

Abstract
Induction of protein-protein interactions is a daunting challenge, but recent studies show promise for small molecules that specifically bring two or more protein molecules together for enhanced or novel biological effect. The first such bifunctional molecules were the rapamycin- and FK506-based “Chemical Inducers of Dimerization”, but the field has since expanded with new molecules and new applications in chemical genetics and cell biology. Examples include coumermycin-mediated gyrase B dimerization, proteolysis targeting chimeric molecules (PROTACS), drug hybrids, and strategies for exploiting multivalency in toxin binding and antibody recruitment. This review discusses these and other advances in the design and use of bifunctional small molecules, and potential strategies for future systems.
Introduction
Given the importance of protein-protein interactions in mediating diverse intracellular processes, inhibition of these interactions with small molecules is one of the “holy grails” of pharmacology (1). However, because of the large, complex surfaces involved in these interfaces, there has been only limited success towards this goal, with some notable exceptions (reviewed in (2–4)). But the opposite challenge – small molecule mediated dimerization or oligomerization of proteins – has progressed much further, relying on bifunctional ligands. Here, we discuss efforts to bring proteins together to induce a specific biological effect.
A bifunctional small molecule can be as simple as a natural product that interacts with two protein molecules simultaneously, such as the antibiotic coumermycin (see below) or the fungal toxin brefeldin A, which inhibits the small G-protein Arf by stabilizing an inactive complex with GDP-Arf and a guanine nucleotide exchange factor (5). However, the majority of successful, synthetic, bifunctional molecules consist of two protein-binding moieties joined via a linker of appropriate length (Figure 1). If these moieties are identical (homobifunctional), the molecule will dimerize two identical protein monomers, but if the moieties differ (heterobifunctional), two different proteins can be brought together. The applications of these molecules are as varied as their design. For example, the juxtaposition of two or more proteins can cause ligand-independent activation of receptors or bipartite transcription factors. It can sequester one protein in the subcellular location of the other, target a protein for destruction, or create a binding surface recognized by other proteins, leading to positive or negative regulation of signaling. Bifunctional molecules can specifically localize toxins, and “drug hybrids” (see below) can influence the activity of two proteins without forming dimers. Notably, in most cases these effects are tunable (by varying small molecule concentration), and reversible (on removal of drug).
Figure 1.

A generic bifunctional molecule brings two protein molecules together to induce a biological effect.
The concept of “chemical inducers of dimerization” (CID) (6) dates to early work by the Schreiber and Crabtree groups (7). These labs took advantage of the specific binding of the natural product FK506 to the protein FKBP12 (FK506 binding protein 12). Normally, this drug acts as a natural heterodimerizer, causing the recruitment of the protein phosphatase calcineurin to the FK506-FKBP12 complex. However, an artificial dimer of FK506 named FK1012 instead promotes FKBP12 homodimerization, allowing small-molecule controlled dimerization of signaling proteins (initially, the cytoplasmic domains of the T-cell receptor ζ subunit) fused to FKBP12 monomers (7). Similarly, the natural product heterodimerizer rapamycin promotes interaction between FKBP12 and the FKBP-rapamycin-binding (FRB) domain of mTor. Thus, rapamycin can be used to heterodimerize protein fusions containing FKBP and FRB domains (8). Alternatively, heterodimerization can be induced with an FK506-cyclosporin fusion, which brings together FKBP12 and cyclophilin proteins (9). Subsequently, variants of these molecules were engineered to bind mutant versions of their target proteins, creating orthogonal dimerizer systems that have been used in transgenic mice (10, 11), although recent evidence suggests that both mutant and wild-type FRB-protein fusions are destabilized (12). Dimerization “kits” based on these systems have been developed by ARIAD Pharmaceuticals, Inc (http://www.ariad.com/wt/page/regulation_kits).
The diversity of applications of FKBP-based CID has been comprehensively reviewed recently (13). One noteworthy variant is the “yeast three-hybrid” system (Figure 2) for identifying protein targets of a small molecule (reviewed in (14)). Similar to the yeast two-hybrid method for detecting protein-protein interactions, this approach involves bringing a transcriptional activation domain fusion into contact with a DNA-binding domain fusion, leading to transcription of a reporter gene. Rather than relying on protein-protein interactions of the two fused proteins to activate transcription, the three-hybrid approach uses a CID, with a moiety that binds to the DNA-binding domain fusion, and a moiety that binds to the activation domain fusion. If the latter moiety is a small molecule of unknown target, a library of activation domain-cDNA fusions can be used to identify possible targets. The first proof-of-principle of this system used the synthetic glucocorticoid dexamethasone fused to FK506 to bring FKBP12 and glucocorticoid receptor fusions together (15). But FKBP12/FRB/cyclophilin are not the only possible targets in three-hybrid, or indeed in other, CID systems; other classes of small-molecule dimerizers have been described and offer viable alternatives to traditional CID. Given the recent review of FK506/rapamycin-based CIDs (13), we focus on those bifunctional molecules that do not incorporate FK506 or rapamycin derivatives.
Figure 2.
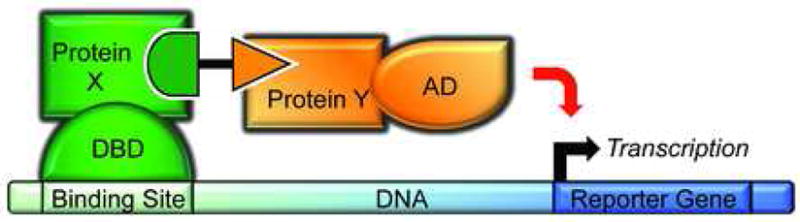
Overview of the yeast three-hybrid technique. A DNA-binding domain (DBD) is fused to Protein X. A heterobifunctional small molecule binds to Protein X, linking it to Protein Y, which in turn is fused to a transcriptional activation domain (AD). The presence of the small molecule brings the two protein fusions together, activating transcription of a reporter gene.
We discuss here three classes of bifunctional molecules, grouped according to their increasing potential for use in whole organisms: those used to date solely in cell-free studies; those used in cells, but requiring genetic manipulation of one or more target proteins; and finally those that target endogenous proteins, i.e., do not require genetic manipulation.
Cell-free studies
Several intriguing small molecule dimerizer systems have unfortunately not yet progressed beyond in vitro experiments (Figure 3). In one of the first departures from FK506/rapamycin-based CID, Kopytek et al. developed, and studied in vitro, a dimerization system based on methotrexate (MTX) (16). This folate analog binds exceptionally strongly to dihydrofolate reductase (DHFR), inhibiting the function of this key enzyme in nucleotide biosynthesis. Since extensive structural information on the MTX-DHFR interaction was available, these authors were able to choose a region of MTX that was not essential for binding, and link it to the same site on a second MTX molecule via an appropriate length linker. The bisMTX 1 caused a concentration-dependent increase in dimerization of purified DHFR. Moreover, binding did not decrease at high bisMTX concentrations, and was tighter to bisMTX than monomeric MTX, suggesting a cooperative binding effect. MTX has since found application as one half of heterobifunctional molecules in other systems (see below).
Figure 3.

Bifunctional molecules used in cell-free studies. Colored shapes represent the regions of each small molecule that bind to the proteins listed. DHFR, dihydrofolate reductase; PLA2, phospholipase A2; SAP, serum amyloid P.
Very recently, a conceptually similar homodimerizer system was presented (17). Human nonpancreatic phospholipase A2 (PLA2) is an enzyme involved in arachidonate and lysophospholipid metabolism, implicated in inflammatory processes. The flat i-face of the enzyme interacts tightly with the phospholipid bilayer. Using a known i-face-binding indole inhibitor of PLA2, Zhou et al. constructed homobifunctional bis-indole ligands with various linkers that were capable of dimerizing purified enzyme. The most effective of these (2) inhibited PLA2 activity five times more effectively than a monomeric indole, offering promise not only as a therapeutic for PLA2-overexpressing rheumatoid arthritis, but also as a novel CID system.
Two other examples build on the CID paradigm, but extend it from a one-small-molecule/two-protein (ternary) system to a multivalent one, leading to a tighter protein-protein interaction. Both cholera toxin B and the human innate immune system protein serum amyloid P (SAP) component exist as pentamers; cholera toxin could potentially be detoxified by aggregation with SAP. Liu et al. reasoned that each cholera toxin pentamer could bind five bifunctional ligands that would each present a moiety to SAP (or vice versa), creating a high avidity interaction (18). Using structure-based design, and known ligands for SAP (D-proline) and cholera toxin (m-nitrophenyl-α-D-galactopyranoside, MNPG), these authors produced a reagent (3) that bound SAP and cholera toxin simultaneously. It also inhibited the binding of cholera toxin to immobilized receptor more effectively than MNPG alone in a SAP-dependent manner.
In a similar vein, Solomon et al. (19) targeted Shiga toxin to SAP. However, rather than relying on binding of five dimerizer molecules, they constructed a five-armed dendrimer (4), with each linker arm ending in a heterobifunctional “glycocluster.” Each glycocluster consisted of a Shiga toxin-binding trisaccharide and a SAP-binding cyclic pyruvate of glycerol. In a Shiga toxin binding competition assay, the dendrimer disrupted Shiga toxin-target binding better than the monomer glycocluster, and addition of SAP further disrupted binding in each case, providing proof-of-principle of this multivalent toxin-SAP dimerizer as a potential therapeutic lead.
Techniques requiring genetic manipulation
Dimerization techniques that have demonstrated function in living cells are substantially closer to whole-organism or clinical deployment than those tested only in vitro. However, many approaches (including the original CID) require genetic manipulation to create chimeric proteins incorporating the dimerizer target and the protein of interest (Figure 4). For this reason, the potential clinical applications of the following are limited, although they are certainly powerful research tools.
Figure 4.

Bifunctional molecules used in techniques requiring genetic manipulation. Colored shapes represent the regions of each small molecule that bind to the fusion proteins listed. GFP, green fluorescent protein; ER, estrogen receptor; DHFR, dihydrofolate reductase; GR, glucocorticoid receptor.
One of the first examples of the CID paradigm to go beyond FK506/rapamycin took advantage of the ability of the well-studied, non-toxic natural product coumermycin (5) to dimerize bacterial DNA gyrase B (GyrB) domains (20). Farrar et al. (21) used coumermycin to form homodimers of Raf-1-GyrB fusions. These homodimers could activate MEK (although surprisingly not all downstream signaling activities (22)), without the usual Ras-dependent membrane localization, thus providing the first evidence of Raf activation independent of Ras. High coumermycin concentrations attenuated this activation, suggesting that binding site saturation leads to dissociation of the dimers, which was confirmed with novobiocin, a “monomeric” form of coumermycin that does not dimerize Raf-GyrB. Interestingly, a similar system was later used to show that coumermycin-mediated Ras dimerization could activate Raf (23).
Subsequently, the GyrB/coumermycin system has been used to activate several signaling pathways, offering more specific control than receptor activation, which tends to feed into multiple signaling pathways. Most work has been done on Jak/Stat signaling. The system has been used to dimerize Janus kinases Jak2 (24, 25) and Tyk2 (26) to dissect receptor-dependent and -independent signaling. Downstream, coumermycin-mediated dimerization of Stat3-GyrB fusion proteins activated transcription, short-circuiting a normally IL-10 dependent growth inhibition (27); in combination with loss-of-function (dominant negative) experiments, the Stat3-GyrB dimer enabled elucidation of the role of p19INK4D in this process (28). However, Stat5-GyrB dimerization could not confer IL-3 independence to cells except in the presence of activated Ras, highlighting the dependence of Stat5 signaling on Ras (29).
A number of membrane-bound receptors have been fused to GyrB. Dimerization of the lymphocyte adhesion molecule L-selectin via fusion of GyrB to its intracellular domain and coumermycin treatment caused increased binding to an extracellular ligand, which is an important part of the inflammatory response (30). Granulocyte colony stimulating factor receptor-GyrB dimerization has been suggested as a potentially viable method of promoting growth of transduced cells in gene therapy, due to the low toxicity of coumermycin (31). In addition, dimerization-mediated activation of vascular endothelial growth factor receptor (VEGFR) allowed analysis of receptor subtypes 1 or 2 separately (32), which was previously only possible by transfection into cell types lacking VEGFR expression (and thus expression of possible downstream signaling components). Coumermycin-mediated dimerization/oligomerization of the platelet activating factor receptor allowed analysis of oligomerization-driven receptor internalization with and without agonist (33).
The system has also been used to confirm the importance of dimerization for intracellular signal transducers. After observing that the mitogen-activated protein kinase kinase kinase ASK1 is naturally activated by dimerization, Gotoh and Cooper confirmed this phenomenon using ASK1-GyrB (34). Sanz et al. used a TRAF6-GyrB fusion to show that dimerized signal transducer TRAF6 could interact with protein kinase C ζ (35), and Ung et al. demonstrated the importance of protein kinase R dimerization (usually RNA-mediated), by replacing the RNA binding domains with GyrB, and showing coumermycin-dependent activity (36).
Finally, this system has been adapted to control gene expression (37): homodimers of the bacteriophage λ repressor, formed by coumermycin-driven dimerization of λ repressor-GyrB fusions, bind to DNA. When a transcriptional activation domain is also fused to these proteins, coumermycin-dependent transcriptional control is possible. Most interestingly, however, is that addition of novobiocin rapidly shuts off coumermycin-driven transcription, offering a powerful on/off gene regulatory system. As in other studies on this well-used dimerization system, strengths of this work are that both coumermycin and novobiocin are well tolerated in vivo, and that GyrB has no known function in mammalian cells, offering promise that this could be a component of regulated gene therapy systems. In addition, it will be interesting to see if recently-reported, simplified coumarin dimers will be amenable to use in GyrB dimerization systems (38).
An alternative, covalent dimerization system was developed by Athavankar and Peterson (39), using unmodified biotin as the small molecule inducer. This system relies on three proteins: the BirA biotin ligase from Escherichia coli, one target protein fused to streptavidin, and the other target protein fused to AviTag, a 15-mer substrate of BirA. BirA catalyzes the biotinylation of lysine in the AviTag (an otherwise extremely rare lysine modification), yielding adduct 6. The biotinylated AviTag fusion protein then binds the streptavidin fusion protein via the extremely high-affinity avidin-biotin interaction. These authors demonstrated the feasibility of this system in a yeast gene expression activation assay similar to a three-hybrid approach: they used a DNA-binding LexA domain fused to streptavidin, and a B42 transcriptional activation domain fused to green fluorescent protein (GFP) and AviTag. Addition of biotin caused a marked increase in lacZ expression driven by the B42 activator, which was saturable at high biotin concentrations. Unfortunately, the advantages of covalent modification offered by this system are largely mitigated by the need to introduce three separate transgenes. Nonetheless, it has recently found application in two gene expression control systems for mammalian cells. The first fuses the AviTag to the VP16 transcriptional activator. In the presence of BirA and biotin, this fusion interacts with a streptavidin-DNA binding module, activating transcription (which can be shut off by treatment with an antibiotic that releases the DNA binding module from the DNA) (40). Conversely, the second system is built around an AviTag-DNA binding module. In the presence of biotin and BirA, a streptavidin-KRAB transrepression domain fusion is recruited, shutting down transcription. This system can be titrated with excess biotin (41).
The Peterson group has also devised novel heterobifunctional reagents for gene expression regulation, in the context of yeast three-hybrid studies (42, 43). One of these molecules (7) combines estrone, a ligand of the estrogen receptor (ER), with biotin, allowing recruitment of streptavidin-B42 to ER-LexA DNA-binding fusions. Along with earlier, FKBP12-based yeast three-hybrid systems, this work complements that of the Cornish laboratory, which synthesized heterobifunctional molecules containing dexamethasone at one end and MTX at the other, allowing recruitment of a glucocorticoid receptor-B42 fusion to a DHFR-LexA fusion (44). They have since modified this system to reduce potential MTX toxicity (but unfortunately losing some activity) by replacing it with the bacterial-specific DHFR inhibitor trimethoprim (45). Most recently, these latter two systems were combined to demonstrate their use as part of a five-input logic circuit (46). The output of the circuit (lacZ transcription) is predictably controlled by the binary variables of presence/absence of unmodified MTX (which inhibits the system in high concentrations), dexamethasone-MTX, dexamethasone-trimethoprim, glucose, and galactose (since the GAL1 promoter that controls the transgene expression is active in the presence of galactose and repressed in the presence of glucose). This demonstration opens new avenues for the use of heterobifunctional small molecules in biological systems engineering.
The Cornish laboratory has also described a specialized twist on their yeast three-hybrid system termed “chemical complementation” (47). They wished to screen the activity of enzymes, so they introduced a cephalosporin unit into the linker of their dexamethasone-MTX molecule (8). In the presence of cephalosporinase, the molecule was cleaved, ablating the transcriptional response (lacZ gene product) (47). Subsequently, they successfully used this system to screen a mutant library for β-lactamase mutants that could be involved in resistance to the cephalosporin cefotaxime (48). In a reverse application, they synthesized dexamethasone linked to cellobioside and MTX linked to lactosyl fluoride, and screened for glycoside linkage formation by glycosynthases, a class of artificial glycosidases (49, 50). In the presence of an appropriate glycosynthase, transcriptional activation of a LEU2 gene occurred, as evidenced by increased survival on leucine dropout medium. This system has been successfully applied to identify a novel glycosynthase (51).
MTX forms one half of yet another dimerizer system, this one developed by the Johnsson lab (52), which has linked it to an O6-benzylguanine (BG) derivative. BG is a substrate for the enzyme O6-alkylguanine-DNA alkyltransferase (AGT, termed “SNAP-Tag”), which covalently binds to it via an active site cysteine. This substrate-enzyme system has been widely used to label fusion proteins with fluorophores for imaging purposes (53, 54). In the dimerizer context, however, the MTX-BG heterobifunctional molecule (9) connected with either an alkyl or polyethylene glycol linker successfully caused a three-hybrid interaction between AGT-LexA and DHFR-B42 fusions, leading to transcriptional activation. In a logical extension of this work, the same group has created bisBG derivatives (with 3 different linker lengths, all functional) capable of covalently dimerizing two AGT-protein fusions (55). This doubly covalent bonding is very appealing, allowing immunoblot evaluation of dimerization efficiency, and “freezing” protein-protein interactions for detection. The authors demonstrated this in two ways: firstly by targeting AGT fusions to different cellular compartments, resulting in much less dimer formation than with similar fusions expressed in the same compartment; and then by showing increased interaction between AGT-FKBP12 and AGT-FRB in the presence of rapamycin. However, the homobifunctional nature of these molecules limits current utility. This group has recently described the “CLIP-tag,” an engineered AGT mutant that specifically binds O2-benzylcytosine (56); we anticipate that future work will yield a BG-benzylcytosine dimerizer.
Techniques not requiring genetic manipulation
The appeal of developing bifunctional small molecules that can exert their effect without the need for genetic manipulation of the treated cells (Figure 5) lies in the fact that we cannot readily genetically manipulate the cells of patients. Two of the earliest examples of such molecules focused on providing selectivity to compounds that were previously non-selective, both targeting the intracellular estrogen receptor (ER).
Figure 5.

Bifunctional molecules used in techniques that do not require genetic manipulation. Colored shapes represent the regions of each small molecule that bind to the proteins listed. ER, estrogen receptor; MetAP-2, methionine aminopeptidase 2; GFP-AR, green fluorescent protein-androgen receptor; VHL, von Hippel Lindau; AHR, aryl hydrocarbon receptor.
First, Rink and co-workers described a system (10) in which a DNA-alkylating agent was conferred with improved selectivity by tethering it to a moiety that recognizes the ER (57). Nitrogen mustards have long been known to alkylate DNA in a non-specific manner, resulting in potent if indiscriminant cytotoxicity. By contrast, 2-phenylindoles bind specifically to their target, the ER, which is over-expressed in many breast and ovarian tumors. Comparisons were made between toxicity towards the ER-positive MCF7 cell line and the ER-negative MDA-MB-231 cell line, both from breast tumors. Compounds with no affinity for the ER showed similar effects on both cell lines. The synthesized bifunctional molecules that did show affinity for the ER demonstrated higher toxicity for the MCF7 cells than for the MDA-MB-231 cells, indicating that the presence or absence of ER was a contributing factor. Treatment with estradiol following application of the drug resulted in lower toxicity towards MCF7 cells, further supporting the role of ER binding in providing the selectivity. The authors propose various explanations for this selectivity, the most plausible of which is the apparent shielding of the damaged DNA from repair enzymes by the proximity of the 2-phenylindole-bound ER. These authors have subsequently optimized their constructs, varying linker length and using estradiol as the ER ligand (58, 59). Improving the selectivity of mustard-based DNA alkylators has also been achieved by linking them to DNA minor groove binders (60).
The second early targeting of the ER involved modifying the toxicity of geldanamycin, a natural product that normally binds to the molecular chaperone Hsp90 and induces proteasome-dependent degradation of a range of proteins. This unfortunately results in non-selective toxicity and undesirable side effects. A strategy to harness this toxicity towards a particular target, the ER, required the synthesis of a geldanamycin derivative attached to a molecule that would bind to the ER, in this case, estradiol (61).
In MCF7 cells, geldanamycin induced rapid degradation of ER, HER-2, Raf-1 and IGF1R. Construct 11 was found to induce degradation of the ER, but had no effect on levels of Raf-1 or IGF1R, and the rate of HER-2 degradation was partially attenuated, although the activity of the construct was highly dependent on the type and length of linker. A similar approach was taken in a more recent report, wherein estradiol was used to target a photoactivatable porphyrin to ER-positive cells, resulting in selective killing of the targeted cells after exposure to red light (62). These studies demonstrate that the selectivity profile of a toxic compound can be vastly improved by linkage to a targeting moiety.
A similar approach was used to target degradation of the androgen receptor (AR), a nuclear receptor implicated in progression of prostate cancer. In that instance, the geldanamycin hybrid was prepared with the AR ligand testosterone (63). Again, biological activity was linker-dependent, but one bifunctional molecule was found to have cytotoxicity towards LNCaP prostate cancer cells comparable with geldanamycin itself. Testing this molecule against a range of cell lines showed that, in AR-dependent cells, it had an IC50 comparable to that of geldanamycin. By contrast, in AR-independent cells, the bifunctional construct was found to be more than ten times weaker than geldanamycin, thus providing a promising therapeutic window.
One area in which heterobifunctional small molecules have played an important role is ubiquitin-dependent proteolysis: poly-ubiquitination of a given protein results in degradation by the proteasome in a process that occurs as part of normal cellular homeostasis. Ubiquitin is a small, highly conserved protein that is attached to lysine residues on proteins targeted for degradation through the action of ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2) and ubiquitin-protein ligases (E3), the last of which binds directly to the substrate protein and thus confers the specificity of ubiquitination. Successive conjugation of ubiquitin to previously added ubiquitin units on the targeted protein results in a polyubiquitin chain that is recognized by the 26S proteasome, ultimately leading to degradation of the target protein.
A bifunctional molecule capable of hijacking this system and selectively degrading targeted proteins is a powerful tool for chemical genetics. Small molecule-induced knockdown provides a new method of learning about the role and function of proteins through controlled knockdown. This is particularly useful for studying proteins essential for development that cannot be constitutively knocked out. PROteolysis TArgeting Chimeric molecules (PROTACS) make use of the ubiquitin-proteasome pathway and allow for such temporally controlled elimination of proteins in a post-translational manner, operating through simultaneous binding of a target protein and an E3 ligase (Figure 6).
Figure 6.

Overview of the PROTACS system. A PROTAC molecule brings a target protein into contact with an E3 ubiquitin ligase, prompting transfer of ubiquitin (Ub) from an E2 ubiquitin conjugating enzyme, leading to polyubiquitination of the target protein and degradation by the 26S proteasome.
The initial proof-of-concept (64) focused on degrading methionine aminopeptidase-2 (MetAP-2), which cleaves N-terminal methionine residues from nascent polypeptides. Ovalicin is a member of the fumagillin class of angiogenesis inhibitors, known to bind to the active site of MetAP-2 (65), and was thus chosen as one of the two heads in “PROTAC-1” (12). Ovalicin was linked to the E3-ligase binding sequence of IκBα, reported to bind to the SCFβ-TRCP complex that induces ubiquitination. After demonstrating that PROTAC-1 binds to MetAP-2 in vitro and recruits it to the SCFβ-TRCP E3 ligase, Sakamoto et al. (64) then showed that MetAP-2 is indeed ubiquitinated, and that this is dependent on the presence of PROTAC-1. When tested in Xenopus egg extracts, a PROTAC-1-MetAP-2 complex was degraded within thirty minutes, while free MetAP-2 levels remained constant. Saliently, addition of proteasome inhibitors blocked this degradation, indicating that PROTAC-1 was acting via the endogenous ubiquitin-proteasome pathway.
From this in vitro beginning, the PROTAC concept has progressed towards potential use as a therapeutic: a subsequent paper used estradiol and dihydroxytestosterone-based PROTACS to degrade the estrogen and androgen receptors, respectively (66), both of which are implicated in certain types of cancer. The authors microinjected a PROTAC into cells, with the result that degradation of an AR-GFP fusion protein could be monitored by the loss of fluorescence. Cell permeability issues related to the phosphorylated IκBα moiety were circumvented in a third paper (67), where the bifunctional molecule incorporated a polyarginine chain to mimic the HIV Tat protein (known to act as a molecular transporter) and used a 7-amino acid recognition sequence for the von Hippel-Lindau tumor suppressor protein (VHL) as the E3-ligase binding domain. This PROTAC-5 (13) was added to HEK293 cells stably transfected with an AR-GFP fusion protein, the only genetic manipulation involved in this system and used purely for visualization purposes. The proteasome-mediated degradation of AR-GFP in these cells was observed through loss of GFP fluorescence, representing a major step towards therapeutic utility.
Subsequently, the 7-amino acid VHL recognition sequence has been simplified to a highly cell-permeable pentapeptide (68), and the first fully genetic manipulation-free system has been reported (69). The aryl hydrocarbon receptor has also been targeted for degradation by 14, enabling future study of the function of this protein, specifically as related to tumor development (70, 71). Most recently, our group has described an all small-molecule (non-peptidic) PROTAC targeting the AR for degradation using an MDM2-binding nutlin derivative as the E3 ligase-binding moiety (72).
Recent contributions to the field of heterobifunctional molecules have tapped into the power of the immune system, a strategy raised in the early 1990s (73, 74) (Figure 7). Kiessling and co-workers (75) have synthesized a small molecule capable of recruiting a common human antibody exclusively to cells expressing a cell-surface receptor that is over-expressed on cancer cells and largely absent from normal tissue, the αvβ3 integrin receptor. This recruitment renders the cell susceptible to attack by the immune system, leading to elimination of specifically targeted cells.
Figure 7.

Harnessing immune recognition with small molecules. A small molecule ligand for the target protein (brown), known to be expressed on the cells of interest, is linked to an epitope that is recognized by endogenous antibodies (purple).
As an initial step, two ligands exhibiting high potency (low nM range) and good specificity for αvβ3 were chosen to form one end of the bifunctional molecule (75). Crystal structures of one ligand (the cyclic RGD peptide) bound to αvβ3 allowed the design of linker-incorporating derivatives that would not perturb binding. The other end of the molecule required the epitope component (see Figure 7). The carbohydrate-based α-Gal epitope is known to be immunogenic, though it is not displayed in human cells. High levels of anti-Gal antibody are, however, present in humans, due to exposure to α-Gal-displaying bacteria. An appropriately functionalized trisaccharide moiety was chosen for antibody recruitment.
Both conjugates prevented interaction between cells expressing αvβ3 and a known protein ligand for that receptor, fibrinogen, with IC50 values in the nanomolar range (75). By contrast, the IC50 values calculated for cells expressing αvβ5 was in the micromolar range, indicating that the bifunctional molecules possessed the required specificity. The conjugate 15 was then shown to bind simultaneously to both αvβ3-positive cells and anti-Gal IgG from human serum, demonstrating the potential for this concept to provide new therapies that specifically target tumor cells. A follow-up report confirmed that the conjugates were cytotoxic only to cells expressing high levels of αvβ3 receptors (76). Cells with only low receptor levels survived, due to an inability to form multivalent antibody interactions with the required avidity.
In a similar approach, O’Reilly et al. (77) recently linked the antigen nitrophenol (for which endogenous antibodies exist in humans) to a modified trisaccharide ligand for CD22, a B cell surface protein that can be targeted in B cell depletion therapy. The resulting bifunctional molecule (16) was able to form complexes between decameric anti-nitrophenol IgM and clusters of CD22 molecules both in vitro and on B cells, offering an attractive, multivalent way of binding these low-affinity receptors, potentially leading to B cell killing by the immune system.
Using a different approach, Matsuda et al. (78) investigated lymphocyte-endothelial cell interactions, which are potentially important for anti-inflammatory and antimetastatic therapeutic interventions. Both carbohydrate-binding selectins and protein-binding integrins are involved in these interactions, so these authors created a chimeric molecule (17) fusing the P-selectin-binding tetrasaccharide known as sialyl Lewisx with a flexible KG dipeptide linker and the integrin-binding peptide RGDS (78). As well as binding integrin β1 and P-selectin independently, this compound could form a ternary complex with both immobilized P-selectin and integrin β1 in solution. In a preliminary functional test, the heterobifunctional construct inhibited T-cell binding to collagen-coated plates, while the individual tetrapeptide and tetrasaccharide constituents did not, suggesting that this kind of double-headed molecule could have useful biological applications.
The Portoghese lab, moving towards the clinic, and following a long line of research on the roles of opioid receptors (79), has reported the identification of heterobifunctional molecules that are not only fifty times more potent than morphine, but that do not induce the same dependence and tolerance as the well-known drug of addiction when administered to mice (80). The so-called “MDAN” ligands (μ-δ agonist-antagonist) comprise an agonist of the μ opioid receptor (oxymorphone) and an antagonist of the δ receptor (naltrindole). MDAN-21 (18), containing a 21-atom linker between the two pharmacophores, produced neither tolerance nor dependance, but shorter linkers produced either both responses or tolerance alone. The authors conclude that this biological effect is a result of the heterodimer formed between the μ and δ receptors. Further, the potency of these compounds holds promise for developing new pain-killers with improved side-effect profiles.
Neurodegenerative diseases such as Parkinson’s and Alzheimer’s operate by complex mechanisms that are not yet fully understood, leading many to believe that a cocktail of drugs may be necessary to achieve improved therapeutic outcomes (81). Since an alternative to multiple drugs is a single drug agent that performs multiple therapeutic roles, researchers in this and other fields are turning towards bifunctional molecules (Figure 8). Unlike many other bifunctional molecules discussed in this review, these “drug hybrids” tend to incorporate two functional elements into a single framework, resulting in smaller drugs than the linking of two distinct fragments through a long linker. While this strategy is suitable for achieving two distinct biological effects, it is unlikely to succeed in inducing formation of a ternary complex with two biomolecules.
Figure 8.

Drug hybrids combine two pharmacophores in a single molecule. Functional moieties are indicated by colored shapes. MAO, monoamine oxidase; GABA, γ-aminobutyric acid.
Levels of both monoamine oxidase (MAO) activity and iron are increased in the brain as a result of Parkinson’s disease. Exploiting the finding that neuroprotection could be obtained from 8-hydroxyquinoline-based iron chelators as well as from MAO-inhibiting propargylamines, Youdim and co-workers combined these two components into a series of molecules that possessed both properties. The best results were obtained for 19, which exhibited good iron chelation levels in vitro and provided neuroprotection against oxidative stress in P19 cells, despite a relatively weak ability to inhibit MAO activity (82, 83). Similarly, work on Alzheimer’s disease has combined the nitric oxide mimicking potential of nitrate esters with a heterocyclic scaffold present in the simple neuroprotective γ-aminobutyric acid receptor agonist clomethiazole. Nitric oxide signaling controls many processes in the central nervous system, and is thus a factor in neurodegenerative diseases. The product of these bifunctional endeavors is GT 1061 (20), an orally available and blood-brain barrier crossing molecule that has been extensively studied in rats (84) and has been approved for Phase I clinical trials for Alzheimer’s disease (85).
Examples of drug hybrids for malaria treatment were recently highlighted in C&E News (86). As with the neurodegenerative treatments discussed above, these hybrids do not form ternary complexes with two separate biomolecules, but act as bifunctional molecules with two different targets. Whether the hybrids remain intact in vivo can be unclear, although they may have advantages over drug cocktails in terms of dosing, delivery and localization. Using this strategy, several different bifunctional molecules targeting malaria have been produced. Exploitation of the synergy between fast-acting artemisinin and slow-acting quinine provided a hybrid (21) with a lower IC50 than either drug alone or both drugs together, in two different strains of the Plasmodium falciparum parasite (87) (a related approach was reported earlier (88)). Also, the Peyton lab has connected the quinoline moiety of the known antimalarial chloroquine to a tricyclic “reversal agent” known to block the action of a transporter that removes chloroquine from its site of action. This resulted in a hybrid (22) that demonstrated potent growth inhibition of P. falciparum in culture and promising early results in mice (89). Application of drug hybrids in the field of HIV treatment was reviewed recently (90).
Conclusions
The past decade has seen significant advances in the use of heterobifunctional small molecules. While the well-established FK506/rapamycin systems have been most extensively studied and validated, the strategies reviewed here also represent valuable additions to the arsenal of those working at the chemistry-biology interface. The breadth of biological problems to which these molecules have been applied demonstrates their potential to tackle many outstanding questions in biomedical research, and to move towards therapeutic applications in the future. Some of the lessons learned in the research discussed above can help lead this field into the future.
The benefits of a modular synthesis cannot be underestimated: fine-tuning the properties of either end of a heterobifunctional molecule can become tedious if a lengthy linear synthetic route is taken. Many of the reported systems make use of small molecules already known to bind to a specific protein, and thus developing a full synthesis from scratch is rarely necessary. Indeed, the wealth of literature from drug discovery programs highlights the many proteins that can be readily targeted in future studies with heterobifunctional molecules. Further, it is worth noting that small molecules need not necessarily bind to the active site of proteins to be useful: any high-affinity ligand is capable of promoting the desired interaction. It is also appealing to envisage a “toolbox” of dimerizing reagents. Once a ligand for a specific protein is discovered, incorporating it into variants of the heterobifunctional molecules described above may provide useful information about the role of that protein. Such an approach would build towards libraries of bifunctional molecules targeting large sections of the proteome, enabling chemical genomic degradation, mislocalization, sequestration, or even rapid screens of potential synergism.
Linking two molecule fragments, however, provides its own challenges. The site through which the linker is tethered can be crucial. In particular, great care must be taken to avoid disrupting pharmacophores or inducing a change in conformation that compromises binding affinity. Although linker composition is frequently governed by synthetic ease and compound availability, identifying the optimum linker length is vital (this is typically determined empirically, with minor length differences able to cause significant differences in activity). Linkers must be long enough and flexible enough to enable both ends of the molecule to interact freely with their protein targets, but short enough to ensure that the proteins are close enough for whatever dimerization effect is desired. Linkers should not be overly hydrophobic (which would promote self-interaction in aqueous solution), or too reactive (esters, for instance, could be cleaved in vivo). One of the major drawbacks of bifunctional molecules as potential therapeutics is their large size. Bulky molecules are not only likely to be insoluble and therefore orally unavailable, but also prone to be membrane impermeable, leading to correspondingly poor pharmacokinetics. Minimizing ligand and linker size, and structure optimization before clinical deployment of bifunctional molecules in future will be key to overcoming these very real concerns. The successes of drug hybrids show that this is possible.
There is great potential to combine elements of some of the systems described here (and elsewhere (13)) to facilitate the targeting of related proteins, or to induce formation of a different ternary complex to achieve a different biological result. New combinations will inevitably produce new ways of obtaining improved drug selectivity, a key feature in much of this research, moving towards Paul Ehrlich’s “magic bullet” concept of directing a toxin to the origin of a disease. Simultaneously, further improvements in potency can be expected, as exemplified by the drug hybrid approaches and also through minimizing loss of drug to other parts of the cell. Part of the selectivity of these bifunctional molecules derives from the need to establish two separate interactions, but this can also represent a drawback: the affinity of each interaction must be strong enough to sustain the desired biological effect. Covalent binding, in this context, can be highly beneficial, as it eliminates concern over at least one binding affinity (91), as demonstrated in the cases of the SNAP-Tag and the BirA biotin ligase systems. New technologies may find useful applications in this respect: the chloroalkane HaloTag ligand has demonstrated potential as a chemically simple, specific and orthogonal covalent labeling technique (92), which has recently been coupled with the SNAP-Tag to create “covalin,” a bifunctional protein capable of heterodimerizing HaloTag ligand- and BG-tagged proteins (93). HaloTag-based small molecule dimerizer systems might represent a new experimental direction in the future.
The benefits of using heterobifunctional molecules are clear, from new molecular probes to selective and specific therapeutics. The next step will be to consider how these molecules could improve the lives of patients, to push beyond proof-of-principle studies and into whole organisms, and to see that often two heads are better than one.
Acknowledgments
We thank A. Schneekloth and T. Neklesa for helpful comments on the manuscript. C.M.C. gratefully acknowledges the support of the National Institutes of Health (CA118631). N.A. is the American-Australian Association’s Alcoa Foundation Fellow. T.W.C. is the Canadian Institutes of Health Research Jean-François St-Denis Fellow in Cancer Research and a Bisby Fellow.
Keywords
- Bifunctional
Of a small molecule, having two functionalities, such as binding two proteins
- Chemical Inducers of Dimerization (CID)
Small molecules that can specifically cause dimerization of two protein molecules
- Yeast three-hybrid
A technique used to assay small molecule-protein interactions, consisting of a DNA binding domain fusion protein, a transcriptional activation domain fusion protein, and a small molecule that unites them to activate a transcriptional readout
- Multivalent
Displaying multiple copies of attachment sites for ligands or other binding partners
- Ubiquitin-dependent proteolysis
A mechanism of regulated protein degradation driven by the selective linkage of the small protein ubiquitin to a target protein, marking it for destruction by the 26S proteasome
- PROTAC
PROteolysis TArgeting Chimera, a heterobifunctional molecule that links a target protein to an E3 ubiquitin ligase, leading to ubiquitin-dependent proteolysis of the target
- Drug hybrid
A covalent fusion of two or more existing drugs or pharmacophores to create a single molecule with multiple, but not necessarily simultaneous, pharmacological targets
- Magic bullet
A drug that specifically targets a disease-causing agent, such as a microorganism, with no side effects on the patient. The term is attributed to Paul Ehrlich, developer of arsphenamine (Salvarsan), the first modern chemotherapeutic, which selectively kills the spirochete that causes syphilis
References
- 1.Chene P. Drugs targeting protein-protein interactions. Chem Med Chem. 2006;1:400–411. doi: 10.1002/cmdc.200600004. [DOI] [PubMed] [Google Scholar]
- 2.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 3.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 4.Yin H, Hamilton AD. Strategies for targeting protein-protein interactions with synthetic agents. Angew Chem Int Ed. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 5.Mossessova E, Corpina RA, Goldberg J. Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol Cell. 2003;12:1403–1411. doi: 10.1016/s1097-2765(03)00475-1. [DOI] [PubMed] [Google Scholar]
- 6.Pollock R, Clackson T. Dimerizer-regulated gene expression. Curr Opin Biotechnol. 2002;13:459–467. doi: 10.1016/s0958-1669(02)00373-7. [DOI] [PubMed] [Google Scholar]
- 7.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling signal transduction with synthetic ligands. Science. 1993;262:1019–1024. doi: 10.1126/science.7694365. [DOI] [PubMed] [Google Scholar]
- 8.Rivera VM, Clackson T, Natesan S, Pollock R, Amara JF, Keenan T, Magari SR, Phillips T, Courage NL, Cerasoli F, Jr, Holt DA, Gilman M. A humanized system for pharmacologic control of gene expression. Nat Med. 1996;2:1028–1032. doi: 10.1038/nm0996-1028. [DOI] [PubMed] [Google Scholar]
- 9.Belshaw PJ, Ho SN, Crabtree GR, Schreiber SL. Controlling protein association and subcellular localization with a synthetic ligand that induces heterodimerization of proteins. Proc Natl Acad Sci U S A. 1996;93:4604–4607. doi: 10.1073/pnas.93.10.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liberles SD, Diver ST, Austin DJ, Schreiber SL. Inducible gene expression and protein translocation using nontoxic ligands identified by a mammalian three-hybrid screen. Proc Natl Acad Sci U S A. 1997;94:7825–7830. doi: 10.1073/pnas.94.15.7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stankunas K, Bayle JH, Gestwicki JE, Lin YM, Wandless TJ, Crabtree GR. Conditional protein alleles using knockin mice and a chemical inducer of dimerization. Mol Cell. 2003;12:1615–1624. doi: 10.1016/s1097-2765(03)00491-x. [DOI] [PubMed] [Google Scholar]
- 12.Edwards SR, Wandless TJ. The rapamycin-binding domain of the protein kinase mammalian target of rapamycin is a destabilizing domain. J Biol Chem. 2007;282:13395–13401. doi: 10.1074/jbc.M700498200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gestwicki JE, Marinec PS. Chemical control over protein-protein interactions: beyond inhibitors. Comb Chem High Throughput Screen. 2007;10:667–675. doi: 10.2174/138620707782507296. [DOI] [PubMed] [Google Scholar]
- 14.Kley N. Chemical dimerizers and three-hybrid systems: scanning the proteome for targets of organic small molecules. Chem Biol. 2004;11:599–608. doi: 10.1016/j.chembiol.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 15.Licitra EJ, Liu JO. A three-hybrid system for detecting small ligand-protein receptor interactions. Proc Natl Acad Sci U S A. 1996;93:12817–12821. doi: 10.1073/pnas.93.23.12817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopytek SJ, Standaert RF, Dyer JC, Hu JC. Chemically induced dimerization of dihydrofolate reductase by a homobifunctional dimer of methotrexate. Chem Biol. 2000;7:313–321. doi: 10.1016/s1074-5521(00)00109-5. [DOI] [PubMed] [Google Scholar]
- 17.Zhou L, Fang C, Wei P, Liu S, Liu Y, Lai L. Chemically induced dimerization of human nonpancreatic secretory phospholipase A2 by bis-indole derivatives. J Med Chem. 2008;51:3360–3366. doi: 10.1021/jm7010707. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Zhang Z, Tan X, Hol WG, Verlinde CL, Fan E. Protein heterodimerization through ligand-bridged multivalent pre-organization: enhancing ligand binding toward both protein targets. J Am Chem Soc. 2005;127:2044–2045. doi: 10.1021/ja043817r. [DOI] [PubMed] [Google Scholar]
- 19.Solomon D, Kitov PI, Paszkiewicz E, Grant GA, Sadowska JM, Bundle DR. Heterobifunctional multivalent inhibitor-adaptor mediates specific aggregation between Shiga toxin and a pentraxin. Org Lett. 2005;7:4369–4372. doi: 10.1021/ol051529+. [DOI] [PubMed] [Google Scholar]
- 20.Farrar MA, Olson SH, Perlmutter RM. Coumermycin-induced dimerization of GyrB-containing fusion proteins. Methods Enzymol. 2000;327:421–429. doi: 10.1016/s0076-6879(00)27293-5. [DOI] [PubMed] [Google Scholar]
- 21.Farrar MA, Alberol I, Perlmutter RM. Activation of the Raf-1 kinase cascade by coumermycin-induced dimerization. Nature. 1996;383:178–181. doi: 10.1038/383178a0. [DOI] [PubMed] [Google Scholar]
- 22.Farrar MA, Tian J, Perlmutter RM. Membrane localization of Raf assists engagement of downstream effecters. J Biol Chem. 2000;275:31318–31324. doi: 10.1074/jbc.M003399200. [DOI] [PubMed] [Google Scholar]
- 23.Inouye K, Mizutani S, Koide H, Kaziro Y. Formation of the Ras dimer is essential for Raf-1 activation. J Biol Chem. 2000;275:3737–3740. doi: 10.1074/jbc.275.6.3737. [DOI] [PubMed] [Google Scholar]
- 24.Mohi MG, Arai K, Watanabe S. Activation and functional analysis of Janus kinase 2 in BA/F3 cells using the coumermycin gyrase B system. Mol Biol Cell. 1998;9:3299–3308. doi: 10.1091/mbc.9.12.3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R, Liu CB, Mohi MG, Arai K, Watanabe S. Analysis of mechanisms involved in the prevention of gamma irradiation-induced apoptosis by hGM-CSF. Oncogene. 2000;19:571–579. doi: 10.1038/sj.onc.1203364. [DOI] [PubMed] [Google Scholar]
- 26.Mizuguchi R, Hatakeyama M. Conditional activation of janus kinase (JAK) confers factor independence upon interleukin-3-dependent cells - Essential role of Ras in JAK-triggered mitogenesis. J Biol Chem. 1998;273:32297–32303. doi: 10.1074/jbc.273.48.32297. [DOI] [PubMed] [Google Scholar]
- 27.O’Farrell AM, Liu Y, Moore KW, Mui ALF. IL-10 inhibits macrophage activation and proliferation by distinct signaling mechanisms: evidence for Stat3-dependent and -independent pathways. EMBO J. 1998;17:1006–1018. doi: 10.1093/emboj/17.4.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Farrell AM, Parry DA, Zindy F, Roussel MF, Lees E, Moore KW, Mui ALF. Stat3-dependent induction of p19INK4D by IL-10 contributes to inhibition of macrophage proliferation. J Immunol. 2000;164:4607–4615. doi: 10.4049/jimmunol.164.9.4607. [DOI] [PubMed] [Google Scholar]
- 29.Mizuguchi R, Noto S, Yamada M, Ashizawa S, Higashi H, Hatakeyama M. Ras and signal transducer and activator of transcription (STAT) are essential and sufficient downstream components of Janus kinases in cell proliferation. Japan J Cancer Res. 2000;91:527–533. doi: 10.1111/j.1349-7006.2000.tb00977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li X, Steeber DA, Tang MLK, Farrar MA, Perlmutter RM, Tedder TF. Regulation of L-selectin-mediated rolling through receptor dimerization. J Exp Med. 1998;188:1385–1390. doi: 10.1084/jem.188.7.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kume A, Ito K, Ueda Y, Hasegawa M, Urabe M, Mano H, Ozawa K. A G-CSF receptor-gyrase B fusion gene: A new type of molecular switch for expansion of genetically modified hematopoietic cells. Biochem Biophys Res Commun. 1999;260:9–12. doi: 10.1006/bbrc.1999.0859. [DOI] [PubMed] [Google Scholar]
- 32.Knight EL, Warner AJ, Maxwell A, Prigent SA. Chimeric VEGFRs are activated by a small-molecule dimerizer and mediate downstream signalling cascades in endothelial cells. Oncogene. 2000;19:5398–5405. doi: 10.1038/sj.onc.1203915. [DOI] [PubMed] [Google Scholar]
- 33.Perron A, Chen ZG, Gingras D, Dupre DJ, Stankova J, Rola-Pleszczynski M. Agonist-independent desensitization and internalization of the human platelet-activating factor receptor by coumermycin-gyrase B-induced dimerization. J Biol Chem. 2003;278:27956–27965. doi: 10.1074/jbc.M212302200. [DOI] [PubMed] [Google Scholar]
- 34.Gotoh Y, Cooper JA. Reactive oxygen species- and dimerization-induced activation of apoptosis signal-regulating kinase 1 in tumor necrosis factor-α signal transduction. J Biol Chem. 1998;273:17477–17482. doi: 10.1074/jbc.273.28.17477. [DOI] [PubMed] [Google Scholar]
- 35.Sanz L, Diaz-Meco MT, Nakano H, Moscat J. The atypical PKC-interacting protein p62 channels NF-κ B activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–1586. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ung TL, Cao C, Lu JM, Ozato K, Dever TE. Heterologous dimerization domains functionally substitute for the double-stranded RNA binding domains of the kinase PKR. EMBO J. 2001;20:3728–3737. doi: 10.1093/emboj/20.14.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao HF, Boyd J, Jolicoeur N, Shen SH. A coumermycin/novobiocin-regulated gene expression system. Hum Gene Ther. 2003;14:1619–1629. doi: 10.1089/104303403322542266. [DOI] [PubMed] [Google Scholar]
- 38.Burlison JA, Blagg BS. Synthesis and evaluation of coumermycin A1 analogues that inhibit the Hsp90 protein folding machinery. Org Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 39.Athavankar S, Peterson BR. Control of gene expression with small molecules: biotin-mediated acylation of targeted lysine residues in recombinant yeast. Chem Biol. 2003;10:1245–1253. doi: 10.1016/j.chembiol.2003.11.007. [DOI] [PubMed] [Google Scholar]
- 40.Weber W, Bacchus W, Gruber F, Hamberger M, Fussenegger M. A novel vector platform for vitamin H-inducible transgene expression in mammalian cells. J Biotechnol. 2007;131:150–158. doi: 10.1016/j.jbiotec.2007.06.008. [DOI] [PubMed] [Google Scholar]
- 41.Weber W, Bacchus W, Daoud-El Baba M, Fussenegger M. Vitamin H-regulated transgene expression in mammalian cells. Nucleic Acids Res. 2007;35:e116. doi: 10.1093/nar/gkm466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muddana SS, Peterson BR. Facile synthesis of CIDs: biotinylated estrone oximes efficiently heterodimerize estrogen receptor and streptavidin proteins in yeast three hybrid systems. Org Lett. 2004;6:1409–1412. doi: 10.1021/ol0497537. [DOI] [PubMed] [Google Scholar]
- 43.Hussey SL, Muddana SS, Peterson BR. Synthesis of a β-estradiol-biotin chimera that potently heterodimerizes estrogen receptor and streptavidin proteins in a yeast three-hybrid system. J Am Chem Soc. 2003;125:3692–3693. doi: 10.1021/ja0293305. [DOI] [PubMed] [Google Scholar]
- 44.Lin H, Abida WM, Sauer RT, Cornish VW. Dexamethasone-methotrexate: an efficient chemical inducer of protein dimerization in vivo. J Am Chem Soc. 2000;122:4247–4248. [Google Scholar]
- 45.Gallagher SS, Miller LW, Cornish VW. An orthogonal dexamethasone-trimethoprim yeast three-hybrid system. Anal Biochem. 2007;363:160–162. doi: 10.1016/j.ab.2006.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bronson JE, Mazur WW, Cornish VW. Transcription factor logic using chemical complementation. Mol Biosyst. 2008;4:56–58. doi: 10.1039/b713852k. [DOI] [PubMed] [Google Scholar]
- 47.Baker K, Bleczinski C, Lin H, Salazar-Jimenez G, Sengupta D, Krane S, Cornish VW. Chemical complementation: a reaction-independent genetic assay for enzyme catalysis. Proc Natl Acad Sci U S A. 2002;99:16537–16542. doi: 10.1073/pnas.262420099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Carter BT, Lin H, Goldberg SD, Althoff EA, Raushel J, Cornish VW. Investigation of the mechanism of resistance to third-generation cephalosporins by class C β-lactamases by using chemical complementation. Chem Bio Chem. 2005;6:2055–2067. doi: 10.1002/cbic.200500058. [DOI] [PubMed] [Google Scholar]
- 49.Lin H, Tao H, Cornish VW. Directed evolution of a glycosynthase via chemical complementation. J Am Chem Soc. 2004;126:15051–15059. doi: 10.1021/ja046238v. [DOI] [PubMed] [Google Scholar]
- 50.Tao H, Peralta-Yahya P, Lin H, Cornish VW. Optimized design and synthesis of chemical dimerizer substrates for detection of glycosynthase activity via chemical complementation. Bioorg Med Chem. 2006;14:6940–6953. doi: 10.1016/j.bmc.2006.06.034. [DOI] [PubMed] [Google Scholar]
- 51.Tao H, Peralta-Yahya P, Decatur J, Cornish VW. Characterization of a new glycosynthase cloned by using chemical complementation. Chem Bio Chem. 2008;9:681–684. doi: 10.1002/cbic.200700545. [DOI] [PubMed] [Google Scholar]
- 52.Gendreizig S, Kindermann M, Johnsson K. Induced protein dimerization in vivo through covalent labeling. J Am Chem Soc. 2003;125:14970–14971. doi: 10.1021/ja037883p. [DOI] [PubMed] [Google Scholar]
- 53.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 54.Keppler A, Kindermann M, Gendreizig S, Pick H, Vogel H, Johnsson K. Labeling of fusion proteins of O6-alkylguanine-DNA alkyltransferase with small molecules in vivo and in vitro. Methods. 2004;32:437–444. doi: 10.1016/j.ymeth.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 55.Lemercier G, Gendreizig S, Kindermann M, Johnsson K. Inducing and sensing protein--protein interactions in living cells by selective cross-linking. Angew Chem Int Ed. 2007;46:4281–4284. doi: 10.1002/anie.200700408. [DOI] [PubMed] [Google Scholar]
- 56.Gautier A, Juillerat A, Heinis C, Correa IR, Jr, Kindermann M, Beaufils F, Johnsson K. An engineered protein tag for multiprotein labeling in living cells. Chem Biol. 2008;15:128–136. doi: 10.1016/j.chembiol.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 57.Rink SM, Yarema KJ, Solomon MS, Paige LA, Tadayoni-Rebek BM, Essigmann JM, Croy RG. Synthesis and biological activity of DNA damaging agents that form decoy binding sites for the estrogen receptor. Proc Natl Acad Sci U S A. 1996;93:15063–15068. doi: 10.1073/pnas.93.26.15063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma U, Marquis JC, Nicole Dinaut A, Hillier SM, Fedeles B, Rye PT, Essigmann JM, Croy RG. Design, synthesis, and evaluation of estradiol-linked genotoxicants as anti-cancer agents. Bioorg Med Chem Lett. 2004;14:3829–3833. doi: 10.1016/j.bmcl.2004.04.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mitra K, Marquis JC, Hillier SM, Rye PT, Zayas B, Lee AS, Essigmann JM, Croy RG. A rationally designed genotoxin that selectively destroys estrogen receptor-positive breast cancer cells. J Am Chem Soc. 2002;124:1862–1863. doi: 10.1021/ja017344p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bielawski K, Bielawska A. Small-molecule based delivery systems for alkylating antineoplastic compounds. Chem Med Chem. 2008;3:536–542. doi: 10.1002/cmdc.200700229. [DOI] [PubMed] [Google Scholar]
- 61.Kuduk SD, Zheng FF, Sepp-Lorenzino L, Rosen N, Danishefsky SJ. Synthesis and evaluation of geldanamycin-estradiol hybrids. Bioorg Med Chem Lett. 1999;9:1233–1238. doi: 10.1016/s0960-894x(99)00185-7. [DOI] [PubMed] [Google Scholar]
- 62.Swamy N, Purohit A, Fernandez-Gacio A, Jones GB, Ray R. Nuclear estrogen receptor targeted photodynamic therapy: selective uptake and killing of MCF-7 breast cancer cells by a C17α-alkynylestradiol-porphyrin conjugate. J Cell Biochem. 2006;99:966–977. doi: 10.1002/jcb.20955. [DOI] [PubMed] [Google Scholar]
- 63.Kuduk SD, Harris TC, Zheng FF, Sepp-Lorenzino L, Ouerfelli Q, Rosen N, Danishefsky SJ. Synthesis and evaluation of geldanamycin-testosterone hybrids. Bioorg Med Chem Lett. 2000;10:1303–1306. doi: 10.1016/s0960-894x(00)00208-0. [DOI] [PubMed] [Google Scholar]
- 64.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98:8554–8559. doi: 10.1073/pnas.141230798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sin N, Meng L, Wang MQ, Wen JJ, Bornmann WG, Crews CM. The anti-angiogenic agent fumagillin covalently binds and inhibits the methionine aminopeptidase, MetAP-2. Proc Natl Acad Sci U S A. 1997;94:6099–6103. doi: 10.1073/pnas.94.12.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, Deshaies RJ. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Mol Cell Proteomics. 2003;2:1350–1358. doi: 10.1074/mcp.T300009-MCP200. [DOI] [PubMed] [Google Scholar]
- 67.Schneekloth JS, Jr, Fonseca FN, Koldobskiy M, Mandal A, Deshaies R, Sakamoto K, Crews CM. Chemical genetic control of protein levels: selective in vivo targeted degradation. J Am Chem Soc. 2004;126:3748–3754. doi: 10.1021/ja039025z. [DOI] [PubMed] [Google Scholar]
- 68.Zhang D, Baek SH, Ho A, Lee H, Jeong YS, Kim K. Targeted degradation of proteins by small molecules: a novel tool for functional proteomics. Comb Chem High Throughput Screen. 2004;7:689–697. doi: 10.2174/1386207043328364. [DOI] [PubMed] [Google Scholar]
- 69.Bargagna-Mohan P, Baek SH, Lee H, Kim K, Mohan R. Use of PROTACS as molecular probes of angiogenesis. Bioorg Med Chem Lett. 2005;15:2724–2727. doi: 10.1016/j.bmcl.2005.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee H, Puppala D, Choi E-Y, Swanson H, Kim K-B. Targeted degradation of the aryl hydrocarbon receptor by the PROTAC approach: a useful chemical genetic tool. Chem Bio Chem. 2007;8:2058–2062. doi: 10.1002/cbic.200700438. [DOI] [PubMed] [Google Scholar]
- 71.Puppala D, Lee H, Kim KB, Swanson HI. Development of an aryl hydrocarbon receptor antagonist using the proteolysis-targeting chimeric molecules approach: a potential tool for chemoprevention. Mol Pharmacol. 2008;73:1064–1071. doi: 10.1124/mol.107.040840. [DOI] [PubMed] [Google Scholar]
- 72.Schneekloth AR, Pucheault M, Tae HS, Crews CM. Targeted intracellular protein degradation induced by a small molecule: En route to chemical proteomics. Bioorg Med Chem Lett. 2008 doi: 10.1016/j.bmcl.2008.1007.1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bertozzi C, Bednarski M. C-Glycosyl compounds bind to receptors on the surface of Escherichia coli and can target proteins to the organism. Carbohydr Res. 1992;223:243–253. doi: 10.1016/0008-6215(92)80021-r. [DOI] [PubMed] [Google Scholar]
- 74.Shokat KM, Schultz PG. Redirecting the immune response: ligand-mediated immunogenicity. J Am Chem Soc. 1991;113:1861–1862. [Google Scholar]
- 75.Owen RM, Carlson CB, Xu J, Mowery P, Fasella E, Kiessling LL. Bifunctional ligands that target cells displaying the αvβ3 integrin. Chem Bio Chem. 2007;8:68–82. doi: 10.1002/cbic.200600339. [DOI] [PubMed] [Google Scholar]
- 76.Carlson CB, Mowery P, Owen RM, Dykhuizen EC, Kiessling LL. Selective tumor cell targeting using low-affinity, multivalet interactions. ACS Chem Biol. 2007;2:119–127. doi: 10.1021/cb6003788. [DOI] [PubMed] [Google Scholar]
- 77.O’Reilly MK, Collins BE, Han S, Liao L, Rillahan C, Kitov PI, Bundle DR, Paulson JC. Bifunctional CD22 ligands use multimeric immunoglobulins as protein scaffolds in assembly of immune complexes on B cells. J Am Chem Soc. 2008;130:7736–7745. doi: 10.1021/ja802008q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Matsuda M, Nishimura S, Nakajima F, Nishimura T. Heterobifunctional ligands: practical chemoenzymatic synthesis of a cell adhesive glycopeptide that interacts with both selectins and integrins. J Med Chem. 2001;44:715–724. doi: 10.1021/jm000295r. [DOI] [PubMed] [Google Scholar]
- 79.Portoghese PS. From models to molecules: opioid receptor dimers, bivalent ligands, and selective opioid receptor probes. J Med Chem. 2001;44:2259–2269. doi: 10.1021/jm010158+. [DOI] [PubMed] [Google Scholar]
- 80.Daniels DJ, Lenard NR, Etienne CL, Law PY, Roerig SC, Portoghese PS. Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc Natl Acad Sci U S A. 2005;102:19208–19213. doi: 10.1073/pnas.0506627102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mandel S, Amit T, Bar-Am O, Youdim MBH. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Progr Neurobiol. 2007;82:348–360. doi: 10.1016/j.pneurobio.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 82.Zheng H, Weiner LM, Bar-Am O, Epsztejn S, Cabantchik ZI, Warshawsky A, Youdim MBH, Fridkin M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg Med Chem. 2005;13:773–783. doi: 10.1016/j.bmc.2004.10.037. [DOI] [PubMed] [Google Scholar]
- 83.Youdim MB, Fridkin M, Zheng H. Bifunctional drug derivatives of MAO-B inhibitor rasagiline and iron chelator VK-28 as a more effective approach to treatment of brain ageing and ageing neurodegenerative diseases. Mech Ageing Dev. 2005;126:317–326. doi: 10.1016/j.mad.2004.08.023. [DOI] [PubMed] [Google Scholar]
- 84.Bennett BM, Reynolds JN, Prusky GT, Douglas RM, Sutherland RJ, Thatcher GRJ. Cognitive deficits in rats after forebrain cholinergic depletion are reversed by a novel NO mimetic nitrate ester. Neuropsychopharmacology. 2007;32:505–513. doi: 10.1038/sj.npp.1301054. [DOI] [PubMed] [Google Scholar]
- 85.Thatcher GR, Bennett BM, Reynolds JN. NO chimeras as therapeutic agents in Alzheimer’s disease. Curr Alzheimer Res. 2006;3:237–245. doi: 10.2174/156720506777632925. [DOI] [PubMed] [Google Scholar]
- 86.Arnaud CH. Drug hybrids enter the fray. Chem Eng News. 2007;85:46–48. [Google Scholar]
- 87.Walsh JJ, Coughlan D, Heneghan N, Gaynor C, Bell A. A novel artemisinin-quinine hybrid with potent antimalarial activity. Bioorg Med Chem Lett. 2007;17:3599–3602. doi: 10.1016/j.bmcl.2007.04.054. [DOI] [PubMed] [Google Scholar]
- 88.Dechy-Cabaret O, Benoit-Vical F, Robert A, Meunier B. Preparation and antimalarial activities of “trioxaquines”, new modular molecules with a trioxane skeleton linked to a 4-aminoquinoline. Chem Bio Chem. 2000;1:281–283. doi: 10.1002/1439-7633(20001117)1:4<281::AID-CBIC281>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 89.Burgess SJ, Selzer A, Kelly JX, Smilkstein MJ, Riscoe MK, Peyton DH. A chloroquine-like molecule designed to reverse resistance in Plasmodium falciparum. J Med Chem. 2006;49:5623–5625. doi: 10.1021/jm060399n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muhanji CI, Hunter R. Current developments in the synthesis and biological activity of HIV-1 double-drug inhibitors. Curr Med Chem. 2007;14:1207–1220. doi: 10.2174/092986707780597952. [DOI] [PubMed] [Google Scholar]
- 91.Mack ET, Perez-Castillejos R, Suo Z, Whitesides GM. Exact analysis of ligand-induced dimerization of monomeric receptors. Anal Chem. 2008;80:5550–5555. doi: 10.1021/ac800578w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV. HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS Chem Biol. 2008;3:373–382. doi: 10.1021/cb800025k. [DOI] [PubMed] [Google Scholar]
- 93.Chidley C, Mosiewicz K, Johnsson K. A designed protein for the specific and covalent heteroconjugation of biomolecules. Bioconjug Chem. 2008 doi: 10.1021/bc800268j. [DOI] [PubMed] [Google Scholar]