this case was ~8 dynes/cm2.
Measurement of PCL ATP and adenosine (ADO) Concentrations
Pilot studies revealed that it was difficult to sample PCL after >3 h due to the viscosity of PCL but that [nucleotide] after a maneuver reached a new steady state within 15 min. Therefore, at t = 0, 50 μl of PBS ± 5 units of apyrase was placed on the culture surface, the culture placed in the incubator under static or phasic motion conditions, and 5–10 μl of PCL was sampled 1 h later by microcapillary pipettes lowered onto the culture surface with a micromanipulator. ATP was measured by luminometry (22), and ADO was measured by HPLC after etheno-derivatization (23).
Virus Infection and ATPase Activity
CF airway epithelia were inoculated with a recombinant replication-competent RSV encoding a GFP reporter gene (18) 48 h before confocal measurements of PCL height. Parallel cultures had extracellular hydrolysis measured with [γ-32P]ATP or Ap4A by HPLC as previously described (24). For media transfer studies, PCL and culture media was collected with micropipettes, exposed to UV light for 30 min to kill viruses, and transferred to uninfected cultures.
Solutions and Chemicals
During thin film experiments, cultures were bathed serosally in a TES-buffered, modified Ringer solution, and PBS was used as a volume load and for washing the apical surface. Amiloride (300 μM), Ap4A (100 μm), apyrase (5 units/ml), bumetanide (100 μM), 8-SPT (10 μM), and all salts were obtained from Sigma-Al-drich. All fluorescent compounds and DIDS were obtained from Molecular Probes. [γ-32P]ATP (10 μm) was obtained from Amersham Biosciences. PFCs (FC-72 and FC-77) were obtained from 3M and had no effect on PCL height, rotational mucus transport rate, PCL [Cl− ], or Vt as previously reported (8, 20). CFTRinh172 was generously provided by Dr. A. S. Verkman. Bumetanide was dissolved as a concentrated stock in Me2SO and diluted 1000-fold in the serosal bath to give the final concentration (100 μM). DIDS and CFTRinh172 were added to the initial 20 μl of PBS at t = 0 with Texas Red-dextran where appropriate. The other compounds were added as dry powders suspended in PFC (FC-72) (8).
Statistics
Parametric statistics (analysis of variance followed by the Tukey test to determine significant differences among groups) were used where the variances were homogeneously distributed. In the case of non-homogeneity of variance, significance of difference between means was determined by either analysis of variance followed by Dunn’s multiple comparison test, the Mann-Whitney U test, or the Wilcoxon Signed Rank test as appropriate. All values are expressed as the means ± S.E., where n represents the number of cultures (a minimum of three donors provided tissues per experiment).
RESULTS
Standard (Static) Air-Liquid Interface Culture Conditions; NL Versus CF PCL Volume Regulation
We removed accumulated mucus before each experiment to measure PCL volume regulation without the buffering effect of the mucus layer (20) and initiated each experiment by adding an excess volume of PCL in the form of an isotonic solution similar to PCL in vivo (25, 26).
Normal bronchial epithelial cultures absorbed the added (“excess”) PCL volume over a period of ~12 h following which absorption ceased, and PCL volume was maintained at the height of the extended cilium (~7 μm), i.e. the definition of optimal PCL volume/height (Fig. 1, a and b). In contrast, CF bronchial epithelia absorbed PCL more rapidly and did not stop absorbing PCL until a height of ~3 μm was reached (Fig. 1, a and b).
FIGURE 1. Abnormal regulation of PCL height by CF airway epithelia.

a, XZ confocal images of PCL at 0, 6, and 48 h after mucosal addition of 20 μl of PBS containing Texas-red dextran to NL and CF bronchial epithelial cultures. b, mean PCL height with time taken from NL (squares, n = 9) and CF (circles, n = 8) cultures. Note that the blue-shaded region denotes normal height of outstretched cilia (i.e. normal PCL height). c, electron micrographs of bronchial epithelial culture surfaces fixed with OsO4/PFC 48 h after PBS addition. d, bar graphs depicting changes in transepithelial electric potential difference (Vt) in response to bumetanide (10−4 M, basolateral) and amiloride (3 × 10−4 M, apical) in NL (open bars, n = 6) and CF (closed bars, n = 7) cultures at 0 and 48 h. All scale bars are 7 μm. Data shown are the mean ± S.E. *, significantly different from t = 0. †, significantly different between NL and CF cultures.
An electron microscopy technique visualized the PCL in NL versus CF cultures after 48 h of PCL volume regulation (7). The 7-μm height on NL culture surfaces was associated with fully extended cilia and an electron lucent zone that represented PCL (Fig. 1c). In contrast, the cilia of the CF airway epithelia were bent over and tightly packed (Fig. 1c). The ciliary shaft is heavily glycosylated so that the residual 3 μm of liquid on CF airway surfaces is likely associated with the glycocalyx on ciliary shafts and the cell surface and not available for absorption by active ion transport.
The two phases of PCL volume regulation by NL airway epithelia, i.e. rapid removal of liquid from airway surfaces (0–12 h), followed by a steady state volume (12–48 h), suggested that active ion transport systems had been adjusted (Fig. 1b). To identify the ion transport processes involved in this regulation, we measured the transepithelial potential difference (Vt) under basal conditions and following sequential exposure to blockers of Cl− secretion (bumetanide) and Na+ absorption (amiloride). At t = 0, amiloride-sensitive Na+ transport dominated (Fig. 1d). When PCL had achieved a steady state height of ~7 μm (48 h), Vt dropped (−11.2 ± 0.9 mV at t = 0; −7.5 ± 0.7 mV at t = 48 h, p < 0.05, n = 9), suggesting a slowing of total ion transport. Analysis of each component of Vt revealed that amiloride-sensitive Na+ transport fell and bumetanide-sensitive Cl− secretion persisted and became dominant.
The pattern for CF cultures was distinctly different. First, Vt did not decrease over time (−11.4 ± 1.0 mV at t = 0; −10.2 ± 1.3 mV at t = 48 h, n = 6) despite the fact that PCL volume absorption was excessive (Fig. 1, a and b). Furthermore, Vt remained entirely amiloride-sensitive (Fig. 1d), suggesting that, unlike in NLs, Na+ transport rates were not regulated. Second, the amiloride-sensitive Vt was greater in CF than NL cultures at both t = 0 and t = 48 h (Fig. 1d), consistent with an abnormally high rate of CF Na+ transport. Third, no significant bumetanide-sensitive Vt was detected at either t = 0 or 48 h, suggesting that CF cultures could not secrete Cl− basally or in response to a low PCL volume. Thus, despite the excessive loss of PCL volume, CF airway epithelia exhibited an unregulated, abnormally high rate of Na+ transport and no Cl− secretion.
Extracellular Nucleotide/Nucleoside Regulation of PCL Volume
Tri-phosphate nucleotides (e.g. ATP) have been shown both to inhibit Na+ absorption via P2Y2 receptors (16) and stimulate Cl− secretion also via P2Y2-R, perhaps in combination with P2X (17, 27) purino-receptors. Thus, we designed a series of experiments to assess whether extracellular ATP could modulate PCL volume (Fig. 2). Under these static culture conditions, NL PCL [ATP] was 1.2 ± 0.2 nM (n = 6), which is insufficient to activate P2Y2-R. However, we also functionally tested for a role for ATP in PCL regulation by the addition of apyrase, an enzyme that rapidly degrades ATP to AMP (28).
FIGURE 2. Regulation of Na+ absorption/Cl− secretion by signals encoded in the PCL.

a, xz confocal images of PCL immediately (0) and 6 and 48 h after mucosal addition of PBS with 5 units/ml apyrase to NL and CF bronchial epithelia. b, mean PCL height over 48 h for NL cultures exposed to apyrase (5 units/ml; closed squares, n = 9) or apyrase and 8-SPT (10−5 M; triangles, n = 5); CF cultures (n = 7) exposed to apyrase are denoted by red circles. c, change in Vt in response to bumetanide (bumet., 10−4 M, basolateral) and amiloride (amil., 3 × 10−4 M, apical) in NL (open bars, n = 8) and CF (closed bars, n = 4) cultures before (0 h) and 48 h after apyrase-exposure. d, mean PCL height after mucosal the addition of 8-SPT (10−5 M) alone at t = 0 to NL (squares, n = 7) and CF (circles, n = 6) cultures. Data shown as the mean ± S.E. †, data significantly different from t = 0. ‡, data significantly different between NL and CF cultures.
As compared with control rates of PCL volume absorption, the addition of apyrase to the PCL of NL or CF cultures had no detectable effect on the initial rate of volume absorption (compare 0–2 h data in Fig. 2b to Fig. 1b). This result suggests the basal levels of ATP were below those required to inhibit Na+ transport under static conditions (see Fig. 5a). However, apyrase was associated with a greater PCL height at 48 h in NL but not CF epithelia (Fig. 2, a and b). The response of NL epithelia to apyrase led us to speculate that this compound had increased the production of ADO that, interacting with the A2b subclass of P-1 purinoceptors to raise cellular cAMP, had increased the rate of CFTR-dependent Cl− secretion (23, 29, 30). Three observations were consistent with this notion. First, the apyrase-treated cultures exhibited a raised Vt (−14.0 ± 2.0 mV) at 48 h as compared with non-apyrase-treated cultures (Vt −7.5 ± 0.7 mV), consistent with acceleration of electrogenic Cl− secretion (n = 6). Second, the absolute magnitude of the bumetanide-sensitive Vt, i.e. Cl− secretion, was increased in the presence of apyrase (Fig. 2c) versus without apyrase (Fig. 1d, p < 0.05). Third, an adenosine receptor blocker, 8-SPT, completely blocked the apyrase-induced increase in PCL and indeed reduced PCL volume to CF-like depths, i.e. 3 μm (Fig. 2b), strongly supporting a role of increased ADO production as the stimulus for raised CFTR-mediated Cl− secretion in apyrase-treated NL cultures.
FIGURE 5. Effects of phasic motion are mediated by ATP release into PCL.

Mean PCL [ATP] (a) and mean serosal bath [ATP] (b) obtained from CF cultures 1 h after 50 μl of PBS addition under variable phasic motion (both, n = 4). NL PCL [ATP] was not significantly different from CF PCL ATP (1.9 ± 0.6, 12 ± 4, 95 ± 13, and 131 ± 41 nM at 0, 0.006, 0.6, and 6 dynes/cm2, respectively; all n = 4 and p < 0.05). c, mean PCL height after 48 h of phasic motion in the presence of mucosal apyrase (apyr., 5 units/ml) and/or 8-SPT (10 μM) in NL (open bars; n = 6 – 8) and CF (closed bars; n = 5–7). d, bumetanide (bumet.) and amiloride (amil.)-sensitive changes in Vt in NL (open bars, n = 8) and CF (closed bars, n = 4) cultures before (0) and 48 h after PBS addition/phasic motion in the presence of 5 units/ml mucosal apyrase. Note that amiloride and bumetanide values for CF cultures at 48 h are significantly different from the values without apyrase at 48 h (Fig. 3d). e, simultaneous measurements of Vt and intracellular calcium (Ca2+i) in CF cultures perfused bilaterally with Ringer solution. Left, changes in Vt/Ca2+i induced by stopping then restarting mucosal perfusion (denoted by arrows). Right, altered perfusion rates in the presence of mucosal apyrase (5 units/ml). The mean changes in Vt and Ca2+i responses to phasic perfusion with KBR were −7.8 ± 0.3 mV and 198 ± 12 nM, respectively (p < 0.05; n = 7); the changes in each parameter were significantly reduced in the presence of apyrase; ΔVt = −0.2 ± 0.1 mV, ΔCa2+ = 3 ± 2 nM, respectively (p < 0.05; n = 6). Data shown as the mean ± S.E. *, data significantly different between NL and CF cultures. †, data significantly different from t = 0. ‡, data significantly different from apyrase. §, data significantly different from 8-SPT.
The observation that 8-SPT reduced PCL volume to CF-like levels (Fig. 2b) suggested that ADO may control basal CFTR activity (23). The PCL ADO concentration was sufficient (100 ± 19 nM; n = 10) to activate A2b receptors (A2b-R), the dominant adenosine receptor in airway epithelia (29). However, to test this notion directly, we exposed NL airway epithelial cells under basal conditions to 8-SPT and found 8-SPT alone reduced PCL heights into the range of CF cultures (3 μm) (Fig. 2d). Despite similar ADO levels (162 ± 30 nM; n = 7), 8-SPT was without effect on CF cultures (Fig. 2d). These data suggest that the presence of a partially activated CFTR channel is important for regulating PCL volume and that ADO contained within PCL is the major regulator of basal CFTR activity in our airway culture system.
Phasic Motion (Shear Stress) Culture Conditions; Normal Versus CF Bronchial Epithelial PCL Volume Regulation
Breathing imparts shear on pulmonary surfaces by airflow-induced shear stress, cyclic compressive (transmural) shear, and stretch (13). However, calculation of total shear-stress on pulmonary surfaces has been difficult, and comprehensive model systems are unavailable. We focused our studies on the best characterized component of breathing-induced shear, i.e. the shear stress due to tidal oscillations in airflow. Estimates of the magnitude of this shear stress range between ~0.4 and 2 dynes·cm−2 (see Fig. 3a and the supplemental material). We developed a novel cell culture system for epithelia that imparts a phasic rotational shear on the cultures to mimic airflow-induced shear in vivo (see “Experimental Procedures”). We used this system to test whether phasic shear imparted regulation to PCL volume.
FIGURE 3. Phasic motion-induced changes in PCL volume and epithelial bioelectric properties in NL versus CF cultures.

a, airflow induced in vivo shear stress as a function of airway generation at airway walls (see the supplemental material). The trachea is denoted by generation 0. b, xz confocal images of PCL immediately (0) and 48 h after mucosal PBS addition to NL and CF epithelia cultured under phasic motion. c, mean PCL heights after 48 h of phasic motion culture for NL (open bars, n = 7) and CF (closed bars, n = 8). d, bumetanide (bumet.) and amiloride (amil.)-sensitive Vt in NL (open bars, n = 7) and CF (closed bars, n = 4) cultures before (0) and after 48 h of phasic motion. e, rotational mucus transport rates in static CF cultures (closed bars, n = 12) and phasic motion cultures (gray bars, n = 14) 48 h after volume addition. Data are shown as mean ± S.E. *, data significantly different between NL and CF cultures. †, data significantly different from t = 0. ‡, significantly different between static and phasic motion.
Phasic shear (~0.5 dynes·cm−2) had important effects on PCL physiology. The same general pattern of PCL volume homeostasis, i.e. absorption of excess liquid followed by a plateau, was observed in the phasic motion condition in both NL and CF cultures. However, the plateau level was very different for both preparations (Fig. 3, b and c). For NLs, the plateau level approximated 12.5–15 μm, similar to the apyrase values under static conditions (Fig. 2b). Importantly, the plateau for the CF cultures more than doubled and reached the height of “normal” PCL, i.e. 7 μm. Thus, motion triggers signals that allow CF airway epithelia to maintain PCL volume.
We next investigated the ion transport correlates of PCL volume transport under physiologic shear stress conditions Fig. 3d). For NL cultures, the bioelectric properties exhibited subtle differences under phasic shear versus static conditions, i.e. a shift to a more Cl− secretory (and less Na+ absorptive) physiology at 48 h with motion (Fig. 3d). For CF cultures, however, the pattern of Na+ versus Cl− transport was strikingly different under phasic shear versus static conditions. At t = 0, the Vt was almost entirely amiloride-sensitive, consistent with the dominance of Na+ transport (Fig. 3d). However, after 48 h under phasic motion conditions, the CF cultures exhibited a large reduction of the amiloride-sensitive Vt with the appearance of a significant bumetanide-sensitive Vt that, indeed, was dominant. Thus, these data suggest that CF cultures under phasic motion generated a signal that both slowed Na+ transport and induced Cl− secretion.
We have previously shown that rotational mucus transport is preserved in NL cultures but abolished by PCL volume depletion in CF cultures under static culture conditions (7). Accordingly, we asked whether the increased PCL height detected in CF cultures under phasic motion conditions was sufficient to maintain effective mucus transport. As shown in Fig. 3e, rotational mucus transport by CF cultures under static culture conditions ceased by 48 h. In contrast, CF mucus transport was preserved under motion conditions. Thus, the volume of PCL maintained on CF airway surfaces in response to phasic motion appears physiologically significant.
To test whether the increases in PCL height reflected a component of Cl− secretion, we added known Cl− channel inhibitors to the mucosal surface and measured PCL height after 3 h of phasic motion. Mucosal addition of the CFTR antagonist CFTRinh172 (58) caused PCL height to fall from ~16 to 8 μm (Fig. 4), suggesting that the majority of PCL secretion was mediated via CFTR. However, mucosal DIDS addition (in the presence of CFTRinh172) caused PCL height to fall further from ~8 to ~ 5 μm, suggesting that a small amount of PCL secretion was via the Ca2+-activated Cl− conductance (CaCC).
FIGURE 4. Phasic motion-induced PCL secretion requires activation of Cl− channels.
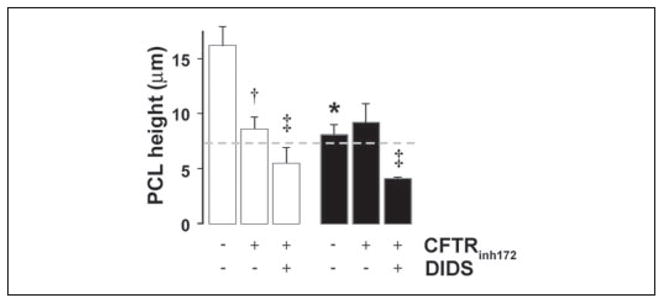
Mean PCL height after 3 h of phasic motion in the presence of a CFTR antagonist (CFTRinh172; 10 μM) or CFTRinh172 and a CaCC antagonist (DIDS; 100 μM). NLs (open bars; n = 6) and CFs (closed bars; n = 6). Data are shown as the mean ± S.E. *, data significantly different between NL and CF cultures. †, data significantly different from control. ‡, data significantly different from CFTRinh172.
Consistent with the lack of CFTR in CF tissues, CFTRinh172 was without effect on CF airway cultures (Fig. 4). However, in the presence of DIDS, CF PCL height was significantly reduced from ~8 to 4 μm, suggesting that CaCC was the sole pathway for Cl− secretion in CF airways under phasic motion (Fig. 4).
Identification of Increased ATP Concentration as the Key Signal Regulating PCL Volume Homeostasis during Phasic Motion Conditions
We first tested whether shear stress released ATP into the PCL in a dose-related manner. Airway epithelia exhibited low levels of ATP in PCL under static conditions, and there was a shear-dependent increase in the PCL ATP concentrations (Fig. 5a). The ATP concentration induced by physiologic shear stress, i.e. 0.5 dynes·cm−2, was in the range where P2Y2-R is predicted to be activated (EC50 = 100 nM). Interestingly, phasic shear selectively promoted ATP release into the PCL (Fig. 5a) but not the serosal media (Fig. 5b), i.e. release was vectorial. Apyrase (5 units/ml) eliminated the motion-induced increases in ATP concentration (shear at 0.5 dyne/cm2 = 41.3 ± 8.3 nM; shear + apyrase = 2.5 ± 0.3 nM; n = 4–8; p < 0.01).
To determine whether the phasic motion-generated signal that regulated ion transport and PCL was ATP, we added apyrase to the PCL of cultures under phasic motion conditions (Fig. 5c). In NL cultures, the pattern of PCL volume responses to liquid addition over time was not appreciably changed. This result plus the observation that 8-SPT addition significantly reduced PCL height (Fig. 5c) suggests that ADO is a key signaling molecule in NL airways during phasic motion as well as under static conditions. A role for ATP signaling in NL airways under phasic motion conditions, however, was revealed by the observation that the combined addition of apyrase and 8-SPT reduced PCL height more than 8-SPT alone (Fig. 5c). In contrast, apyrase had striking effects on CF PCL homeostasis. Apyrase alone produced PCL volume depletion, i.e. eliminated the capacity to regulate PCL height to 7 μm. Importantly, 8-SPT was without additional effect on CF PCL height (Fig. 5c), suggesting that CF cultures rely solely on ATP to rebalance PCL height.
We then sought the bioelectric correlates of these apyrase-mediated effects (Fig. 5d). In NL cultures, Vt did not decrease with time (−11.4 ± 0.9 mV at t = 0; −10.2 ± 1.0 mV at t = 48 h; n = 8), and the majority of Vt at 48 h was bumetanide-sensitive, suggesting that phasic motion with apyrase produced sufficient ADO to accelerate Cl− secretion. However, in apyrase-treated CF airway epithelia, Vt at 48 h was dominated by Na+ absorption (large amiloride-sensitive Vt), and Cl− secretion (bumetanide-sensitive Vt) was absent. Together, these data strongly suggest that the inhibition of Na+ transport and the induction of Cl− secretion in CF observed under phasic motion conditions was mediated by increased extracellular ATP interacting with P2 receptors (16, 17, 31) (Fig. 5d).
We performed one additional study to test the link between phasic motion-induced ATP release and regulation of ion transport. CF cultures were placed in a modified Ussing chamber to simultaneously measure Cl− secretion and intracellular Ca2+ (Ca2+i), a parameter that reflects the signal transduction pathway utilized by ATP signaling in airway epithelia (21). Both the initiation and the cessation of mucosal bathing solution flow (shear) produced increases in Vt (Cl− secretion) and increases in Ca2+i that relaxed with time (Fig. 5e). These data suggest that it is the rate of change of shear, not constant shear, that is the important stimulus for ion transport regulation. To test for a role of released ATP in these responses, we included apyrase in the mucosal perfusate. Both the Vt and the Ca2+i responses to phasic flow were abolished by this maneuver. Thus, these data strongly suggest that changes in mucosal flow (shear) induced mucosal ATP release that activated apical P2Y2-Rs and/or P2X-Rs to increase Ca2+i and initiate Cl− secretion.
An Initiating Event for the Development of CF Lung Disease; Viral Infections Break-down CF PCL Volume Homeostatic Mechanisms
If tidal breathing stimulates sufficient ATP release in vivo to sustain mucus clearance, then why do CF patients get chronic bacterial infections? Because acute insults, including viral infections, have been associated with initiation of CF bacterial infections in neonates, we studied interactions between RSV (11) and PCL autoregulation. Cultures under phasic motion were infected with a recombinant RSV encoding GFP, which is not cytotoxic over 48 h (18). In the presence of RSV-gfp and phasic motion, PCL height in NL cultures was reduced but maintained at a physiologically relevant level (9 ± 2 μm; n = 5). In contrast, PCL height was reduced in CF cultures to ~4.5 μm, a level that fails to sustain mucus transport, suggesting that the protective effect of phasic motion had been breached (Fig. 6, a and b).
FIGURE 6. RSV infections inhibit nucleotide-dependent PCL homeostasis in CF cultures.
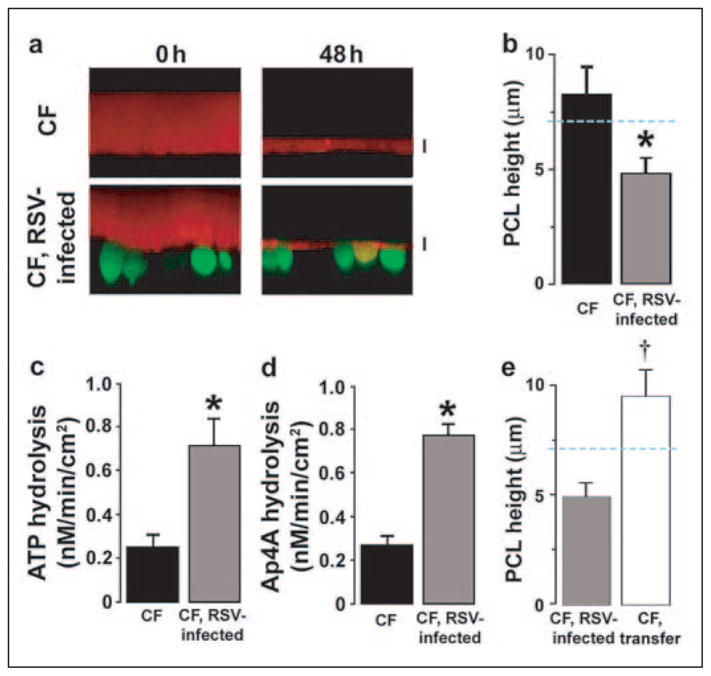
a, XZ confocal images of PCL (red) covering control or RSV-gfp-infected ciliated cells (green) before (0) and 48 h after mucosal PBS addition to CF cultures under phasic motion conditions. b, PCL height 48 h after the addition of PBS for control CF cultures (closed bars; n = 10) and CF cultures infected with RSV-gfp (gray bars, n = 10). Also shown are mucosal ATP (c) and Mucosal Ap4A (d) hydrolysis rates in CF epithelia in control (closed bars) and RSV-infected cultures (gray bars). Both, n = 3. e, PCL height 48 h after the addition of PBS for CF cultures infected with RSV-gfp (gray bars, n = 5), and for cultures exposed to PCL and serosal media, but not virus, from RSV-infected cultures for 48 h under phasic motion conditions (CF transfer, open bars, n = 5). Data are shown as the mean ± S.E. *, difference (p < 0.05) between control and RSV-infected CF epithelia. †, difference (p < 0.05) between RSV-infected and CF-RSV media-exposed epithelia.
We have previously shown that infection/inflammation triggers the expression of extracellular ATPases that lower ATP concentrations in PCL (32). Accordingly, we first measured total ecto-ATPase activity post-RSV infection and found it was raised significantly (Fig. 6c). Kinetic analyses revealed that ecto-nucleotide pyrophosphatase phosphohydrolases (eNPPs) are the dominant enzymes hydrolyzing ATP in the concentrations detected under phasic motion (22). Bioinformatics analyses of the phosphohydrolase promoter identified NFκB and AP-1 sites that would be predicted to be activated by an RSV infection of airway epithelia (33, 34). Therefore, we tested for RSV up-regulation of ecto-nucleotide pyrophosphatase phosphohydrolase (eNPP) activity with the eNPP-specific substrate Ap4A post-RSV infection (Fig. 6). Ecto-nucleotide pyrophosphatase phosphohydrolase (eNPP) activity was increased 3-fold on the mucosal surface of CF bronchial cultures post-RSV infection (Fig. 6d), suggesting that RSV infection depletes PCL volume via an increase in eNPP-mediated extracellular ATP hydrolysis.
To test whether the inhibition of PCL homeostasis was due to release of inflammatory mediators versus a direct effect of the viruses on the epithelia, we infected CF cultures with RSV virus for 48 h. After this time we then lavaged the apical surface of these cultures with 100 μl of PBS (1 h at 37 °C followed by UV exposure) and transferred 20 μl of the lavage per culture to a second group of uninfected cultures. We also transferred the entire serosal volume to the uninfected cultures (2 ml/culture) and added fresh media to the infected cultures. Forty-eight hours after these manipulations, the RSV-infected cultures again exhibited a reduced PCL height under phasic motion (Fig. 6e), whereas uninfected cultures containing the transferred media/lavage were still capable of regulating PCL height to ~7 μm (Fig. 6e). These data suggest that ecto-nucleotide pyrophosphatase phosphohydrolase up-regulation was due to a direct effect of the virus on the epithelia rather than a release of cytokines.
DISCUSSION
Mechanical clearance of mucus is the dominant system that protects airways from infection by inhaled bacteria, and the 7-μm-deep PCL is critical for promoting both ciliary-dependent and cough-dependent mucus clearance (4). The volume of PCL in an airway region reflects the balance between movement onto the region from distal surfaces and local regulatory activities (35, 36). The fact that PCL moves cephalad along the surfaces of converging airways suggests that typically there will be a small excess of PCL at the convergence point of two airways that must be absorbed to maintain an optimally functional (7 μm) layer (6). Our experimental strategy was to mimic this situation by starting each experiment with a modest excess of liquid to evaluate how PCL volume was regulated by NL and CF airway epithelia under conventional cell culture (static) conditions and phasic motion conditions that mimicked the dynamic activity of respiration.
The Normal Control of PCL Volume (Fig. 7, a and c)
FIGURE 7. Schema describing PCL height regulation by active ion transport.
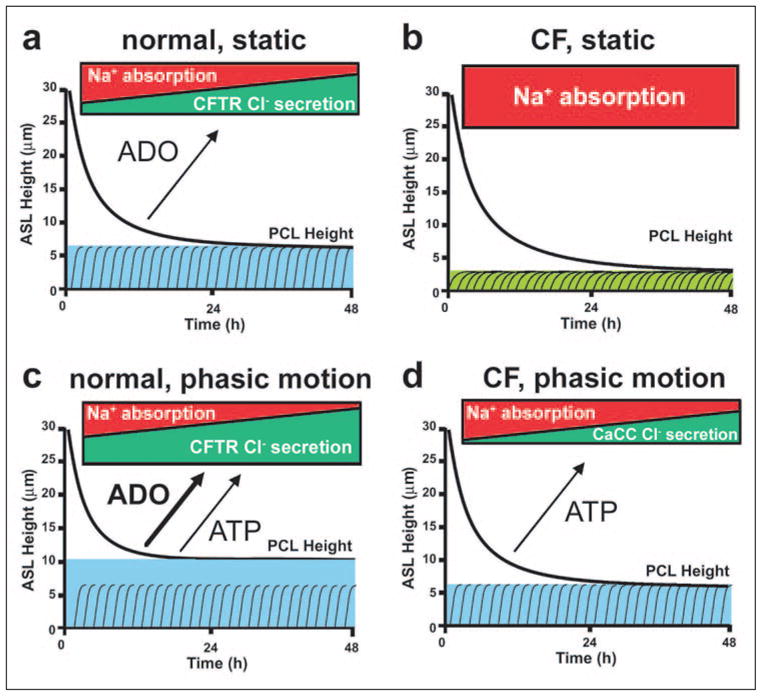
a, NL airway epithelia under static conditions coordinately regulate the rates of Na+ absorption and Cl− secretion to adjust PCL volume from an “excessive” PCL height (25 μm) to the physiologic PCL height with time. The blue color depicts PCL height as referenced to extended cilia. b, in CF epithelia, the higher basal rate of Na+ absorption, the failure to inhibit Na+ transport rates, and the failure to initiate Cl− secretion under static conditions lead to PCL depletion (note “flattened” cilia). c, NL airway epithelia under phasic motion respond to increased nucleotide/nucleoside release into PCL by shifting the balance toward Cl− secretion via CFTR (and CaCC; not shown) and a higher PCL. d, CF cultures under phasic motion conditions release sufficient ATP into the PCL to inhibit Na+ absorption and initiate CaCC-mediated Cl− secretion to restore PCL to a physiologically adequate height.
Because airway epithelia are highly water-permeable (37, 38), the PCL is determined by the amount of salt on the surfaces of airway epithelia. Our data demonstrate that normal airway epithelia coordinate active absorptive and secretory ion flows to finely control the quantity of salt on airway surfaces and, hence, PCL volume. Na+ absorption is greatest (Fig. 1, b and d) when the PCL volume excess is greatest, and as the PCL approaches an optimal depth (7 μm) Na+ absorption is inhibited, generating electrochemical driving forces for CFTR-dependent Cl− secretion (Fig. 1d) (39).
The regulation of the relative rates of Na+ versus Cl− transport occurs in the apical domain of the airway epithelial cell. The relevant ion channels, e.g. epithelial Na+ channel and CFTR, and most of their intra-cellular regulators, are localized within the apical plasma membrane (23). However, the signals that “describe” the PCL volume status to these ion channels and regulators had not been identified. Our data demonstrate that key signals are the concentrations of nucleotide/nucleoside contained within the PCL itself.
Under static conditions Na+ absorption is inhibited (regulated) by activation of CFTR (40), and our data show that CFTR activity is governed under basal conditions by the ADO contained in PCL (Fig. 2). Under phasic motion conditions, sufficient ATP is released into PCL to regulate the Na+ transport via a second mechanism, i.e. via P2-R regulation.
With respect to Cl− secretion, our data demonstrate that phasic motion exerts its effects primarily through CFTR (Fig. 4). It is likely that the PCL concentration of ADO regulates the contribution of this pathway to PCL volume homeostasis. The observation that PCL volume in normal cultures under static conditions could not be maintained in the presence of an ADO receptor antagonist (8-SPT) (Fig. 2d) supports a role for CFTR-mediated Cl− secretion in static PCL volume control (23, 41). With phasic motion, the Ca2+-activated Cl− conductance was also active (Fig. 4), due to the increase in ATP release in addition to the regulation provided by ADO (Fig. 5). We speculate that the increased liquid on airway culture surfaces observed during phasic motion is selectively added to the mucus layer and acts as a buffer for periods of relative PCL depletion (20).
CF PCL Volume Control; Static Conditions (Fig. 7b)
CF airway epithelia absorbed PCL more rapidly from airway surfaces and could not maintain a functional PCL height/volume under basal conditions (Fig. 1). Indeed, abnormal CF airway ion transport removed all of the “available” liquid from airway surfaces under static conditions (Fig. 1c).
The rapid absorption of PCL and the failure to preserve a functional PCL under static conditions by CF epithelia reflected abnormalities in the basal activities of both the Na+ and Cl− transport paths. The raised basal Na+ transport rates in CF bronchial cultures (larger Δamiloride Vt; see Fig. 1d) reflects the absence of the tonic inhibitory influence of apical membrane CFTR on the epithelial Na+ channel (40, 42, 43). Note, the absence of 8-SPT effects despite levels of ADO in PCL sufficient to activate A2b reflects the absence of CFTR and not A2b signaling capacity.
Phasic Motion Conditions (Fig. 7d)
Remarkable differences in CF ion transport physiology under phasic motion versus static conditions were observed. Under conditions of phasic (shear) motion, CF airway epithelia maintained PCL at a physiologically relevant height (7 μm), as evidenced by the capacity to sustain mucus transport over prolonged periods relative to static conditions (Fig. 3e). The bioelectric changes in CF cultures under shear conditions were consistent with the actions of increased ATP release, e.g. inhibition of Na+ transport (reduced amiloride sensitivity of Vt) and stimulation of bumetanide-sensitive Cl− secretion (Fig. 3d). Our studies with flow chambers, measuring simultaneously Cl− secretion and intracellular Ca2+ responses to phasic flow, strongly suggest that the Cl− secretion activated by motion in CF epithelia involved a Ca2+-activated pathway regulating the CaCC rather than CFTR (Fig. 5e) (21).
Multiple experiments suggested that this rebalancing of CF ion transport reflected the phasic motion-induced release of ATP and activation of CaCC. First, PCL volume was DIDS-sensitive (Fig. 4). Second, there was an increase in the measured PCL [ATP] into ranges predicted to regulate ion transport in response to phasic shear (Fig. 5a). Third, the effects of phasic motion to maintain an adequate PCL, inhibit Na+ transport, and induce Cl− transport in CF epithelia were abolished by the addition of apyrase to the luminal bath (Fig. 5, c and d). Finally, the capacity to induce CaCC-mediated Cl− secretion in response to phasic flow was also blocked by the addition of apyrase to the luminal bath (Fig. 5e).
Relevance of Phasic Motion to Normal Lung Defense
Phasic motion is a key feature of pulmonary physiology. For example, it has been long appreciated that tidal volume breathing coupled with intermittent large volume breaths (sighs) is important for maintaining alveolar health/patency, presumably by stimulating surfactant secretion (45). Heretofore, it has not been clear that phasic motion is important for airways defense. Surprisingly, airflow-induced shear-stresses are roughly the same throughout the respiratory tree; airflow velocity decreases dramatically in distal airways, but airway radius does as well (see Fig. 3a and the supplemental material). Therefore, the shear-stress stimulus used in this study is likely relevant to both bronchial and bronchiolar regions of the lung.
The link between the phasic motion of pulmonary ventilation and airways defense appears to be mediated by shear stress-induced release of ATP and ATP-dependent regulation of MCC rates. As an example, in a previous study designed to measure airflow-dependent clearance of mucus, it was not clear why increased inspiratory flow was equally effective as increased expiratory flow in accelerating MCC (46). Our data suggest that increased airflow-induced shear stress, irrespective of direction of airflow, released sufficient ATP to up-regulate ion transport rates and increase mucus transport. Interestingly, in the contest between host and pathogen, it has been reported that shear stimulates bacterial changes that promote bacterial adherence (47). Here, we show that phasic shear arms the host to promote the clearance of inhaled pathogens. Indeed, during cough, flow-induced shear can approach 1700 dynes·cm−2, which is predicted to release sufficient ATP to double basal mucus transport rates and maximize pathogen clearance.
Relevance of Phasic Motion to CF Pathogenesis in Vivo
Our observations may have relevance to three important facets of CF pathogenesis in vivo. First, studies of mucus clearance have not revealed a complete absence of MCC in the lower respiratory tract of young CF patients in vivo (48). In the CF nose, where airflow-induced shear stress is greatest, MCC is reported to be normal in vivo (49). Motion-induced ATP release could support MCC in vivo, as in vitro (Fig. 3e), until generalized or local insults to airways occur (see below).
Second, it has been reported that exercise is important in promoting mucus clearance and slowing the progression of CF lung disease (50, 51). Furthermore, recent studies have suggested that post-exercise, the Na+ transport path in CF airway epithelia is inhibited (52, 53). Our data showing that phasic motion induced ATP release (Fig. 5a) and that nucleotides inhibited Na+ transport and initiated Cl− secretion (Fig. 5d) provide one mechanism to account for amelioration of the severity of CF lung disease by exercise.
Third, there has always been the perplexing question in the natural history of CF lung disease whereby the upper lobes develop disease before the lower lobes (54). Because of gravitational effects, the upper lobes are ventilated less (by ~50%) than the lower lobes (55). The reduced upper lobe tidal volume would reduce airflow-induced shear on airway epithelia, leading to less local nucleotide release. We speculate that the reduction in shear-induced ATP release in the upper lobes reduces the PCL concentration of the sole regulator of CF mucus clearance (ATP) and contributes to the vulnerability of this region to infection.
How Might Viral Infections Trigger CF Bacterial Infection?
Clinical studies have suggested that CF lung disease progresses by intermittent clinical “exacerbations” that are often initiated by a defined insult, e.g. viral infections (11, 12). Many viruses have been implicated, including RSV (56). RSV can produce the mucus plugging that leads to bacterial infection via nonspecific mechanisms, including cytotoxic effects on cells ciliary damage and cytokine release. However, the importance of the PCL in mucus clearance (4) led us to investigate whether RSV-infected CF airway epithelial cells also lose the capacity to preserve an effective PCL. Indeed, RSV infection itself, not release of cytokines directly depleted PCL volume under phasic motion conditions without inducing detectable epithelial damage (Fig. 6). Our data suggest this effect was mediated by RSV-induced up-regulation of the key extracellular ATPases that metabolize ATP levels in the range observed under phasic motion conditions (Fig. 6d). This observation suggests that in CF patients in vivo, reduction of PCL ATP concentrations by increased metabolism may contribute to bacterial infection by reduction of PCL volume and, hence, the promotion of mucus stasis and adherence to airway surfaces.
Nucleotide metabolism has also been implicated in abnormal PCL volume regulation after RSV infection in an in vivo murine model (59). In contrast, these investigators found that RSV infection decreased alveolar fluid clearance by an up-regulation of nucleotide levels, resulting in inhibition of Na+ absorption. It is likely that RSV infection has different consequences in different regions of the lung (e.g. alveolar versus bronchial regions).
Conclusions and Therapeutic Implications
In normal airways, PCL volume is regulated by redundant mechanisms. ADO alone is effective under static conditions, whereas under phasic motion both ADO and ATP signaling are important (Fig. 7, a and c). In CF, PCL volume regulation is crippled by the absence of CFTR-mediated Na+ transport regulation and Cl− secretion (Fig. 7b). Thus, under static conditions ADO is ineffective as a regulator of CF PCL volume. Importantly, our data demonstrate that CF epithelia can maintain an adequate PCL and mucus transport under phasic motion conditions (Fig. 7d) but rely solely on motion-induced ATP release to slow Na+ transport and initiate CaCC-dependent Cl− secretion to maintain these functions. Note that although our emphasis has been on the role of superficial epithelia in regulating PCL volume, reductions in submucosal gland volume may also be important in CF pathogenesis. Reduction in gland secretion onto airway surfaces would serve to exacerbate the problem of PCL depletion (57, 58).
These studies describe a mechanism by which the CF lung at birth and, indeed, many lung regions during life are functionally normal with respect to mucus clearance. However, the unique aspect of the CF lung is its vulnerability to viruses or other insults that excessively slow or, indeed, abolish mucus clearance and set the stage for progressive bacterial infections. These data point to the importance of initiating and/or accelerating therapy designed to promote mucus clearance during viral infections.
Finally, our studies provide a mechanism, i.e. ATP release, to explain how maneuvers that increase motion/shear in the lung, e.g. exercise (50, 51), physiotherapy (60), and external flutter devices (61), facilitate clearance of secretions from the lungs of CF patients. Indeed, our in vitro culture system provides a tool to identify the maximally effective parameters for ATP release, i.e. shear rates and frequencies, which may assist in the refinement of these therapeutic maneuvers. Perhaps vigorous therapy can forestall the onset of vicious cycles of mucus stasis that, once in place, generates the persistent infection of the CF lung.
Supplementary Material
Acknowledgments
We thank the CF Center Histology and Tissue Cores, Dr. Mu-Ming Poo (University of California-Berkeley) for use of a confocal microscope, Drs. Jack Stutts and Bill Davis for critical review of the manuscript, and Drs. Garrett Matthews, Richard Superfine, and Scot Winters for useful discussions on shear stress.
Footnotes
This work was supported by National Institutes of Health Grants HL34322, HL60280, HL074158, CFF R026, and TARRAN01I0.
The on-line version of this article (available at http://www.jbc.org) contains supplemental material.
The abbreviations used are: PCL, periciliary liquid; HPLC, high performance liquid chromatography; 8-SPT, 8(p-sulfophenyl)theophylline; DIDS, 4,4′-diisothiocya-nostilbene-2,2′-disulfonic acid; CaCC, Ca2+-activated Cl− conductance; MCC, mucociliary clearance; CF. cystic fibrosis; CFTR, transmembrane conductance regulator; RSV, respiratory syncitial virus; GFP, green fluorescent protein; NL, normal; PBS, phosphate-buffered saline; PFC, perfluorocarbon; TES, 2-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}ethanesulfonic acid; Vt, transepithelial potential difference; ADO, adenosine; Ap4A, diadenosine tetraphosphate; -R, receptor.
References
- 1.Wine JJ. J Clin Investig. 1999;103:309–312. doi: 10.1172/JCI6222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guggino WB. Nat Med. 2001;7:888–889. doi: 10.1038/90914. [DOI] [PubMed] [Google Scholar]
- 3.Verkman AS. Am J Physiol. 2001;281:L306–L308. doi: 10.1152/ajplung.2001.281.2.L306. [DOI] [PubMed] [Google Scholar]
- 4.Knowles MR, Boucher RC. J Clin Investig. 2002;109:571–577. doi: 10.1172/JCI15217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Puchelle E, de Bentzmann S, Zahm JM. Respiration. 1995;62(Suppl 1):2–12. doi: 10.1159/000196486. [DOI] [PubMed] [Google Scholar]
- 6.Matsui H, Randell SH, Peretti SW, Davis CW, Boucher RC. J Clin Investig. 1998;102:1125–1131. doi: 10.1172/JCI2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- 8.Tarran R, Grubb BR, Parsons D, Picher M, Hirsh AJ, Davis CW, Boucher RC. Mol Cell. 2001;8:149–158. doi: 10.1016/s1097-2765(01)00286-6. [DOI] [PubMed] [Google Scholar]
- 9.Gibson RL, Burns JL, Ramsey BW. Am J Respir Crit Care Med. 2003;168:918–951. doi: 10.1164/rccm.200304-505SO. [DOI] [PubMed] [Google Scholar]
- 10.Robinson M, Bye PTB. Pediatr Pulmonol. 2002;33:293–306. doi: 10.1002/ppul.10079. [DOI] [PubMed] [Google Scholar]
- 11.Armstrong D, Grimwood K, Carlin JB, Carzino R, Hull J, Olinsky A, Phelan PD. Pediatr Pulmonol. 1998;26:371–379. doi: 10.1002/(sici)1099-0496(199812)26:6<371::aid-ppul1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 12.Hiatt PW, Grace SC, Kozinetz CA, Raboudi SH, Treece DG, Taber LH, Piedra PA. Pediatrics. 1999;103:619–626. doi: 10.1542/peds.103.3.619. [DOI] [PubMed] [Google Scholar]
- 13.Basser PJ, McMahon TA, Griffith P. J Biomech Eng. 1989;111:288–297. doi: 10.1115/1.3168381. [DOI] [PubMed] [Google Scholar]
- 14.Burnstock G. Clin Med. 2002;2:45–53. doi: 10.7861/clinmedicine.2-1-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guyot A, Hanrahan JW. J Physiol (Lond) 2002;545:199–206. doi: 10.1113/jphysiol.2002.030148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mason SJ, Paradiso AM, Boucher RC. Br J Pharmacol. 1991;103:1649–1656. doi: 10.1111/j.1476-5381.1991.tb09842.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zsembery A, Fortenberry JA, Liang L, Bebok Z, Tucker TA, Boyce AT, Braunstein GM, Welty E, Bell PD, Sorscher EJ, Clancy JP, Schwiebert EM. J Biol Chem. 2004;279:10720–10729. doi: 10.1074/jbc.M313391200. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. J Virol. 2002;76:5654–5666. doi: 10.1128/JVI.76.11.5654-5666.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tarran R, Boucher RC. Methods Mol Med. 2002;70:479–492. doi: 10.1385/1-59259-187-6:479. [DOI] [PubMed] [Google Scholar]
- 20.Tarran R, Grubb BR, Gatzy JT, Davis CW, Boucher RC. J Gen Physiol. 2001;118:223–236. doi: 10.1085/jgp.118.2.223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paradiso AM, Ribeiro CMP, Boucher RC. J Gen Physiol. 2001;117:53–68. doi: 10.1085/jgp.117.1.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazarowski ER, Boucher RC, Harden TK. J Biol Chem. 2000;275:31061–31068. doi: 10.1074/jbc.M003255200. [DOI] [PubMed] [Google Scholar]
- 23.Huang P, Lazarowski ER, Tarran R, Milgram SL, Boucher RC, Stutts MJ. Proc Natl Acad Sci U S A. 2001;98:14120–14125. doi: 10.1073/pnas.241318498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Picher M, Boucher RC. J Biol Chem. 2003;278:11256–11264. doi: 10.1074/jbc.M208071200. [DOI] [PubMed] [Google Scholar]
- 25.Knowles MR, Robinson JM, Wood RE, Pue CA, Mentz WM, Wager GC, Gatzy JT, Boucher RC. J Clin Investig. 1997;100:2588–2595. doi: 10.1172/JCI119802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Caldwell RA, Grubb BR, Tarran R, Boucher RC, Knowles MR, Barker PM. J Gen Physiol. 2002;119:3–14. doi: 10.1085/jgp.119.1.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devor DC, Bridges RJ, Pilewski JM. Am J Physiol. 2000;279:C461–C479. doi: 10.1152/ajpcell.2000.279.2.C461. [DOI] [PubMed] [Google Scholar]
- 28.Parr CE, Sullivan DM, Paradiso AM, Lazarowski ER, Burch LH, Olsen JC, Erb L, Weisman GA, Boucher RC, Turner JT. Proc Natl Acad Sci U S A. 1994;91:3275–3279. doi: 10.1073/pnas.91.8.3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazarowski ER, Mason SJ, Clarke LL, Harden TK, Boucher RC. Br J Pharmacol. 1992;106:774–782. doi: 10.1111/j.1476-5381.1992.tb14412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clancy JP, Ruiz FE, Sorscher EJ. Am J Physiol. 1999;276:C361–C369. doi: 10.1152/ajpcell.1999.276.2.C361. [DOI] [PubMed] [Google Scholar]
- 31.Devor DC, Pilewski JM. Am J Physiol. 1999;276:C827–C837. doi: 10.1152/ajpcell.1999.276.4.C827. [DOI] [PubMed] [Google Scholar]
- 32.Picher M, Boucher RC. Am J Respir Cell Mol Biol. 2000;23:255–261. doi: 10.1165/ajrcmb.23.2.4088. [DOI] [PubMed] [Google Scholar]
- 33.Carpenter LR, Moy JN, Roebuck KA. BMC Infect Dis. 2002;2:5–12. doi: 10.1186/1471-2334-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mastronarde JG, Monick MM, Mukaida N, Matsushima K, Hunninghake GW. J Infect Dis. 1998;177:1275–1281. doi: 10.1086/515279. [DOI] [PubMed] [Google Scholar]
- 35.Kilburn KH. Am Rev Respir Dis. 1968;98:449–463. doi: 10.1164/arrd.1968.98.3.449. [DOI] [PubMed] [Google Scholar]
- 36.Boucher RC. J Physiol (Lond) 1999;516:631–638. doi: 10.1111/j.1469-7793.1999.0631u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsui H, Davis CW, Tarran R, Boucher RC. J Clin Investig. 2000;105:1419–1427. doi: 10.1172/JCI4546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Song Y, Jayaraman S, Yang B, Matthay MA, Verkman AS. J Gen Physiol. 2001;117:573–582. doi: 10.1085/jgp.117.6.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Boucher RC. Am J Respir Crit Care Med. 1994;150:271–281. doi: 10.1164/ajrccm.150.1.8025763. [DOI] [PubMed] [Google Scholar]
- 40.Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 41.Lazarowski ER, Tarran R, Grubb BR, van Heusden CA, Okada S, Boucher RC. J Biol Chem. 2004;279:36855–36864. doi: 10.1074/jbc.M405367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunzelmann K, Schreiber R, Nitschke R, Mall M. Pfluegers Arch Eur J Physiol. 2000;440:193–201. doi: 10.1007/s004240000255. [DOI] [PubMed] [Google Scholar]
- 43.Ji HL, Chalfant ML, Jovov B, Lockhart JP, Parker SB, Fuller CM, Stanton BA, Benos DJ. J Biol Chem. 2000;275:27947–27956. doi: 10.1074/jbc.M002848200. [DOI] [PubMed] [Google Scholar]
- 44.Deleted in proof
- 45.Dietl P, Haller T, Mair N, Frick M. News Physiol Sci. 2001;16:239–243. doi: 10.1152/physiologyonline.2001.16.5.239. [DOI] [PubMed] [Google Scholar]
- 46.Bennett WD, Foster WM, Chapman WF. J Appl Physiol. 1990;69:1670–1675. doi: 10.1152/jappl.1990.69.5.1670. [DOI] [PubMed] [Google Scholar]
- 47.Thomas WE, Trintchina E, Forero M, Vogel V, Sokurenko EV. Cell. 2002;109:913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 48.Regnis JA, Robinson M, Bailey DL, Cook P, Hooper P, Chan HK, Gonda I, Bautovich G, Bye PT. Am J Respir Crit Care Med. 1994;150:66–71. doi: 10.1164/ajrccm.150.1.8025774. [DOI] [PubMed] [Google Scholar]
- 49.McShane D, Davies JC, Wodehouse T, Bush A, Geddes D, Alton EW. Eur Respir J. 2004;24:95–100. doi: 10.1183/09031936.04.00097503. [DOI] [PubMed] [Google Scholar]
- 50.Schneiderman-Walker J, Pollock SL, Corey M, Wilkes DD, Canny GJ, Pedder L, Reisman JJ. J Pediatr. 2000;136:304–310. doi: 10.1067/mpd.2000.103408. [DOI] [PubMed] [Google Scholar]
- 51.Bradley J, Moran F. Cochrane Database Syst Rev. 2002:CD002768. doi: 10.1002/14651858.CD002768. http://www3.interscience.wiley.com/cgi-bin/mrwhome/106568753/HOME. [DOI] [PubMed]
- 52.Alsuwaidan S, Wan Po LA, Morrison G, Redmond A, Dodge JA, McElnay J, Stewart E, Stanford CF. Thorax. 1994;49:1249–1250. doi: 10.1136/thx.49.12.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hebestreit A, Kersting U, BPCL er B, Jeschke R, Hebestreit H. Am J Respir Crit Care Med. 2001;164:443–446. doi: 10.1164/ajrccm.164.3.2007168. [DOI] [PubMed] [Google Scholar]
- 54.Santis G, Hodson ME, Strickland B. Clin Radiol. 1991;44:20–22. doi: 10.1016/s0009-9260(05)80220-x. [DOI] [PubMed] [Google Scholar]
- 55.Milic-Emili J, Henderson JA, Dolovich MB, Trop D, Kaneko K. J Appl Physiol. 1966;21:749–759. doi: 10.1152/jappl.1966.21.3.749. [DOI] [PubMed] [Google Scholar]
- 56.Abman SH, Ogle JW, Butler-Simon N, Rumack CM, Accurso FJ. J Pediatr. 1988;113:826–830. doi: 10.1016/s0022-3476(88)80008-8. [DOI] [PubMed] [Google Scholar]
- 57.Joo NS, Irokawa T, Wu JV, Robbins RC, Whyte RI, Wine JJ. J Biol Chem. 2002;277:50710–50715. doi: 10.1074/jbc.M208826200. [DOI] [PubMed] [Google Scholar]
- 58.Thiagarajah JR, Song Y, Haggie PM, Verkman AS. FASEB J. 2004;18:875–877. doi: 10.1096/fj.03-1248fje. [DOI] [PubMed] [Google Scholar]
- 59.Davis IC, Sullender WM, Hickman-Davis JM, Lindsey JR, Matalon S. Am J Physiol. 2004;286:L112–L120. doi: 10.1152/ajplung.00218.2003. [DOI] [PubMed] [Google Scholar]
- 60.Reisman JJ, Rivington-Law B, Corey M, Marcotte J, Wannamaker E, Harcourt D, Levison H. J Pediatr. 1988;113:632–636. doi: 10.1016/s0022-3476(88)80370-6. [DOI] [PubMed] [Google Scholar]
- 61.Gondor M, Nixon PA, Mutich R, Rebovich P, Orenstein DM. Pediatr Pulmonol. 1999;28:255–260. doi: 10.1002/(sici)1099-0496(199910)28:4<255::aid-ppul4>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 62.Lechner JF, LaVeck MA. J Tiss Cult Meth. 1985;9:43–48. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.