

Abstract
Androgens can protect neurones from injury, but androgen neuroprotection is not well characterised in terms of either specificity or mechanism. Here, we compared the ability of androgens to protect neurones against a panel of insults, empirically determined to induce cell death by apoptotic or non-apoptotic mechanisms. Three criteria defining, but not inclusive of apoptosis are: protection by caspase inhibition, protection by protein synthesis inhibition, and presence of pyknotic nuclei. According to these criteria, β-amyloid, staurosporine, and Apoptosis Activator II induced cell death involving apoptosis, while hydrogen peroxide (H2O2), iron, calcium ionophore, and 3-nitropropionic acid induced cell death featuring non-apoptotic characteristics. Pretreatment of hippocampal neurones with testosterone or dihydrotestosterone attenuated cell death induced by β-amyloid, staurosporine, and Apoptosis Activator II, but none of the other insults. The anti-oxidant Trolox did not reduce cell death induced by β-amyloid, staurosporine, and Apoptosis Activator II, but did protect against H2O2 and iron. Similarly, a supra-physiological concentration of oestrogen reduced cell death induced by H2O2 and iron, an effect not observed with androgens. We also show that activation of oestrogen pathways was not necessary for androgen neuroprotection. These data suggest that androgens directly activate a neuroprotective mechanism specific to inhibition of cell death involving apoptosis. Determining the specificity of androgen neuroprotection may enable the development of androgen compounds for the treatment of neurodegenerative disorders.
Keywords: Androgens, apoptosis, dihydrotestosterone, neuroprotection, oestrogen, testosterone
INTRODUCTION
Men experience a significant decrease in levels of testosterone in blood (1) and brain (2) due to normal aging. Age-related androgen loss in men adversely affects muscle and bone mass, sexual arousal, sperm production, and brain functions such as mood, memory, and cognition (1). Recent data also suggest that low levels of testosterone in aging men may be one of several risk factors for the development of Alzheimer’s disease (AD) (2,3). Androgens have many beneficial actions in the CNS, which the loss of may contribute to age-related neurological deficits and AD. For example, testosterone decreases levels of β-amyloid (Aβ), a protein that is a key contributor to AD pathogenesis (4,5). In addition, androgens are positive regulators of neuronal plasticity in the spinal nucleus of the bulbocavernosus (6), excitability in the CA1 region of hippocampus (7), and spine density in hippocampus (8). Androgens also prevent retraction (9) or increase the length (10) and size (11) of neurites from motor neurones. Other neurotrophic effects of testosterone include cell differentiation (12), neurogenesis (13–15), and development of neurones in the hippocampus (16) and motor (17–19) and autonomic (20) systems.
One important cellular action of androgens is regulation of neurone viability. During development, testosterone and its oestrogen (17β-oestradiol) and androgen (dihydrotestosterone) metabolites determine neurone number in specific sexually dimorphic nuclei via regulation of apoptosis (21, 22). Further, androgens regulate survival of central and peripheral motoneurones following injury (23). For example, androgens increase the survival of motor neurones in newborn rats after cranial nerve crush (24). In addition, androgens increase the speed of regeneration of injured axons of motor neurones in young and adult rats (25, 26). Androgens are also endogenous regulators of viability in neurones challenged with toxic insults in adult animals. Adult male rats and mice depleted of endogenous androgens by orchidectomy exhibit increased vulnerability to hippocampal neurone loss induced by excitotoxins (27, 28), an effect that can be reversed by treatment with DHT (28). In primary neurone culture paradigms, testosterone and related androgens attenuate cell death induced by serum deprivation (29), Aβ (30–32), and hydrogen peroxide (H2O2) (33). However, androgens can fail to protect neurons and even exacerbate injury in response to some forms of injury such as ischemia (34,35), mitochondrial toxin 3-nitropropionic acid (3-NP) (36), and muscimol-induced excitotoxicity (37).
Why androgens are neuroprotective against some insults, but not others is unclear. Our previous findings show that androgen neuroprotection involves a mitogen-activated protein kinase/extracellular signal-regulated kinase(MAPK/ERK) signalling pathway that functionally inactivates the pro-apoptosis factor Bcl-2-associated death protein (Bad) (32). These data suggest that androgens regulate vulnerability to apoptosis -related mechanisms and thus, androgens may selectively protect neurones against insults that induce cell death involving apoptosis pathways. To investigate this hypothesis, we evaluated the effects of androgens on the viability of neurones challenged with a panel of insults, some of which were empirically determined to induce an apoptotic type of cell death. Specifically, we used seven different neurotoxins: i) Aβ, an aggregating peptide involved in the initiation and promotion of neurodegeneration in AD (38); ii) staurosporine, a general protein kinase inhibitor commonly used to induce apoptosis (39); iii) Apoptosis Activator II, a cell permeable compound that promotes apoptosis by activating caspases in a cytochrome c- and Apaf-1-dependent manner (32,40); iv) the pro-oxidant hydrogen peroxide (H2O2) (41, 42); v) iron (ferrous/ferric chloride, FeCl2/3), an oxidant that acts via Fenton chemistry (41, 43); vi) calcium ionophore A23187 (44); and vii) 3-nitropropionic acid (3-NP), an irreversible inhibitor of succinate dehydrogenase/mitochondrial complex II (45).
MATERIALS AND METHODS
Materials
Testosterone, dihydrotestosterone (DHT), 17β-oestradiol (E2), and 5α-androstane-3β, 17β-diol (3β-diol) were acquired from Steraloids (Newport, RI). zVAD-fmk, cycloheximide, Trolox, staurosporine, hydrogen peroxide (H2O2), ferrous/ferric chloride (FeCl2/3), A23187, and 3-nitropropionic acid (3-NP) were purchased from Sigma-Aldrich (St. Louis, MO). β-Amyloid (Aβ) peptide 25–35 was obtained from Bachem (Torrance, CA) and Apoptosis Activator II was acquired from Calbiochem (San Diego, CA).
Neurone cultures
Neurones (~ 95% neuronal, ~5% astroglial) were cultured from embryonic day 18 Sprague-Dawley rat pups (n ≥ 6 pups per preparation) using a standard protocol, as earlier described (32, 46). In brief, hippocampi were dissected, and then dissociated both enzymatically (0.125% trypsin-EDTA, 37°C, 10 min) and mechanically. The cell suspension was filtered through a 40 μm strainer (Falcon, Franklin Lakes, NJ), and then diluted in serum-free Dulbecco’s modified Eagle medium (DMEM) with 20 mM HEPES, 100μg/ml transferrin, 5μg/ml insulin, 100 μM putrescine, and 30 nM selenium added. Cells were plated at a density of 3.75 × 104 cells/cm2 in 48-well plates (NUNC, Naperville, IL) previously treated with poly-L-lysine (0.05 mg/ml) and used for experiments beginning 3 d after plating. Cultures were kept at 37°C in a humidified incubator with 95% room air/5% CO2.
Culture treatments
Cultures were treated with the steroid hormones testosterone (10 nM or 10 μM), DHT (10 nM or 10 μM), or E2 (10 μM) or vehicle (0.1% ethanol) for 2 h before and during 24 h exposure to toxin. Some cultures were similarly treated with the following inhibitors, delivered at previously established effective concentrations, or vehicle (0.1% dimethyl sulfoxide, DMSO): zVAD-fmk (50 μM) (47, 48), cycloheximide (10 μg/ml) (49), Trolox 250 (μM) (50). The toxins included aggregated Aβ25–35 (0–50 μM), prepared as previously described (32,46), staurosporine (0–0.5 μM), Apoptosis Activator II (0–7 μM), H2O2 (0–25 μM), FeCl2/3 (0–2.5μM), A23187 (0–350 nM), and 3-NP (0–2.5 mM). Steroids, toxins, and inhibitors were solubilised in 100% ethanol or DMSO, and diluted in culture medium to a final ethanol or DMSO concentration of ≤ 0.1%; vehicle controls consisted of ethanol and/or DMSO at 0.1%.
Cell viability
Neuronal viability was determined by standard cell-counting procedures previously described (32, 46, 51). All viable cells within the defined field of a microscope reticle grid (final magnification 300X) were counted using a manual mechanical counter by an experimenter blinded to condition. Cells were scored viable on the basis of both positive staining with the vital dye calcein acetoxymethyl ester (Molecular Probes, Eugene, OR) and the morphological criterion of a smooth, spherical soma. Counts of viable cells were made in four non-overlapping fields per culture well (in a predetermined pattern maintained across all experiments) with each condition represented by 3 separate wells. The number of viable cells counted per well for vehicle-treated control conditions ranged from 100–200. All experiments were repeated in at least 3 independent culture preparations. Raw cell count data were statistically analyzed with one-way ANOVA, followed by between group comparisons using the Fisher LSD test (significance indicated by P < 0.05). Cell viability is presented graphically as a percentage of live cells in the vehicle-treated control condition.
Assessment of nuclear morphology
Nuclear morphology was assessed with the membrane permeable, nucleic acid stain SYTO 11 (Molecular Probes). Briefly, cultures were treated with 1 μM SYTO 11, incubated for 30 min at 37°C in the dark, and nuclear morphology examined using fluorescence microscopy. Neurones were observed for pyknotic nuclei, features of cells undergoing apoptosis (52,53). Degenerating neurones that had condensed nuclei and did not display these features of apoptosis (53) were classified as non-apoptotic. Cultures were evaluated for nuclear morphology by a researcher blinded to experimental condition.
RESULTS
Aβ, staurosporine, and Apoptosis Activator II induce neuronal apoptosis
To begin investigating the hypothesis that androgens selectively protect against apoptosis, we determined whether apoptosis contributes to cell death induced by seven different toxins: Aβ, staurosporine, Apoptosis Activator II, H2O2, FeCl2/3, A23187, and 3-NP. First, we evaluated the effect of two pharmacological inhibitors of apoptosis on the extent of cell death. Specifically, we tested the ability of the general caspase inhibitor zVAD-fmk and the translation inhibitor cycloheximide to attenuate cell death because apoptosis generally involves both activation of caspases and induction of protein synthesis (54,55). Exposure of cultures to 50 μM zVAD-fmk for 2 h before and during insult exposure significantly attenuated cell death due to Aβ, staurosporine, and Apoptosis Activator II (Fig. 1A). In contrast, zVAD-fmk treatment failed to inhibit cell death induced by H2O2, FeCl2/3, A23187, and 3-NP (Fig. 1B). Treatment of cultures with 10μg/ml cyclohexamide also significantly protected against Aβ, staurosporine, and Apoptosis Activator II toxicity (Fig. 1C), but not against H2O2, FeCl2/3, A23187, and 3-NP (Fig. 1D).
Fig. 1.

Pharmacologic inhibition of caspases and protein synthesis attenuates cell death induced by Aβ, staurosporine, and Apoptosis Activator II. Neuroneal cultures were treated with 50 μM zVAD-fmk, 10 μg/ml cycloheximide (CHX), or vehicle for 2 h, then exposed to 50 μM Aβ, 0.4 μM staurosporine (STS), 3 μM Apoptosis Activator II (AAII), 25 μM H2O2, 2.5 μM FeCl2/3, 200 nM A23187, 2.5 mM 3-NP, or vehicle for 24 h, and processed for cell viability. Caspase inhibition with zVAD-fmk reduces cell death induced by (a) Aβ, staurosporine, and Apoptosis Activator II, but not (b) H2O2, FeCl2/3, A23187, and 3-NP. Similarly, cycloheximide attenuates neurotoxicity caused by (c) Aβ, staurosporine, and Apoptosis Activator II, but not (d) H2O2, FeCl2/3, A23187, and 3-NP. Data show mean cell viability (± SEM) pooled from 3 independent experiments (n = 3). * P < 0.05 relative to the vehicle-treated control condition.
To further investigate the contribution of apoptosis in cell death induced by the different toxins, we qualitatively assessed cultures for the presence of pyknotic nuclei, a morphological characteristic of apoptosis (52). Using the nucleic acid stain SYTO 11 to examine nuclear morphology, we found that vehicle-treated neurones exhibited round, smooth, and large nuclei with uniform staining (Fig. 2A). As cells undergo apoptosis, the chromatin condenses and the nuclei break down into small pyknotic spheres (52). After 24 h exposure to 50 μM Aβ (Fig. 2B), 0.4 μM staurosporine (Fig. 2C), or 3 μM Apoptosis Activator II (Fig. 2D), cultures showed numerous neurones with pyknotic nuclei suggestive of cell death involving apoptosis. In contrast, 24 h treatment with 25 μM H2O2 (Fig. 2E), 2.5 μM FeCl2/3 (Fig. 2F), 200 nM calcium ionophore A23187 (Fig. 2G), or 2.5 mM 3-NP (Fig. 2H) resulted in predominately condensed nuclei and essentially no pyknotic nuclei.
Fig. 2.
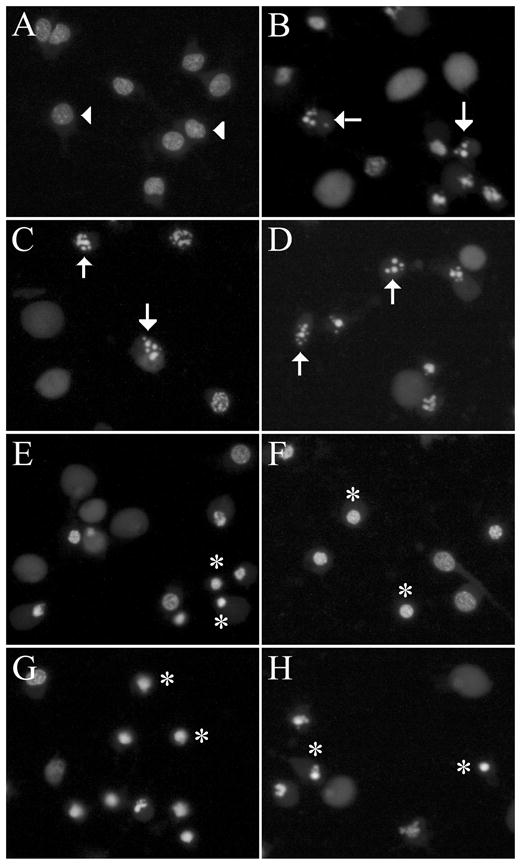
Cell death induced by Aβ, staurosporine, and Apoptosis Activator II is characterised by pyknotic nuclei. Cultures were treated with vehicle, 50 μM Aβ, 0.4 μM staurosporine (STS), 3 μM Apoptosis Activator II (AAII), 25 μM H2O2, 2.5 μM FeCl2/3, 200 nM A23187, or 2.5 mM 3-NP for 24 h. Representative images show nuclear changes visualised with membrane-permeable nucleic acid stain SYTO 11. (a) Vehicle-treated control neurones show normal morphology (arrowheads). (b) Aβ, (c) staurosporine, and (d) Apoptosis Activator II treatment induce pyknotic nuclei in neurones (arrows), while (e) H2O2, (f) FeCl2/3, (g) A23187, and (h) 3-NP induce condensed nuclei (asterisks).
Androgens are neuroprotective against insults that induce apoptosis
If androgen neuroprotection is specific to cell death that involves apoptosis, then only those insults empirically determined to induce apoptosis (i.e., Aβ, staurosporine, Apoptosis Activator II) should be attenuated by androgen treatment. To investigate this prediction, we compared the ability of testosterone to reduce cell death induced by the panel of seven toxins. Cultures were pretreated with 10 nM testosterone, a physiological concentration that we have previously determined exerts maximum levels of neuroprotection (30). Following 2 h testosterone pretreatment, cultures were exposed to increasing concentrations of each toxin, and then analyzed for cell viability by counts of live cells. We observed that androgens protected against neuronal death induced by Aβ, staurosporine, and Apoptosis Activator II across a range of toxin concentrations. Exposure for 24 h to Aβ (0–25 μM), staurosporine (0–0.5 μM), Apoptosis Activator II (0–7 μM), H2O2 (0–25 μM), FeCl2/3 (0–2.5 μM), A23187 (0–350 nM), or 3-NP (0–2.5 mM) decreased cell viability with increasing concentration of toxin compared to vehicle-treated controls (Fig. 3). Pretreatment with 10 nM testosterone significantly reduced toxicity caused by Aβ (Fig. 3A), staurosporine (Fig. 3B), and Apoptosis Activator II (Fig. 3C) at most insult concentrations. In contrast, testosterone pretreatment did not attenuate cell death induced by H2O2 (Fig. 3D), FeCl2/3 (Fig. 3E), A23187 (Fig. 3F), or 3-NP (Fig. 3G).
Fig. 3.

Testosterone is neuroprotective against cell death involving apoptosis. Cultures were treated with 10 nM testosterone (T) or vehicle for 2 h, exposed to increasing concentrations of (a) Aβ25–35, (b) staurosporine (STS), (c) Apoptosis Activator II (AAII), (d) H2O2, (e) FeCl2/3, (f) A23187, or (g) 3-NP for 24 h, and processed for cell viability. Data show mean cell viability (± SEM) pooled from 3 independent experiments (n = 3). * P < 0.05 relative to the matched vehicle-treated condition.
In addition to acting directly on androgen receptors, testosterone is a prohormone that is metabolised in brain into several biologically active hormones. In particular, testosterone is converted to the oestrogen 17β-oestradiol (E2), which acts on oestrogen receptors, and the androgen dihydrotestosterone (DHT), which activates androgen but not oestrogen receptors. Further, DHT can be metabolised to 5α-androstane-3β, 17β-diol (3β-diol), which has been shown to interact with and activate oestrogen receptor β (ERβ) with an affinity similar to E2 (56). Our prior work has shown that testosterone neuroprotection does not involve oestrogen pathways, but rather is dependent upon activation of androgen receptors (30,32), which are expressed in our hippocampal neuron system (32) (and unpublished observations). To confirm an androgen mechanism, we first evaluated the ability of DHT to protect against the seven toxins. We observed that 10 nM DHT induced a pattern of protection parallel to that of testosterone, significantly reducing cell death induced by Aβ, staurosporine, and Apoptosis Activator II (Fig. 4A), but not affecting toxicity resulting from H2O2, FeCl2/3, A23187, or 3-NP (Fig. 4B). Although we found that 10 nM E2 also significantly protected against the insults Aβ and Apoptosis Activator II (Fig. 4D), the DHT metabolite 3β-diol at 1 nM and 10 nM concentrations was not neuroprotective (Fig. 4C). Further, addition of the oestrogen receptor antagonist ICI 182,780 1 h prior to DHT pretreatment did not attenuate DHT protection (Fig. 4E).
Fig. 4.

DHT protects against cell death involving apoptosis by an oestrogen-independent mechanism. To examine neuroprotection, cultures were treated with hormone or vehicle for 2 h, exposed to 50 μM Aβ, 0.4 μM staurosporine (STS), 3 μM Apoptosis Activator II (AAII), 25 μM H2O2, 2.5 μM FeCl2/3, 200 nM A23187, 2.5 mM 3-NP, or vehicle for 24 h, and processed for cell viability. Pretreatment with 10 nM DHT reduced neuronal death induced by apoptosis activators (a) Aβ, staurosporine, and Apoptosis Activator II, but not the non-apoptotic insults (b) H2O2, FeCl2/3, A23187, and 3-NP. In parallel experiments, cell death by the apoptosis activators Aβ, staurosporine, and Apoptosis Activator II was significantly reduced by pretreatment with 10 nM 17β-oestradiol (E2) (d) but not by 1 nM (grey bars) or 10 nM (black bars) concentrations of the DHT metabolite (3β-diol) (c). (e) Neuroprotection afforded by 10 nM DHT was not affected by 1 h pretreatment with 1 μM ICI 182,780, (ICI) an oestrogen receptor antagonist. Data show mean cell viability (± SEM) pooled from 3 independent experiments (n = 3). * P < 0.05 relative to the matched vehicle-treated condition.
Androgens do not protect against oxidative stressors
To investigate the role of oxidative stress in the mechanism of toxin-induced cell death, we assessed the ability of the free radical scavenger Trolox to reduce toxicity induced by the different toxins. Confirming its well established antioxidant properties, treatment of cultures with 250 μM Trolox for 2 h before and during toxin exposure attenuated cell death induced by oxidative stressors H2O2 and FeCl2/3 (Fig. 5B), but not by Aβ, staurosporine, and Apoptosis Activator II (Fig. 5A) nor by A23187 and 3-NP (Fig. 5B).
Fig. 5.

Antioxidants attenuate neuronal death induced by H2O2 and FeCl2/3. Cultures were treated with 250 μM Trolox or vehicle for 2 h, exposed to 50 μM Aβ, 0.4 μM staurosporine (STS), 3 μM Apoptosis Activator II (AAII), 25 μM H2O2, 2.5 μM FeCl2/3, 200 nM A23187, 2.5 mM 3-NP, or vehicle for 24 h, and processed for cell viability. Trolox reduced cell death caused by the oxidative insults (b) H2O2 and FeCl2/3, but not by (a) Aβ, staurosporine, and Apoptosis Activator II nor by (b) A23187 and 3-NP. Data show mean cell viability (± SEM) pooled from 3 independent experiments (n = 3). * P < 0.05 relative to the matched vehicle-treated condition.
Interestingly, oestrogen exerts neuroprotection at micromolar levels by a direct anti-oxidant mechanism (57, 58). To determine if micromolar concentrations of androgens similarly protects, we tested the ability of 10 μM testosterone and 10 μM DHT to attenuate neuronal death induced by oxidative stressors. Consistent with prior reports (57, 58), we found that treatment of cultures with 10 μM E2 for 2 h before and during insult exposure resulted in protection against toxicity induced by H2O2 and FeCl2/3 (Fig. 6D), but not by the other insults (Fig. 6A, D). In comparison, 10 μM testosterone and 10 μM DHT were not protective against the oxidative stressors H2O2 and FeCl2/3 in our paradigm (Fig. 6E, F). The supraphysiological, 10 μM concentration of testosterone and DHT did not increase the efficacy of neuroprotection against apoptosis activator Aβ, staurosporine, and Apoptosis Activator II (Fig. 6B, C) in comparison to the physiological 10 nM concentration (Fig. 3A–C, 4A).
Fig. 6.

Micromolar concentrations of oestrogen, but not androgens can protect against cell death induced by oxidative stressors. Cultures were pretreated for 2 h with vehicle or a supra-physiological concentration (10 μM) of 17β-oestradiol (E2) (a, d), testosterone (b, e), or dihydrotestosterone (DHT), (c, f) then exposed to 50 μM Aβ, 0.4 μM staurosporine (STS), 3 μM Apoptosis Activator II (AAII), 25 μM H2O2, 2.5 μM FeCl2/3, 200 nM A23187, 2.5 mM 3-NP, or vehicle for 24 h, and processed for cell viability. E2 reduced cell death induced by (d) oxidative stressor H2O2, FeCl2/3, but not by (a) apoptotic (Aβ, staurosporine, Apoptosis Activator II) or other non-apoptotic (d) (A23187, 3-NP) insults. Testosterone protection against (b) apoptotic (Aβ, staurosporine, Apoptosis Activator II) and (d) non-apoptotic (H2O2, FeCl2/3, A23187, 3-NP) insults is not enhanced at supra-physiological concentration. Similar results are seen with DHT against (c) apoptotic and (f) non-apoptotic insults. Data show mean cell viability (± SEM) pooled from 3 independent experiments (n = 3). * P < 0.05 relative to the matched vehicle-treated condition.
DISCUSSION
Although androgens are established regulators of neurone survival, the specificity of this action is unclear. In this study, we sought to further characterise androgen neuroprotection by determining what types of insults androgens protect against. Because our prior work demonstrated that androgen neuroprotection involves MAPK/ERK-dependent regulation of the apoptosis-related protein Bad (32), we hypothesised that androgen protection may be specific to cell death involving apoptosis. Consistent with this possibility, our results demonstrate that testosterone and its active androgen metabolite DHT selectively attenuate neuronal death induced by the apoptosis-inducing insults Aβ, staurosporine, and Apoptosis Activator II. Exposure of primary hippocampal neurone cultures to Aβ, staurosporine, or Apoptosis Activator II resulted in pyknotic nuclei and was dependent upon caspase activation and protein synthesis, as evidenced by inhibition of cell death with zVAD-fmk and cycloheximide, respectively. Pyknotic nuclei (52) and involvement of caspases and protein synthesis (53–55) are established components of apoptosis. Conversely, neither testosterone nor DHT reduced cell death caused by exposure to the insults H2O2, FeCl2/3, A23187, and 3-NP. In our paradigm, these insults induced a form of cell death inconsistent with apoptosis as indicated by an absence of pyknotic nuclei and lack of protection resulting from inhibition of caspase activity and protein synthesis. Interestingly, cell death may involve multiple pathways that are not completely blocked by targeted protective agents, as evidenced by our findings that toxicity induced by Aβ, staurosporine, and Apoptosis Activator II was only partially reduced by caspase inhibition, protein synthesis inhibition, and androgens.
Whether specific insults induce apoptosis varies across different culture paradigms. The role of apoptotic pathways may depend not only on the specific insult, but also on insult severity and cellular factors including energy level (59). Consistent with our results, previous studies showed that apoptosis is the predominant form of cell death induced by Aβ (60), staurosporine (39), and Apoptosis Activator II (40). In some paradigms, Aβ can induce non-apoptotic cell death (61). Although H2O2, FeCl2/3, A23187, and 3-NP induced non-apoptotic cell death in our paradigm, these insults can induce apoptosis depending on culture and treatment conditions. For example, calcium ionophore A23187 induces apoptosis at nanomolar concentrations, but necrosis at micromolar concentrations (44). 3-NP can also induce apoptotic and non-apoptotic death in cultured neurones depending on the presence or absence of astrocytes (45) and the level of glutamate (62). Similarly, H2O2 (41,42) and FeCl2/3 (41) induce apoptosis in some culture paradigms. Thus, our classification of insults as either involving apoptosis is expected to vary according to paradigm. Consequently, we predict that the ability of androgens to protect against Aβ, staurosporine, Apoptosis Activator II, H2O2, FeCl2/3, A23187, 3-NP, and other insults may similarly vary across paradigms depending on the relative involvement of apoptosis. Thus, we conclude that androgen neuroprotection is not necessarily insult-specific, but rather specific to cell death that predominantly involves apoptosis.
Despite abundant evidence that androgens are neuroprotective (3), there are also situations in which androgens do not increase neurone viability. For example, although oestrogen protects female mice from injury of nigrostriatal dopaminergic neurones caused by methamphetamines (63) or methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine (MPTP) (64), parallel studies with male mice fail to show a neuroprotective effect of testosterone (63, 64). One explanation for this apparent discrepancy is evidence that classic apoptosis may not be the primary degenerative mechanism resulting from methamphetamine (65) or MPTP (66), although other data suggest apoptotic mechanisms (67, 68). In the absence of a caspase-dependent apoptotic mechanism, our findings would predict that androgens would not protect against dopaminergic neuronal injury. Of course other factors, including regional or cell population differences in androgen responsiveness, may also contribute to observed differences in androgen neuroprotective ability and mechanism. For example, androgens regulate cell number through the apoptotic gene Bax in certain neurones of the bed of nucleus of the stria terminalis, spinal nucleus of the bulbocavernosus, and anteroventral periventricular nucleus of the hypothalamus, but not via Bax in vasopressin-expressing neurones of those regions (69, 70).
Interestingly, a few reports indicate that androgens can actually exacerbate some types of neural injury. In neurone culture paradigms, testosterone can be directly toxic at supraphysiological, micromolar concentrations (71) and increase excitotoxic injury at both micromolar (72) and nanomolar (37) concentrations. Toxic androgen actions may show sex specificity with more robust effects in neuron cultures derived from males (37); our use of cultures from a mixture of male and female rat pups precludes evaluation of whether protective androgen actions are also sex-specific. In animal models, striatal 3-NP lesions are increased by testosterone and reduced by oestrogen in ovariectomised female rats (36). Also, testosterone worsens neural injury caused by middle cerebral artery occlusion model of ischemia-reperfusion (73). Cheng et al. (35) found that DHT increases ischemic infarction volume, perhaps through DHT-regulation of apoptosis-related genes sphingosine kinase 1, B-cell leukemia/lymphoma 2 related proteinA1, and inhibitor of apoptosis protein 1. However, infarction volume may be dose-dependent, with lower androgen doses protecting against and higher doses exacerbating ischemic insults (74). One possibility is that the same signaling pathways that contribute to androgen neuroprotection against some insults may potentiate other insults. For example, our prior work demonstrated that androgen neuroprotection involves activation of a MAPK/ERK signaling pathway (32). However, activation of MAPK/ERK signaling can also drive neuronal cell death in several paradigms (75), including ischemia-reperfusion models (76). Androgen activation of protective versus cell death-promoting pathways may also depend on the relative contributions of intracellular versus membrane-associated androgen receptors (77). Alternatively, as in the case of spinal and bulbar muscular atrophy, androgen-dependent degeneration may result from androgen interaction with androgen receptors containing extended polyglutamine repeat (78). Thus, although our data suggest a straightforward association of androgen neuroprotection with apoptotic insults, the relationship is likely more complex.
Our results also suggest that androgen neuroprotection may not involve direct anti-oxidant action previously demonstrated with oestrogen. Consistent with earlier reports (57,79), we found that supraphysiological, micromolar concentrations of oestrogen are neuroprotective against established oxidative insults. In contrast, micromolar concentrations of testosterone and DHT neither enhanced androgen neuroprotection nor protected against cell death induced by the oxidative stressors H2O2 and FeCl2/3. The observed difference between oestrogen and the androgens testosterone and DHT in terms of anti-oxidant action may reflect the presence of a phenol group on the A ring of oestrogen (79) that is not found on either testosterone or DHT. A phenol-containing structure is common to many anti-oxidants, including vitamin E, and contributes to the neutralization of reactive oxygen species. A prior report demonstrated that testosterone protected cultured cerebellar neurones from cell death caused by the pro-oxidant H2O2 (33). Importantly, the data did not indicate a direct anti-oxidant androgen action, but rather a genomic mechanism involving increased expression of the anti-oxidant enzyme catalase (33). However, whether H2O2 induced apoptosis in this cerebellar neurone paradigm was not determined, and therefore we cannot discount the idea that androgens can protect against oxidative injury that involves apoptosis as the primary mechanism of cell death. Nonetheless, it is noteworthy that androgen neuroprotection may involve regulation of oxidative stress via induction of antioxidant defenses (80–82).
An important cellular function of androgens is promotion of neurone viability, not only during development (21,83), but also in adulthood (28). Apoptosis plays a key role in the regulation of neuronal survival in both development and age-related neurodegenerative diseases. In this study, we have established that androgens selectively protect cultured neurones from cell death involving apoptosis. As evidence accumulates linking age-related androgen depletion with enhanced risk for neurodegenerative disorders (3), continued understanding of androgen neuroprotection will be necessary to successfully exploit the therapeutic potential of androgen compounds for the treatment and or prevention of neurodegenerative diseases.
Acknowledgments
This study was supported by a grant from the National Institute on Aging (AG023739).
References
- 1.Morley JE. Androgens and aging. Maturitas. 2001;38:61–71. doi: 10.1016/s0378-5122(00)00192-4. discussion 71–73. [DOI] [PubMed] [Google Scholar]
- 2.Rosario ER, Chang L, Stanczyk FZ, Pike CJ. Age-related testosterone depletion and the development of Alzheimer disease. JAMA. 2004;292:1431–1432. doi: 10.1001/jama.292.12.1431-b. [DOI] [PubMed] [Google Scholar]
- 3.Pike CJ, Carroll JC, Rosario ER, Barron AM. Protective actions of sex steroid hormones in Alzheimer’s disease. Front Neuroendocrinol. 2009;30:239–258. doi: 10.1016/j.yfrne.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gouras GK, Xu H, Gross RS, et al. Testosterone reduces neuronal secretion of Alzheimer’s beta-amyloid peptides. Proc Natl Acad Sci U S A. 2000;97:1202–1205. doi: 10.1073/pnas.97.3.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramsden M, Nyborg AC, Murphy MP, et al. Androgens modulate beta-amyloid levels in male rat brain. J Neurochem. 2003;87:1052–1055. doi: 10.1046/j.1471-4159.2003.02114.x. [DOI] [PubMed] [Google Scholar]
- 6.Matsumoto A, Prins GS. Androgenic regulation of expression of androgen receptor protein in the perineal motoneurons of aged male rats. J Comp Neurol. 2002;443:383–387. [PubMed] [Google Scholar]
- 7.Pouliot WA, Handa RJ, Beck SG. Androgen modulates N-methyl-D-aspartate-mediated depolarization in CA1 hippocampal pyramidal cells. Synapse. 1996;23:10–19. doi: 10.1002/(SICI)1098-2396(199605)23:1<10::AID-SYN2>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 8.Leranth C, Hajszan T, MacLusky NJ. Androgens increase spine synapse density in the CA1 hippocampal subfield of ovariectomized female rats. J Neurosci. 2004;24:495–499. doi: 10.1523/JNEUROSCI.4516-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ottem EN, Beck LA, Jordan CL, Breedlove SM. Androgen-dependent regulation of brain-derived neurotrophic factor and tyrosine kinase B in the sexually dimorphic spinal nucleus of the bulbocavernosus. Endocrinology. 2007;148:3655–3665. doi: 10.1210/en.2007-0308. [DOI] [PubMed] [Google Scholar]
- 10.Marron TU, Guerini V, Rusmini P, et al. Androgen-induced neurite outgrowth is mediated by neuritin in motor neurones. J Neurochem. 2005;92:10–20. doi: 10.1111/j.1471-4159.2004.02836.x. [DOI] [PubMed] [Google Scholar]
- 11.Brooks BP, Merry DE, Paulson HL, Lieberman AP, Kolson DL, Fischbeck KH. A cell culture model for androgen effects in motor neurons. J Neurochem. 1998;70:1054–1060. doi: 10.1046/j.1471-4159.1998.70031054.x. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L, Chang YH, Barker JL, et al. Testosterone and estrogen affect neuronal differentiation but not proliferation in early embryonic cortex of the rat: the possible roles of androgen and estrogen receptors. Neurosci Lett. 2000;281:57–60. doi: 10.1016/s0304-3940(99)00942-8. [DOI] [PubMed] [Google Scholar]
- 13.Spritzer MD, Galea LA. Testosterone and dihydrotestosterone, but not estradiol, enhance survival of new hippocampal neurons in adult male rats. Dev Neurobiol. 2007;67:1321–1333. doi: 10.1002/dneu.20457. [DOI] [PubMed] [Google Scholar]
- 14.Zhang JM, Konkle AT, Zup SL, McCarthy MM. Impact of sex and hormones on new cells in the developing rat hippocampus: a novel source of sex dimorphism? Eur J Neurosci. 2008;27:791–800. doi: 10.1111/j.1460-9568.2008.06073.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang JM, Tonelli L, Regenold WT, McCarthy MM. Effects of neonatal flutamide treatment on hippocampal neurogenesis and synaptogenesis correlate with depression-like behaviors in preadolescent male rats. Neuroscience. 2010 Apr 21; doi: 10.1016/j.neuroscience.2010.03.029. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nunez JL, McCarthy MM. Resting intracellular calcium concentration, depolarizing Gamma-Aminobutyric Acid and possible role of local estradiol synthesis in the developing male and female hippocampus. Neuroscience. 2009;158:623–634. doi: 10.1016/j.neuroscience.2008.09.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto A. Hormonally induced neuronal plasticity in the adult motoneurons. Brain Res Bull. 1997;44:539–547. doi: 10.1016/s0361-9230(97)00240-2. [DOI] [PubMed] [Google Scholar]
- 18.Jones KJ, Brown TJ, Damaser M. Neuroprotective effects of gonadal steroids on regenerating peripheral motoneurons. Brain Res Brain Res Rev. 2001;37:372–382. doi: 10.1016/s0165-0173(01)00107-2. [DOI] [PubMed] [Google Scholar]
- 19.Huppenbauer CB, Tanzer L, DonCarlos LL, Jones KJ. Gonadal steroid attenuation of developing hamster facial motoneuron loss by axotomy: equal efficacy of testosterone, dihydrotestosterone, and 17-beta estradiol. J Neurosci. 2005;25:4004–4013. doi: 10.1523/JNEUROSCI.5279-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keast JR, Saunders RJ. Testosterone has potent, selective effects on the morphology of pelvic autonomic neurons which control the bladder, lower bowel and internal reproductive organs of the male rat. Neuroscience. 1998;85:543–556. doi: 10.1016/s0306-4522(97)00631-3. [DOI] [PubMed] [Google Scholar]
- 21.Nunez JL, Sodhi J, Juraska JM. Ovarian hormones after postnatal day 20 reduce neuron number in the rat primary visual cortex. J Neurobiol. 2002;52:312–321. doi: 10.1002/neu.10092. [DOI] [PubMed] [Google Scholar]
- 22.Schwarz JM, McCarthy MJ. Cellular mechanisms of estradiol-mediated masculinization of the brain. J Steroid Biochem Mol Biol. 2008;109:300–306. doi: 10.1016/j.jsbmb.2008.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fargo KN, Foecking EM, Jones KJ, Sengelaub DR. Neuroprotective actions of androgens on motoneurons. Front Neuroendocrinol. 2009;30:130–141. doi: 10.1016/j.yfrne.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu WH. Administration of testosterone attenuates neuronal loss following axotomy in the brain-stem motor nuclei of female rats. J Neurosci. 1989;9:3908–3914. doi: 10.1523/JNEUROSCI.09-11-03908.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yu WH, Srinivasan R. Effect of testosterone and 5 alpha-dihydrotestosterone on regeneration of the hypoglossal nerve in rats. Exp Neurol. 1981;71:431–435. doi: 10.1016/0014-4886(81)90101-1. [DOI] [PubMed] [Google Scholar]
- 26.Kujawa KA, Kinderman NB, Jones KJ. Testosterone-induced acceleration of recovery from facial paralysis following crush axotomy of the facial nerve in male hamsters. Exp Neurol. 1989;105:80–85. doi: 10.1016/0014-4886(89)90174-x. [DOI] [PubMed] [Google Scholar]
- 27.Azcoitia I, Sierra A, Veiga S, Honda S, Harada N, Garcia-Segura LM. Brain aromatase is neuroprotective. J Neurobiol. 2001;47:318–329. doi: 10.1002/neu.1038. [DOI] [PubMed] [Google Scholar]
- 28.Ramsden M, Shin TM, Pike CJ. Androgens modulate neuronal vulnerability to kainate lesion. Neuroscience. 2003;122:573–578. doi: 10.1016/j.neuroscience.2003.08.048. [DOI] [PubMed] [Google Scholar]
- 29.Hammond J, Le Q, Goodyer C, Gelfand M, Trifiro M, LeBlanc A. Testosterone-mediated neuroprotection through the androgen receptor in human primary neurons. J Neurochem. 2001;77:1319–1326. doi: 10.1046/j.1471-4159.2001.00345.x. [DOI] [PubMed] [Google Scholar]
- 30.Pike CJ. Testosterone attenuates beta-amyloid toxicity in cultured hippocampal neurons. Brain Res. 2001;919:160–165. doi: 10.1016/s0006-8993(01)03024-4. [DOI] [PubMed] [Google Scholar]
- 31.Zhang Y, Champagne N, Beitel LK, Goodyer CG, Trifiro M, LeBlanc A. Estrogen and androgen protection of human neurons against intracellular amyloid beta1–42 toxicity through heat shock protein 70. J Neurosci. 2004;24:5315–5321. doi: 10.1523/JNEUROSCI.0913-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nguyen TV, Yao M, Pike CJ. Androgens activate mitogen-activated protein kinase signaling: Role in neuroprotection. J Neurochem. 2005;94:1639–1651. doi: 10.1111/j.1471-4159.2005.03318.x. [DOI] [PubMed] [Google Scholar]
- 33.Ahlbom E, Prins GS, Ceccatelli S. Testosterone protects cerebellar granule cells from oxidative stress-induced cell death through a receptor mediated mechanism. Brain Res. 2001;892:255–262. doi: 10.1016/s0006-8993(00)03155-3. [DOI] [PubMed] [Google Scholar]
- 34.Yang SH, Perez E, Cutright J, et al. Testosterone increases neurotoxicity of glutamate in vitro and ischemia-reperfusion injury in an animal model. J Appl Physiol. 2002;92:195–201. doi: 10.1152/jappl.2002.92.1.195. [DOI] [PubMed] [Google Scholar]
- 35.Cheng J, Alkayed NJ, Hurn PD. Deleterious effects of dihydrotestosterone on cerebral ischemic injury. J Cereb Blood Flow Metab. 2007;27:1553–1562. doi: 10.1038/sj.jcbfm.9600457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishino H, Nakajima K, Kumazaki M, et al. Estrogen protects against while testosterone exacerbates vulnerability of the lateral striatal artery to chemical hypoxia by 3-nitropropionic acid. Neurosci Res. 1998;30:303–312. doi: 10.1016/s0168-0102(98)00010-8. [DOI] [PubMed] [Google Scholar]
- 37.Nunez JL, McCarthy MM. Androgens predispose males to GABAA-mediated excitotoxicity in the developing hippocampus. Exp Neurol. 2008;210:699–708. doi: 10.1016/j.expneurol.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature. 1999;399:A23–31. doi: 10.1038/399a023. [DOI] [PubMed] [Google Scholar]
- 39.Harms C, Lautenschlager M, Bergk A, et al. Differential mechanisms of neuroprotection by 17 beta-estradiol in apoptotic versus necrotic neurodegeneration. Journal of Neuroscience. 2001;21:2600–2609. doi: 10.1523/JNEUROSCI.21-08-02600.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang X, Kim HE, Shu H, et al. Distinctive roles of PHAP proteins and prothymosin-alpha in a death regulatory pathway. Science. 2003;299:223–226. doi: 10.1126/science.1076807. [DOI] [PubMed] [Google Scholar]
- 41.Bao F, Liu D. Hydroxyl radicals generated in the rat spinal cord at the level produced by impact injury induce cell death by necrosis and apoptosis: protection by a metalloporphyrin. Neuroscience. 2004;126:285–295. doi: 10.1016/j.neuroscience.2004.03.054. [DOI] [PubMed] [Google Scholar]
- 42.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 43.Burkitt MJ, Gilbert BC. Model studies of the iron-catalysed Haber-Weiss cycle and the ascorbate-driven Fenton reaction. Free Radic Res Commun. 1990;10:265–280. doi: 10.3109/10715769009149895. [DOI] [PubMed] [Google Scholar]
- 44.Gwag BJ, Canzoniero LM, Sensi SL, et al. Calcium ionophores can induce either apoptosis or necrosis in cultured cortical neurons. Neuroscience. 1999;90:1339–1348. doi: 10.1016/s0306-4522(98)00508-9. [DOI] [PubMed] [Google Scholar]
- 45.Ohgoh M, Shimizu H, Ogura H, Nishizawa Y. Astroglial trophic support and neuronal cell death: influence of cellular energy level on type of cell death induced by mitochondrial toxin in cultured rat cortical neurons. J Neurochem. 2000;75:925–933. doi: 10.1046/j.1471-4159.2000.0750925.x. [DOI] [PubMed] [Google Scholar]
- 46.Pike CJ, Burdick D, Walencewicz AJ, Glabe CG, Cotman CW. Neurodegeneration induced by beta-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1687. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gottron FJ, Ying HS, Choi DW. Caspase inhibition selectively reduces the apoptotic component of oxygen-glucose deprivation-induced cortical neuronal cell death. Mol Cell Neurosci. 1997;9:159–169. doi: 10.1006/mcne.1997.0618. [DOI] [PubMed] [Google Scholar]
- 48.Bilsland J, Roy S, Xanthoudakis S, et al. Caspase inhibitors attenuate 1-methyl-4-phenylpyridinium toxicity in primary cultures of mesencephalic dopaminergic neurons. J Neurosci. 2002;22:2637–2649. doi: 10.1523/JNEUROSCI.22-07-02637.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mattson MP, Furukawa K. Anti-apoptotic actions of cycloheximide: blockade of programmed cell death or induction of programmed cell life? Apoptosis. 1997;2:257–264. doi: 10.1023/a:1026433019210. [DOI] [PubMed] [Google Scholar]
- 50.Pike CJ, Ramezan-Arab N, Cotman CW. Beta-amyloid neurotoxicity in vitro: evidence of oxidative stress but not protection by antioxidants. J Neurochem. 1997;69:1601–1611. doi: 10.1046/j.1471-4159.1997.69041601.x. [DOI] [PubMed] [Google Scholar]
- 51.Pike CJ, Vaughan PJ, Cunningham DD, Cotman CW. Thrombin attenuates neuronal cell death and modulates astrocyte reactivity induced by beta-amyloid in vitro. J Neurochem. 1996;66:1374–1382. doi: 10.1046/j.1471-4159.1996.66041374.x. [DOI] [PubMed] [Google Scholar]
- 52.Weil M, Jacobson MD, Coles HS, et al. Constitutive expression of the machinery for programmed cell death. J Cell Biol. 1996;133:1053–1059. doi: 10.1083/jcb.133.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria? Genes Cells. 1998;3:697–707. doi: 10.1046/j.1365-2443.1998.00223.x. [DOI] [PubMed] [Google Scholar]
- 55.Colussi PA, Kumar S. Targeted disruption of caspase genes in mice: what they tell us about the functions of individual caspases in apoptosis. Immunol Cell Biol. 1999;77:58–63. doi: 10.1046/j.1440-1711.1999.00788.x. [DOI] [PubMed] [Google Scholar]
- 56.Weihua Z, Lathe R, Warner M, Gustafsson J. An endrocrine pathway in the prostate, ERβ, AR, 5α-androstane-3β, 17β-diol, and CYP7B1, regulates prostrate growth. Proc Natl Acad Sci U S A. 2002;99:13589–13594. doi: 10.1073/pnas.162477299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Behl C, Skutella T, Lezoualc’h F, et al. Neuroprotection against oxidative stress by estrogens: Structure-Activity relationship. Mol Pharmacol. 1997;51:535–541. [PubMed] [Google Scholar]
- 58.Perez E, Liu R, Yang SH, Cai ZY, Covey DF, Simpkins JW. Neuroprotective effects of an estratriene analog are estrogen receptor independent in vitro and in vivo. Brain Res. 2005;1038:216–222. doi: 10.1016/j.brainres.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 59.Roy M, Sapolsky R. Neuronal apoptosis in acute necrotic insults: why is this subject such a mess? Trends Neurosci. 1999;22:419–422. doi: 10.1016/s0166-2236(99)01435-6. [DOI] [PubMed] [Google Scholar]
- 60.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci U S A. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Behl C, Davis JB, Klier FG, Schubert D. Amyloid beta peptide induces necrosis rather than apoptosis. Brain Res. 1994;645:253–264. doi: 10.1016/0006-8993(94)91659-4. [DOI] [PubMed] [Google Scholar]
- 62.Pang Z, Bondada V, Sengoku T, Siman R, Geddes JW. Calpain facilitates the neuron death induced by 3-nitropropionic acid and contributes to the necrotic morphology. J Neuropathol Exp Neurol. 2003;62:633–643. doi: 10.1093/jnen/62.6.633. [DOI] [PubMed] [Google Scholar]
- 63.Gao X, Dluzen DE. The effect of testosterone upon methamphetamine neurotoxicity of the nigrostriatal dopaminergic system. Brain Res. 2001;892:63–69. doi: 10.1016/s0006-8993(00)03221-2. [DOI] [PubMed] [Google Scholar]
- 64.Dluzen DE. Effects of testosterone upon MPTP-induced neurotoxicity of the nigrostriatal dopaminergic system of C57/B1 mice. Brain Res. 1996;715:113–118. doi: 10.1016/0006-8993(95)01566-3. [DOI] [PubMed] [Google Scholar]
- 65.Pereira FC, Lourenco ES, Borges F, et al. Single or multiple injections of methamphetamine increased dopamine turnover but did not decrease tyrosine hydroxylase levels or cleave caspase-3 in caudate-putamen. Synapse. 2006;60:185–193. doi: 10.1002/syn.20285. [DOI] [PubMed] [Google Scholar]
- 66.Smith PD, Mount MP, Shree R, et al. Calpain-regulated p35/cdk5 plays a central role in dopaminergic neuron death through modulation of the transcription factor myocyte enhancer factor 2. J Neurosci. 2006;26:440–447. doi: 10.1523/JNEUROSCI.2875-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deng X, Wang Y, Chou J, Cadet JL. Methamphetamine causes widespread apoptosis in the mouse brain: evidence from using an improved TUNEL histochemical method. Brain Res Mol Brain Res. 2001;93:64–69. doi: 10.1016/s0169-328x(01)00184-x. [DOI] [PubMed] [Google Scholar]
- 68.Viswanath V, Wu Y, Boonplueang R, et al. Caspase-9 activation results in downstream caspase-8 activation and bid cleavage in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinson’s diease. J Neurosci. 2001;21:9519–9528. doi: 10.1523/JNEUROSCI.21-24-09519.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Forger NG, Rosen GJ, Waters EM, Jacob D, Simerly RB, de Vries GJ. Deletion of Bax eliminates sex differences in the mouse forebrain. Proc Natl Acad Sci U S A. 2004;101:13666–13671. doi: 10.1073/pnas.0404644101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Holmes MM, McCutcheon J, Forger NG. Sex differences in NeuN- and androgen receptor-positive cells in the bed nucleus of the stria terminalis are due to Bax-dependent cell death. Neuroscience. 2009;158:1251–1256. doi: 10.1016/j.neuroscience.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Estrada M, Varshney A, Ehrlich BE. Elevated testosterone induces apoptosis in neuronal cells. J Biol Chem. 2006;281:25492–25501. doi: 10.1074/jbc.M603193200. [DOI] [PubMed] [Google Scholar]
- 72.Orlando R, Caruso A, Molinaro G, et al. Nanomolar concentrations of anabolic-androgenic steroids amplify excitotoxic neuronal death in mixed mouse cortical cultures. Brain Res. 2007;1165:21–29. doi: 10.1016/j.brainres.2007.06.047. [DOI] [PubMed] [Google Scholar]
- 73.Hawk T, Zhang YQ, Rajakumar G, Day AL, Simpkins JW. Testosterone increases and estradiol decreases middle cerebral artery occlusion lesion size in male rats. Brain Res. 1998;796:296–298. doi: 10.1016/s0006-8993(98)00327-8. [DOI] [PubMed] [Google Scholar]
- 74.Uchida M, Palmateer JM, Herson PS, DeVries AC, Cheng J, Hurn PD. Dose-dependent effects of androgens on outcome after focal cerebral ischemia in adult male mice. J Cereb Blood Flow Metab. 2009;29:1454–1462. doi: 10.1038/jcbfm.2009.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhuang S, Schnellmann RG. A death-promoting role for extracellular signal-regulated kinase. J Pharmacol Exp Ther. 2006;319:991–997. doi: 10.1124/jpet.106.107367. [DOI] [PubMed] [Google Scholar]
- 76.Li F, Omori N, Sato K, et al. Coordinate expression of survival p-ERK and proapoptotic cytochrome c signals in rat brain neurons after transient MCAO. Brain Res. 2002;958:83–88. doi: 10.1016/s0006-8993(02)03465-0. [DOI] [PubMed] [Google Scholar]
- 77.Gatson JW, Kaur P, Singh M. Dihydrotestosterone differentially modulates the mitogen-activated protein kinase and the phosphoinositide 3-kinase/Akt pathways through the nuclear and novel membrane androgen receptor in C6 cells. Endocrinology. 2006;147:2028–2034. doi: 10.1210/en.2005-1395. [DOI] [PubMed] [Google Scholar]
- 78.Suzuki E, Zhao Y, Ito S, et al. Aberrant E2F activation by polyglutamine expansion of androgen receptor in SBMA neurotoxicity. Proc Natl Acad Sci U S A. 2009;106:3818–3822. doi: 10.1073/pnas.0809819106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Prokai L, Simpkins JW. Structure-nongenomic neuroprotection relationship of estrogens and estrogen-derived compounds. Pharmacol Ther. 2007;114:1–12. doi: 10.1016/j.pharmthera.2007.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chisu V, Manca P, Lepore G, Gadau S, Zedda M, Farina V. Testosterone induces neuroprotection from oxidative stress. Effects on catalase activity and 3-nitro-L-tyrosine incorporation into alpha-tubulin in a mouse neuroblastoma cell line. Arch Ital Biol. 2006;144:63–73. [PubMed] [Google Scholar]
- 81.Chisu V, Manca P, Zedda M, Lepore G, Gadau S, Farina V. Effects of testosterone on differentiation and oxidative stress resistance in C1300 neuroblastoma cells. Neuro Endocrinol Lett. 2006;27:807–812. [PubMed] [Google Scholar]
- 82.Schmidt AJ, Krieg JC, Vedder H. Differential effects of glucocorticoids and gonadal steroids on glutathione levels in neuronal and glial cell systems. J Neurosci Res. 2002;67:544–550. doi: 10.1002/jnr.10146. [DOI] [PubMed] [Google Scholar]
- 83.Davis EC, Popper P, Gorski RA. The role of apoptosis in sexual differentiation of the rat sexually dimorphic nucleus of the preoptic area. Brain Res. 1996;734:10–18. [PubMed] [Google Scholar]