

Abstract
The Pd-catalyzed cross coupling of terminal allylic carbonates and allylB(pin) is described. The regioselectivity of this reaction is sensitive to the bite angle of the ligand with small bite angle ligands favoring the branched substitution product. This mode of regioselection is consistent with a reaction that operates by a 3,3′ reductive elimination reaction. In the presence of appropriate chiral ligands, this reaction is rendered enantioselective and applies to both aromatic and aliphatic allylic carbonates.
The Pd-catalyzed cross-coupling of organic electrophiles with organometallic reagents, especially organoboron derivatives, is broadly developed and has made a significant impact on the way complex molecules are prepared.1 One exception to this generalization is the cross-coupling of substituted allylmetal reagents. This reaction has received less attention but holds significant promise for asymmetric synthesis;2 recently reported Pd-catalyzed enantioselective couplings of crotyl boronates and aryl electrophiles are illustrative.3 Similarly, the cross coupling of allylmetal reagents with allyl electrophiles is attractive because it has the capacity to establish two new stereocenters with concomitant formation of an sp3-sp3 carbon-carbon bond.4 This catalytic reaction is also not well developed, perhaps because of the propensity for π-allyl intermediates to undergo β-hydride elimination5 (delivering 1,3-dienes) and perhaps because these reactions often lack regioselectivity or favor achiral products. Indeed, unlike the isoelectronic decarboxylative allylation of allyl enol carbonates6 and Tsuji-Trost reaction with preformed enolates,7 the allyl-allyl coupling does not benefit from an inherent regiocontrol bias. In this report, we present a paradigm for regiocontrol in allyl-allyl coupling reactions and use it to establish highly regio- and enantioselective catalytic variants.
Recent studies in our laboratory have focused on the transition metal catalyzed enantioselective addition of allylboronates to unsaturated carbonyls.8 These reactions proceed by way of oxygenated bis(allyl)metal species with a 3,3′-reductive elimination being responsible for construction of the C-C bond. The 3,3′-reductive elimination mechanism, as put forward by Echavarren, operates when coordination of a ligand to a bis(allyl)metal species causes both allyl groups to adopt the η1 bonding mode (B, Scheme 1) instead of the more common η3 mode (as in A).4j,9 We surmised that in the case of simple allyl-allyl cross coupling, bidentate ligands would be most effective at prompting the bis(η1-allyl) bonding mode and that, if the allyl groups are able to rapidly isomerize at some point on the reaction pathway, the least hindered bis(η1-allyl) (C) might predominate when substitution is present. In such a situation, the selectivity for formation of the chiral, branched 3,3′ elimination product relative to the linear 1,1′ elimination product should be sensitive to the bite angle of the ligand, and this feature provides a potential control element for the reaction.10
Scheme 1.

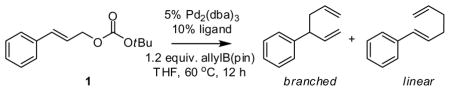
Inspired by observations of Miyaura, our initial experiments probed the reactivity of allylB(pin) and allylic carbonates.11 Efficient reactivity was observed with carbonate 1. As depicted in Table 1, in the presence of Pd(0) and bidentate ligands the allyl-allyl cross coupling reaction occurs and, with small bite angle ligands, complete conversion and high selectivity is observed. From the data in Table 1 it is clear that smaller bite angle ligands favor the branched product whereas larger bite angle ligands are non-selective. This is consistent with the reaction proceeding through an intermediate such as C (Scheme 1); the increased C1-C1′ separation that accompanies small bite angle ligands,12 may disfavor the 1,1′ elimination pathway relative to the 3,3′ path.13
Table 1.
Pd-Catalyzed Branch-Selective Cross-Coupling.
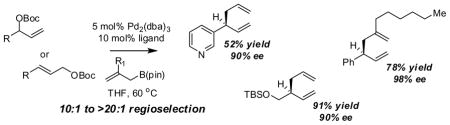
With an effective means to control the regioselectivity in allyl-allyl cross-coupling reactions, a survey was undertaken to gage the ability of small-bite-angle chiral bidentate ligands to control enantioselectivity in the process. 2,2-Bis(difurylphosphino)-6,6-dimethoxybiphenyl14 (A, Table 2) was found to deliver the allyl-allyl cross coupling product with high regio- and enantioselectivity. As depicted in Table 2, the reaction is effective with a number of allyl carbonates, generally delivering the branched substitution product with excellent regio- and enantioselectivity. As shown in entries 1 and 2, both tert-butyl and methyl carbonates can be used interchangeably in the reaction. Also of note, the corresponding internal racemic carbonate (entry 3) reacts with levels of selectivity that are comparable to those of primary alcohol-derived substrates (entries 1 and 2). This observation has mechanistic implications (vide infra) and is also of practical importance; substrates such as that depicted in entry 3 can be more accessible than substrates such as in entries 1 and 2. Entries 4–7 and 9 show that sterically encumbered substrates and those possessing halogens or heteroatoms are also suitable partners for the reaction. Remarkably, as the example in entry 9 indicates, unactivated alcohols are reactive leaving groups in the coupling reaction although this feature currently appears most effective with electron-rich substrates (additional data not shown). Currently, one limitation to the reaction is that an electron-deficient allyl electrophile was observed to react with diminished levels of selectivity (entry 8).
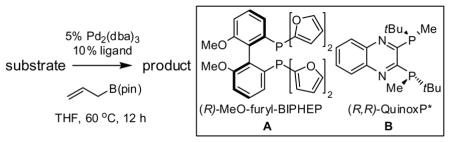
Table 2.
Catalytic Enantioselective Allyl-Allyl Cross-Coupling.a
 | ||||||
|---|---|---|---|---|---|---|
| entry | substrate | product | ligand | yield | regio. | ee |
| 1 | 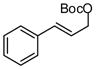 |
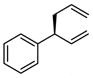 |
A | 72 | >20:1 | 91 |
| 2 | 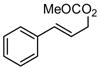 |
 |
A | 69 | >20:1 | 91 |
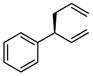
| 3 | 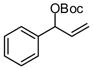 |
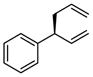 |
A | 75 | >20:1 | 91 |
| 4 | 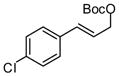 |
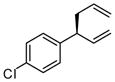 |
A | 59 | >20:1 | 90 |
| 5b | 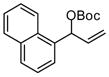 |
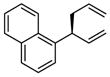 |
A | 87 | >20:1 | 90 |
| 6 | 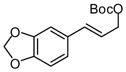 |
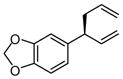 |
A | 83 | >20:1 | 91 |
| 7 | 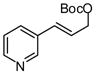 |
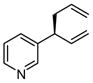 |
A | 52 | >20:1 | 90 |
| 8 | 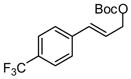 |
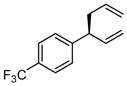 |
A | 60 | >20:1 | 74 |
| 9c | 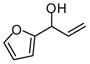 |
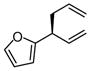 |
A | 53 | >20:1 | 87 |
| 10 | 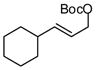 |
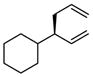 |
A B |
35 9 |
>20:1 3:1 |
83 94 |
| 11 | 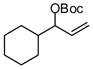 |
 |
B | 63 | 10:1 | 91 |
| 12 | 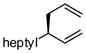 |
B | 80 | 11:1 | 84 | |
| 13d | B | 91 | >20:1 | 90 | ||
| 14 | B | 75 | >20:1 | 85 | ||
Unless otherwise noted, reactions employ 1.2 equivalents of allylB(pin).
Reaction with 1.5 equiv. of allylB(pin).
2.0 equiv. allylB(pin), 36h.
1.2 equivalents of Cs2CO3 added.
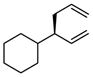
A notable observation with respect to synthesis utility is that aliphatic substrates also participate in the allyl-allyl coupling reaction: while the example in entry 10 occurred with moderate selectivity using (R)-MeO-furyl-BIPHEP, the selectivity could be improved substantially by employing (R,R)-QuinoxP*15 as ligand. The problematic low yield in entry 10 was easily ameliorated by employing the regioisomeric internal carbonate depicted in entry 11. It merits mention that linear alkyl substrates as well as those bearing allylic and homoallylic oxygen functionality are readily processed in the reaction (entry 12–14) and deliver the branched product efficiently and with good regio- and enantioselectivity.
The observation that isomeric allylic carbonates (i.e. entries 1–3 and entries 10–11) react with similar levels of enantioselectivity suggests that oxidative addition of the allyl carbonate to Pd(0) provides a common intermediate (such as D, Scheme 2). Subsequent reaction with allylB(pin) might occur by transmetalation from boron to Pd followed by inner sphere 3,3′ reductive elimination as alluded to in the introduction (see C, Scheme 2). Alternatively, the reaction might occur by backside outer sphere attack of the allylboronate on a η3 Pd-allyl (E, Scheme 2).16
Scheme 2.

To distinguish between the above-described mechanisms, isotopically-labeled allylB(pin) was employed as the substrate (Scheme 3) in the allyl-allyl coupling reaction. When this experiment was conducted in the presence of the chiral catalyst, the deuterium label was found at both allyl termini in the reaction product (recovered allylBpin is unscrambled). Considering that nucleophilic addition involving allylmetal reagents often occurs by an SE2′ reaction mechanism,17 it might be expected that the outer sphere pathway would proceed without isotope scrambling. Alternatively, SE2′ transmetalation8b,18 followed by equilibration of the bis(allyl)Pd complex may account for the reaction outcome. Along these lines, it should be noted that while four-coordinate bis(allyl)Pd complexes most often adopt the bis(η1) bonding mode (such as in C, Scheme 2), (η1-methallyl)(η3-methallyl)Pd(dcpe) is a known compound, the existence of which supports the accessibility of intermediates required for isotope scrambling.19
Scheme 3.

To further validate to proposed inner-sphere reductive elimination pathway, enantiomerically-enriched (S)-Z-10 (Scheme 4) was prepared and subjected to the asymmetric cross-coupling with (R)-MeO-furyl-BIPHEP as the ligand. This reaction provided (S)-E-11 as the sole product in 91% ee. Assuming that conversion of (S)-Z-10 to the derived π-allyl complex occurs by an anti displacement of the leaving group (to give F) as is known for related systems,20 it can be concluded that (S)-E-11 can only be produced by C-C bond formation syn to palladium after the metal has migrated to the opposite face of the cinnamyl-derived allyl fragment (G or related); this outcome is only consistent with an inner-sphere reductive elimination.
Scheme 4.

Substituted allyl boronates are readily prepared by Miyaura borylation of the corresponding allyl acetates.21 Using this strategy, both 2-methyl and 2-hexyl substituted allyl boronates were prepared and employed in the allyl-allyl cross coupling reaction. With (R)-MeO-furyl-BIPHEP as the ligand, both compounds underwent smooth cross-coupling with the cinnamyl alcohol-derived carbonate and provided products in excellent levels of selectivity (eq. 1 and 2, Scheme 5). To further probe the practical utility of the allyl-allyl cross-coupling reaction, the experiment in equation 3 was carried out. It was found that the catalyst loading could be lowered to 2.5 mol% palladium and the THF solvent typically employed in the cross-coupling could be replaced with ethyl acetate. Under these conditions, the allyl-allyl coupling could be executed on larger scale and still furnished the product in good enantioselectivity albeit with some erosion of efficiency.
Scheme 5.

In conclusion, a highly regio- and enantioselective allyl-allyl coupling reaction is described. The regiochemical outcome of the reaction and the described isotope labeling experiments are consistent with a pathway involving 3,3′-reductive elimination of bis(allyl)Pd complexes. An important feature of the reaction is that it provides enantiomerically-enriched vicinal π-π systems that are not accessible by the Cope rearrangement and this may find use in organic synthesis.
Supplementary Material
Acknowledgments
Support by the NIGMS (GM-64451) and the NSF (DBI-0619576, BC Mass. Spec. Center) is gratefully acknowledged. PZ is grateful for a LaMattina Fellowship. We thank Frontier Scientific for a generous donation of allylB(pin).
Footnotes
Supporting Information Available: Characterization and procedures. This information is available free of charge through the internet at http://pubs.acs.org.
References
- 1.de Meijere A, Diederich F, editors. Metal-Catalyzed Cross-Coupling Reactions. Wiley-VCH; New York: 2004. [Google Scholar]
- 2.For cross coupling of aryl electrophiles with prochiral allyl metal reagents, see: Sn: Echavarren AM, Stille JK. J Am Chem Soc. 1987;109:5478.Obora Y, Tsuji Y, Kobayashi M, Kawamura T. J Org Chem. 1995;60:4647.Si: Hatanaka Y, Ebina Y, Hiyama T. J Am Chem Soc. 1991;113:7075.Hatanaka Y, Goda K, Hiyama T. Tetrahedron Lett. 1994;35:1279.Hatanaka Y, Goda K, Hiyama T. Tetrahedron Lett. 1994;35:6511.Boron: Kalinin VN, Denisov FS, Bubnov YN. Mendeleev, Commun. 1996. p. 206.Yamamoto Y, Takada S, Miyaura N. Chem Lett. 2006;35:704.
- 3.(a) Yamamoto Y, Takada S, Miyaura N. Chem Lett. 2006;35:1368. [Google Scholar]; (b) Yamamoto Y, Takada S, Miyaura N, Iyama T, Tachikawa H. Organometallics. 2009;28:152. [Google Scholar]
- 4.Allylstannanes: Trost BM, Keinan E. Tetrahedron Lett. 1980;21:2595.Godschalx J, Stille JK. Tetrahedron Lett. 1980;21:2599.Keinan E, Peretz M. J Org Chem. 1983;48:5302.Trost BM, Pietrusiewicz KM. Tetrahedron Lett. 1985;26:4039.Goliaszewski A, Schwartz J. Tetrahedron. 1985;41:5779.Keinan E, Bosch E. J Org Chem. 1986;51:4006.Cuerva JM, Gómez-Bengoa E, Méndez M, Echavarren AM. J Org Chem. 1997;62:7540.van Heerden FR, Huyser JJ, Williams DBG, Holzapfel CW. Tetrahedron Lett. 1998;39:5281.Nakamura H, Bao M, Yamamoto Y. Angew Chem Int Ed. 2001;40:3208. doi: 10.1002/1521-3773(20010903)40:17<3208::AID-ANIE3208>3.0.CO;2-U.Méndez M, Cuerva JM, Gómez-Bengoa E, Cárdenas DJ, Echavarren AM. Chem Eur J. 2002;8:3620. doi: 10.1002/1521-3765(20020816)8:16<3620::AID-CHEM3620>3.0.CO;2-P.While this work was in progress, the cross-coupling of allyl boronates and allyl carbonates appeared: Flegeau EF, Schneider U, Kobayashi S. Chem Eur J. 2009;15:12247. doi: 10.1002/chem.200902221.
- 5.Keinan E, Kumar S, Dangur V, Vaya J. J Am Chem Soc. 1994;116:11151. [Google Scholar]
- 6.Shimizu I, Yamada T, Tsuji J. Tetrahedron Lett. 1980;21:3199.Tsuji J, Minami I, Shimizu I. Tetrahedron Lett. 1983;24:1793.Tsuji J, Yamada T, Minami I, Yuhara M, Nisar M, Shimizu I. J Org Chem. 1987;52:2988.Tsuji J, Minami I. Acc Chem Res. 1987;20:140.Asymmetric version was developed by Stoltz and Trost. Review: Mohr JT, Stoltz BM. Chem Asian J. 2007;2:1476. doi: 10.1002/asia.200700183.
- 7.Review: Braun M, Meier T. Angew Chem Int Ed. 2006;45:6952. doi: 10.1002/anie.200602169.
- 8.(a) Sieber JD, Liu S, Morken JP. J Am Chem Soc. 2007;129:2214. doi: 10.1021/ja067878w. [DOI] [PubMed] [Google Scholar]; (b) Sieber JD, Morken JP. J Am Chem Soc. 2008;130:4978. doi: 10.1021/ja710922h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Zhang P, Morken JP. J Am Chem Soc. 2009;131:12550. doi: 10.1021/ja9058537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cárdenas DJ, Echavarren AM. New J Chem. 2004;28:338.For a related experimentally observable η1-allyl-η1-carboxylate, see: Sherden NH, Behenna DC, Virgil SC, Stoltz BM. Angew Chem Int Ed. 2009;48:6840. doi: 10.1002/anie.200902575.
- 10.Reviews: Birkholz MN, Freixa Z, van Leeuwen PWNM. Chem Soc Rev. 2009;38:1099. doi: 10.1039/b806211k.van Leeuwen PWNM, Kamer PCJ, Reek JNH, Dierkes P. Chem Rev. 2000;100:2741. doi: 10.1021/cr9902704.
- 11.Ishiyama T, Ahiko T, Miyaura N. Tetrahedron Lett. Vol. 37. 1996. p. 6889.For related observations, see: Sebelius S, Wallner OA, Szabó KJ. Org Lett. 2003;5:3065. doi: 10.1021/ol035052i.Sebelius S, Olsson VJ, Szabó KJ. J Am Chem Soc. 2005;127:10478. doi: 10.1021/ja052885q.
- 12.Moloy has noted that the Cl-Cl distance diminishes with increasing bite angle in (diphosphine)PdCl2 complexes. See: Marcone JE, Moloy KG. J Am Chem Soc. 1998;120:8527.
- 13.In support of this hypothesis, the calculated transition states for 1,1′ and 3,3′ elimination from (H3P)2Pd(η1-allyl)2 exhibit P-Pd-P angles of 104.9 and 96.6, respectively. See supporting information for reference 4j.
- 14.Broger EA, Foricher J, Heiser B, Schmid R. 5274125. US Patent. 1993
- 15.Imamoto T, Sugita K, Yoshida K. J Am Chem Soc. 1995;127:11934. doi: 10.1021/ja053458f. [DOI] [PubMed] [Google Scholar]
- 16.For decarboxylative allylation, recent studies suggest that both outer-sphere attack of an unstabilized enolate on an η3 π-allyl and inner sphere reductive elimination pathway are plausible. See: Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343. doi: 10.1021/ja9053948.Keith JA, Behenna DC, Mohr JT, Ma S, Marinescu SC, Oxgaard J, Stoltz BM, Goddard WA., III J Am Chem Soc. 2007;129:11876. doi: 10.1021/ja070516j.
- 17.(a) Yamamoto Y, Yatagai H, Maruyama K. J Am Chem Soc. 1981;103:1969. [Google Scholar]; (b) Hayashi T, Konishi M, Kumada M. J Am Chem Soc. 1982;104:4963. [Google Scholar]; (c) Hayashi T, Kabeta K, Yamamoto T, Tamao K, Kumada M. Tetrahedron Lett. 1983;24:5661. [Google Scholar]; (d) Wickham G, Kitching W. J Org Chem. 1983;48:614. [Google Scholar]; (e) Buckle MJC, Fleming I, Gil S, Pang KLC. Org, Biomol Chem. 2004;2:749. doi: 10.1039/b313446f. and references cited therein. [DOI] [PubMed] [Google Scholar]
- 18.(a) Hayashi T, Konishi M, Kumada M. J Chem Soc Chem Commun. 1983:736. [Google Scholar]; (b) Naruta Y, Nishigaichi Y, Maruyama K. Tetrahedron Lett. 1989;45:1067. [Google Scholar]; (c) (e) Hiyama T, Matsuhashi H, Fujita A, Tanaka M, Hirabayashi K, Shimizu M, Mori A. Organometallics. 1996;15:5762. [Google Scholar]
- 19.Jolly PW. Angew Chem Int Ed Engl. 1985;24:283. [Google Scholar]
- 20.(a) Hayashi T, Hagihara T, Konishi M, Kumada M. J Am Chem Soc. 1983;105:7767. [Google Scholar]; (b) Trost BM, Verhoeven TR. J Am Chem Soc. 1980;102:4730. doi: 10.1021/ja00418a063. [DOI] [PubMed] [Google Scholar]
- 21.Ishiyama T, Ahiko T, Miyaura N. Tetrahedron Lett. 1996;38:6889. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.