

Summary
The PI3 kinase family of lipid kinases promotes cell growth and survival by generating the second messenger phosphatidylinositol-3,4,5-trisphosphate. To define targets critical for cancers driven by activation of PI3 kinase, we screened a panel of potent and structurally diverse drug-like molecules that target this enzyme family. Surprisingly, a single agent (PI-103) effected proliferative arrest in glioma cells, despite the ability of many compounds to block PI3 kinase signaling through its downstream effector, Akt. The unique cellular activity of PI-103 was traced directly to its ability to inhibit both PI3 kinase α and mTOR. PI-103 showed significant activity in xenografted tumors with no observable toxicity. These data demonstrate an emergent efficacy due to combinatorial inhibition of mTOR and PI3 kinase α in malignant glioma.
Introduction
PI3 kinases are lipid kinases that are activated by a wide range of receptor tyrosine kinases to generate phosphatidylinositol-3,4,5-trisphosphate (PIP3). PIP3 couples PI3 kinase to down- stream effectors such as Akt, a serine-threonine kinase that signals to suppress apoptosis, promote cell growth, and drive cell proliferation. PIP3 also indirectly activates the protein kinase mTOR, which is critical for cell growth and contains a PI3 kinase homology domain but itself has no lipid kinase activity. The lipid phosphatase PTEN antagonizes PI3 kinase signaling, as it shows frequent inactivation in a broad range of tumors (Cantley and Neel, 1999).
The eight mammalian PI3 kinases are divided into three classes according to their structure, regulation, and substrate specificity (Vivanco and Sawyers, 2002). Among these enzymes, the three class 1A PI3 kinases (p110α, p110β, and p110δ) have been identified as the most critical for cell growth and survival (Vivanco and Sawyers, 2002). Class 1A PI3 kinases are heterodimers of a catalytic p110 lipid kinase subunit and an adaptor p85 subunit that recruits PI3 kinase to tyrosine phosphorylated membrane docking sites (Vivanco and Sawyers, 2002). Gain-of-function mutations in the p110α gene (PIK3CA) are found in cancers of the colon, breast, and brain (Bachman et al., 2004; Broderick et al., 2004; Samuels et al., 2004). Nevertheless, the unique physiological roles of the class 1A isoforms remain poorly understood, and the biochemical basis for signaling specificity among these kinases is unknown.
It is increasingly clear that many of the most effective targeted cancer therapies owe their activity to unexpected synergy through inhibition of multiple targets. The requirement for inhibition of multiple targets likely reflects the complexity of signaling underlying malignant transformation and the ability of tumor cells to dynamically adapt to stress. For this reason, there is a growing consensus that inhibition of individual targets is unlikely to succeed as a therapeutic strategy in solid tumors. Indeed, even in the simplest hematopoietic malignancies such as chronic myelogenous leukemia, it has been argued that the efficacy of imatinib requires inhibition of targets other than Bcr-Abl, the predominant kinase driving this disease (Wong et al., 2004). Unfortunately, it remains challenging to predict which target combinations will be most effective in a specific cancer, and genetic inactivation of these targets rarely predicts the efficacy of the corresponding small molecule drugs (Knight and Shokat, 2005).
As the PI3 kinase family represents an important emerging class of drug targets, we undertook a pharmacological approach to define which PI3 kinase family members are critical for proliferation in malignant glioma. To this end, we have recently synthesized a series of isoform-selective inhibitors of the PI3 kinase family, defined the structural basis for their specificity, and systematically enumerated their biochemical targets relative to all proteins in the family (Knight et al., 2006). These agents, in conjunction with other recently described isoform-selective inhibitors of PI3 kinase (Jackson et al., 2005; Knight et al., 2004; Sadhu et al., 2003), collectively represent, to our knowledge, the first new tools available in a decade for analysis of this important signaling pathway. As this inhibitor panel includes representatives from the majority of chemotypes and target selectivities currently in preclinical development by the pharmaceutical industry, it previews the biological activities of the drugs that will ultimately enter clinical trials. Using this chemical array, we identify a small molecule inhibitor (PI-103) with unique activity against genetically diverse glioma cell lines. The activity of this compound was traced to its ability to selectively block p110α and mTOR at nanomolar concentrations. Combinatorial inhibition of p110α and mTOR was well tolerated in vivo and was highly effective against glioma xenografts. These data suggest that combinatorial inhibition of mTOR and p110α represents a safe and effective therapy in the treatment of cancers driven by aberrant signaling through PI3 kinase.
Results
Inhibitors of p110α or p110β block activation of the downstream effector Akt
To understand which PI3 kinase isoforms are critical for the proliferation of malignant glioma, we evaluated a panel of ten structurally diverse inhibitors that target these enzymes, recognizing distinct but overlapping target sets (Knight et al., 2006). For each agent, we screened a panel of six glioma cell lines varying in mutational status at PTEN or p53 (Table 1; Figure S5 in the Supplemental Data available with this article online), both of which are frequently inactivated in gliomas (Rao and James, 2004). This screen demonstrated that inhibitors that target p110α or p110β were effective in blocking phosphorylation of Akt (which signals downstream of PI3 kinase), inhibitors selective for p110δ were less active, and none of these compounds significantly affected signaling through Erk kinase (Figures 1A and 1B). The p110α inhibitors PI-103 and PIK-90, and the p110β inhibitor TGX-286, were the most potent of these compounds in blocking phosphorylation of Akt. Although the IC50 measurements of compounds against p110α generally correlated with efficacy against Akt in cells treated in cell culture (Figures 1A and 1B), this correlation was less precise with inhibitors of p110β, likely reflecting differences in bioavailability among these chemotypes.
Table 1.
Antiproliferative activity of isoform-selective p110 inhibitors
| Compound | Most potent isoforms targeted | Cell cycle distribution |
||
|---|---|---|---|---|
| G0G1 | S | G2M | ||
| DMSO | — | 52 | 31 | 17 |
| LY294002 | α, β, δ, γ | 59 | 19 | 22 |
| Rapamycin | mTOR | 48 | 36 | 16 |
| PIK-90 | α | 60 | 31 | 10 |
| PIK-112 | PI-103 inactive analog | 55 | 27 | 18 |
| PI-103 | α | 70 | 22 | 8 |
| PIK-64 | α | 53 | 32 | 15 |
| PIK-85 | α | 52 | 34 | 13 |
| PIK-93 | α | 50 | 36 | 14 |
| PIK-124 | α | 53 | 31 | 16 |
| TGX-115 | β, δ | 53 | 32 | 15 |
| PIK-73 | TGX-286 inactive analog | 53 | 31 | 16 |
| TGX-286 | β, δ | 53 | 31 | 16 |
| PIK-108 | β, δ | 53 | 30 | 15 |
| PIK-39 | δ | 53 | 32 | 15 |
LN229 glioma cells were treated with isoform-selective inhibitors (0.5 μM), rapamycin (0.5 nM), or LY294002 (10 μM) for 24 hr and examined by flow cytometry. Figure S5 shows results for additional glioma cell lines.
Figure 1. Activity and toxicity of isoform-selective inhibitors of p110 in comparison with LY294002.
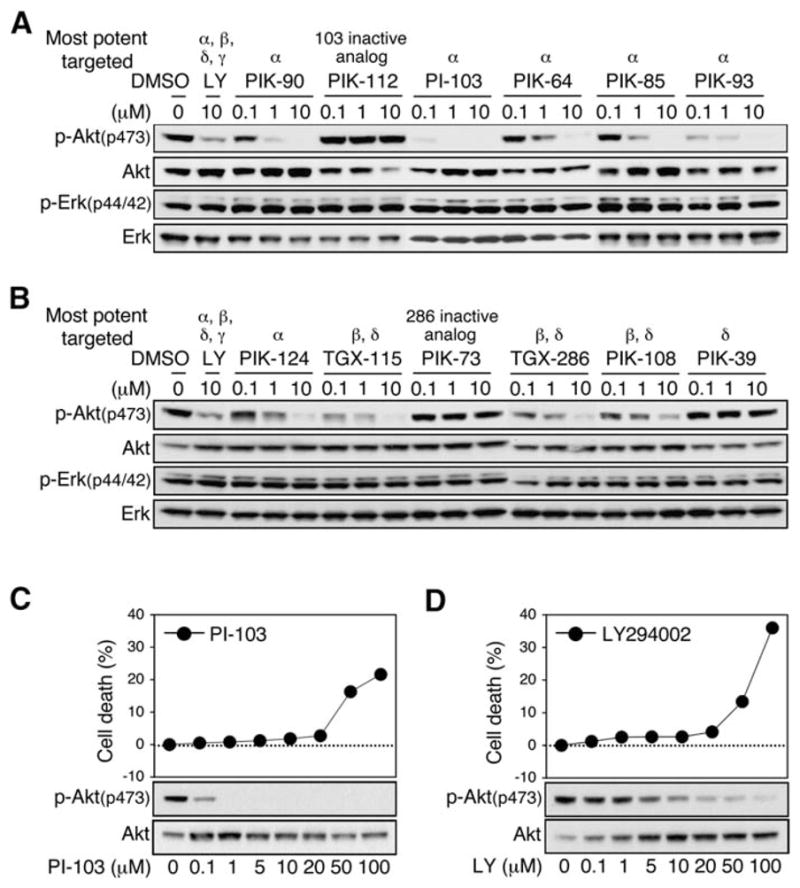
All inhibitors were screened against six glioma cell lines.
A and B: Representative results from U373MG cells treated with selective inhibitors of p110 catalytic subunits at doses indicated (6 hr). LY294002 served as positive control, and PIK-112 and PIK73 served as inactive controls. PI-103 showed the highest potency against p-Akt and was therefore further compared with LY294002. None of the compounds tested impacted activation of Erk kinase in a dose-dependent manner.
C and D: U87MG cells were treated with PI-103 (C) or LY294002 (D) for 24 hr. Graphs show measurements of cell death by LDH release (three 12-well plates per experimental point). Immunoblot shows levels of phosphorylated and total Akt proteins. PI-103 blocked p-Akt at dosages far below the toxic range, whereas toxic and efficacious dosages for LY294002 were overlapping.
Selective inhibition of PI3 kinase signaling shows reduced toxicity in comparison with broad-spectrum inhibitors
Signaling through PI3 kinase is critical to fundamental processes in diverse cell types (Wymann et al., 2003). In part for this reason, the broad-spectrum PI3-K inhibitors wortmannin and LY294002 are associated with significant cellular toxicity. A second potential toxicity relates to inhibition of the PI3 kinase related kinases ATR, ATM, and DNA-PK, which regulate cell cycle checkpoints and DNA repair. Two lines of investigation were therefore used to assess potential toxicities of the most active inhibitor identified in our screen (PI-103).
To address whether PI-103 could induce DNA repair checkpoints, leading to an increased mutation rate, we treated glioma cells with PI-103 and assessed the activation of p53, and of histone γH2AX, which is typically phosphorylated and localized to nuclear foci in response to DNA damage (Lowndes and Toh, 2005). Neither γH2AX nor p53 was activated by treatment of glioma cells with PI-103 (Figure S1), suggesting that this compound does not activate DNA damage to an extent measurable by these assays.
To test whether selective inhibition of p110 isoforms resulted in decreased cellular toxicity in comparison with broad-spectrum PI3 kinase inhibitors, we compared PI-103 with LY294002. U87MG cells were treated with increasing concentrations of PI-103 or LY294002 for 24 hr, and the activity of these inhibitors was measured by monitoring phosphorylation of Akt. PI-103 inhibited phosphorylation of Akt with an IC95 100-fold lower than that for LY294002 (Figures 1C and 1D). In parallel experiments, cytoxicity associated with PI-103 or LY294002 was quantitated by measuring LDH release as an indicator of membrane integrity (Korzeniewski and Callewaert, 1983) and compared with dosages required to block p-Akt. For LY294002, the IC95 for inhibiting phosphorylation of Akt overlapped with the cytotoxic dose. In contrast, the IC95 for PI-103 was more than 10-fold lower than the cytotoxic dose (Figures 1C and 1D). These data indicate that PI-103 can block PI3 kinase signaling at a dose much lower than that which causes general toxicity, suggest that a safe therapeutic index may be possible for inhibitors of this target class, and validate the approach of linking increased specificity to decreased toxicity within this class of compounds.
Inhibition of p110α, but not p110β, blocks proliferation of glioma cells in vitro
Since several chemotypes blocked activation of Akt, we expected that many compounds would inhibit proliferation in glioma cell lines. Surprisingly, only the p110α inhibitors PI-103, and to a lesser extent PIK-90, induced proliferative arrest in a panel of glioma cell lines assayed by flow cytometry (Table 1; Figure S2). Two lines of evidence suggest that p110α is the critical target recognized by these inhibitors. First, although these compounds inhibit p110δ and p110γ to varying extents in addition to p110α, Western blot analysis demonstrated no expression of these isoforms in any of the glioma cell lines tested (data not shown). It is therefore unlikely that inhibition of p110δ or p110γ contributed to the antiproliferative effect of PI-103 or PIK-90. Second, inhibitors of p110β and p110δ (e.g., TGX-286 and PIK-39) had no effect on proliferation of glioma cells (Table 1), despite the fact that inhibitors selective for p110β could block phosphorylation of Akt (Figure 1B). Collectively, these observations suggest that the p110α is functionally the most important isoform of PI3 kinase for cell growth and transformation in glioma.
Inhibition of p110α cooperates with inhibition of mTOR in malignant glioma
The increased activity of PI-103 (relative to other p110α inhibitors) in blocking proliferation of glioma cell lines led us to ask whether the activity of this compound might require inhibition of other proteins within the PI3 kinase family. In contrast to PIK-90 and PIK-85, PI-103 does show more potent inhibition of p110β (Knight et al., 2006). However, addition of a p110β inhibitor (TGX115) did not augment the proliferative arrest mediated by the p110α inhibitors PIK-90 or PIK-85 (Table 2). These data suggest that blockade of p110β does not contribute significantly to the antiproliferative effects of p110α inhibitors.
Table 2.
Combining inhibitors of PI3 kinase and of mTOR: Effects on proliferation
| Compound | Most potent isoforms targeted | Cell cycle distribution |
||
|---|---|---|---|---|
| G0G1 | S | G2M | ||
| DMSO | — | 60 | 28 | 12 |
| Rapamycin | mTOR | 77 | 15 | 8 |
| PIK-90 | α | 73 | 18 | 9 |
| PIK-90 + rapamycin | α + mTOR | 85 | 9 | 6 |
| PI-85 | α | 70 | 22 | 8 |
| PIK-85 + rapamycin | α + mTOR | 84 | 10 | 6 |
| TGX-115 | β | 62 | 26 | 12 |
| TGX-115 + rapamycin | β + mTOR | 74 | 19 | 7 |
| TGX-286 | β | 63 | 26 | 11 |
| TGX-286 + rapamycin | β + mTOR | 75 | 16 | 9 |
| PIK-90 + TGX-115 | α + β | 76 | 15 | 9 |
| PIK-85 + TGX-115 | α + β | 71 | 20 | 9 |
U87MG cells were treated for 24 hr with indicated isoform-selective inhibitors (0.5 μM), rapamycin (0.5 nM), or the combinations shown. Proliferation was measured by flow cytometry.
In addition to the eight bona fide PI3 kinases, the PI3 kinase family also includes six protein kinases and two PI4 kinases (Wymann et al., 2003). We therefore determined the extended selectivity profile of PI-103 and the other compounds in our panel by measuring biochemical IC50 values against the remaining proteins in the PI3-K family and 36 protein kinases (Knight et al., 2006). This analysis revealed that PI-103 uniquely and potently inhibits both complexes of mTOR (Hara et al., 2002; Kim et al., 2002; Loewith et al., 2002; Zheng et al., 1995): the rapamycin-sensitive mTORC1 (IC50 = 0.02 μM) and the rapamycin-insensitive mTORC2 (IC50 = 0.083 μM). PI-103 inhibited mTOR in vitro over 50-fold more potently than any other compound in our panel. While rapamycin is a natural product that only inhibits mTORC1, PI-103 represents, to our knowledge, the first synthetic compound that potently inhibits both mTOR complexes. The biochemical activity of this compound against mTOR in vitro was also observed in cells, where PI-103 (at low nanomolar doses; IC50 < 0.1 μM) was unique among these compounds in blocking the phosphorylation of p70 S6 kinase, ribosomal protein S6, and 4E-BP1, downstream markers of mTOR signaling (Figure 2A).
Figure 2. Activity of isoform-selective inhibitors of p110 against mTOR.
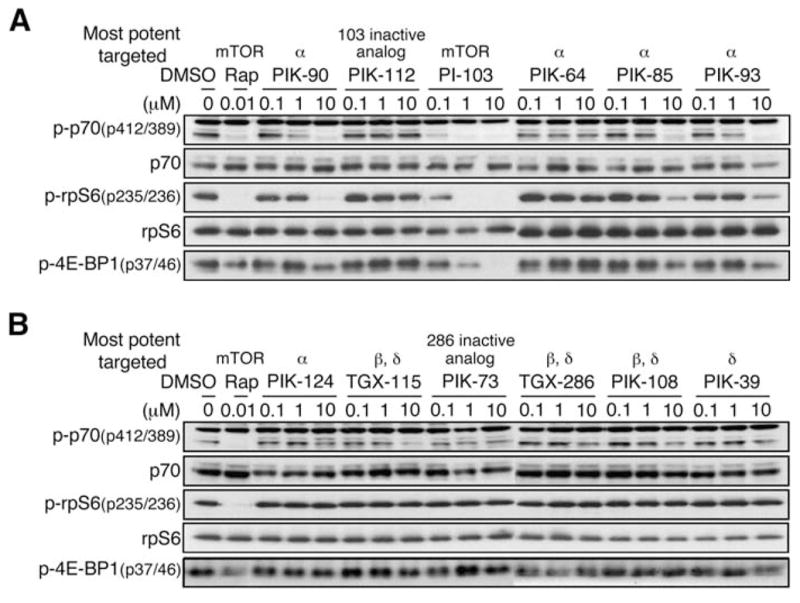
A and B: Inhibitors were screened as in Figure 1A. Proteins signaling downstream from mTOR include p70 S6 kinase, S6 ribosomal protein, and 4E-BP1. Each was used to read out mTOR activity. Representative results from U373MG cells are shown.
As mTOR plays a critical role in controlling cell growth (Aoki and Vogt, 2004; Inoki et al., 2005), these biochemical data raised the possibility that cooperative inhibition of p110α and mTOR may underlie the efficacy of PI-103 in glioma cells. To test this hypothesis, we asked whether it might be possible to phenocopy PI-103 by combining an mTOR inhibitor with an inhibitor of p110α. PIK-90 inhibits p110α with the same affinity as PI-103 (IC50 values 8.2 versus 11 nM) and has a similar selectivity profile against other members of the PI3 kinase family, with the exception of mTOR. As a single agent, PIK-90 induced a modest G0G1 arrest (Figure 3A) at a concentration (0.5 μM) sufficient to inhibit phosphorylation of Akt substantially (Figure 3B). Rapamycin, a widely used allosteric inhibitor of mTORC1 (Hara et al., 2002; Kim et al., 2002; Loewith et al., 2002; Zheng et al., 1995), induced a similar partial G0G1 arrest at a concentration (0.5 nM) sufficient to block phosphorylation of S6 (Figure 3B). Combining these agents induced a profound cell cycle arrest that was indistinguishable from PI-103 treatment alone (Figure 3A). These data argue strongly that the unique cellular activity of PI-103 is due to cooperative inhibition of two PI3 kinase family members, p110α and mTORC1.
Figure 3. Cooperative inhibition of p110α and mTOR arrests growth of human glioma cells.

U87MG cells were treated with the p110α inhibitor PIK-90, the p110β inhibitor TGX-286, the mTOR inhibitor rapamycin, or the dual p110α-mTOR inhibitor PI-103.
A: Rapamycin cooperated with the p110α inhibitor PIK-90 to block proliferation (p < 0.0001 by Student’s t test for combination therapy at either concentration, versus rapamycin or versus PIK-90 monotherapy) and was equivalent in efficacy to PI-103 monotherapy. Error between triplicate measurements was 0.8% or less for each value shown.
B: Rapamycin treatment increased phosphorylation of Akt. Inhibition of mTOR therefore blocked one output of p-Akt signaling, at the expense of activating multiple other outputs. Combining inhibitors of mTOR with inhibitors of p110α blocks all p-Akt-driven outputs, offering a mechanistic rationale for combining inhibitors of mTOR and of p110α in glioma.
C: In contrast to results combining inhibitors of p110α with rapamycin, combination therapy with the p110β inhibitor TGX-286 and rapamycin failed to potentiate the proliferation block observed with rapamycin alone and was less effective than PI-103 monotherapy. Error between triplicate measurements was 0.9% or less for each value shown.
D: Cells treated with TGX-286 in combination with rapamycin showed increased levels of p-Akt, compared with levels of p-Akt using TGX-286 monotherapy (compare lanes 5–6 with lane 4). These data, which stand in contrast to those observed using the more effective dual p110α mTOR inhibitor PI-103 (lane 7), suggest that activation of PI3 kinase by rapamycin is dependent on p110α rather than p110β and argue that combining inhibitors of mTOR and of p110α will show efficacy in glioma.
Interestingly, rapamycin monotherapy activated signaling through PI3 kinase, presumably due to inhibition of an mTOR-dependent retrograde signal (Figure 3B). This observation, which has also been made by others (Sun et al., 2005), suggests that rapamycin blocks one output of Akt signaling (mTOR), at the expense of activating other outputs. The ability of PI-103 to inhibit mTOR, and to block activation of p-Akt in response to mTOR inhibition, offers a mechanistic rationale for the efficacy of this combination.
siRNA against p110α blocked phosphorylation of Akt and proliferation, consistent with our findings using small molecule inhibitors (Figure S3). Surprisingly, we also observed a proliferative block using siRNA directed against p110β. As multiple small molecule inhibitors of p110β blocked phosphorylation of Akt but had no effect on proliferation either alone or in combination with inhibitors of mTOR (Table 2), we investigated whether this discrepancy might reflect differences between how small molecule inhibitors and siRNAs perturb the PI3 kinase pathway. Mouse knockout and siRNA studies within the PI3 kinase family highlight the fact that genetic disruption of the pathway can induce compensatory or unanticipated alterations in the activities of key regulatory proteins; for this reason, kinase-dead and kinase-absent mutations often induce different phenotypes (reviewed in Hennessy et al., 2005). The p85 regulatory subunits have been identified as an important component of this compensatory response. Deletion of catalytic p110 subunits has been shown to affect levels of p85, altering the ratio of p85 to p110, which controls negative feedback loops within this pathway (Brachmann et al., 2005). Furthermore, levels of free p85 also impact the activation of regulatory phosphatases (Gupta et al., 1999) and Jnk (Aguirre et al., 2000; Ueki et al., 2002), which participate in the complex regulation of PI3 kinase signaling. Three further experiments were therefore done to clarify the disparities between small molecule and siRNA experiments directed against p110β. Collectively, these additional experiments demonstrate that p110α is the major isoform driving proliferation in glioma cells.
First, siRNA against p110β led to decreased levels of p85 (Figure S3), consistent with data by others that manipulating the level of p110β also affects other key regulators of PI3 kinase signaling (Brachmann et al., 2005), thereby complicating the interpretation of this experiment.
Second, although the mTOR inhibitor rapamycin cooperated with small molecule inhibitors of p110α, similar cooperative efficacy was not observed using small molecule inhibitors of p110β (Figure 3C; Table 2).
Third, in contrast to our studies using small molecule inhibitors of p110α, small molecule inhibitors of p110β failed to block the activation of PI3 kinase observed in response to inhibition of mTOR (Figure 3D).
Taken together, these observations argue that p110α is critical to the proliferation of glioma cells in vitro, that activation of PI3 kinase in response to mTOR inhibitors is dependent on p110α to the exclusion of p110β, and that cooperative inhibition both of p110α and of mTOR underlies the efficacy of PI-103.
PI-103 shows activity in glioma cell lines irrespective of PTEN, p53, and EGFR status
A number of experiments demonstrate that PI-103 is active in a broad range of glioma cell lines and that this activity persists even in the background of mutations, such as PTEN, that activate signaling through PI3 kinase.
First, the dose response for PI-103 in blocking phosphorylation of Akt and in inducing proliferative arrest did not change as a function of PTEN status. We treated U87MG (PTEN mutant) and LN229 (PTEN wt) cells with increasing concentrations of PI-103 and analyzed proliferation, cell cycle distribution, and levels of phosphorylated Akt by immunoblot (Figure S4). The IC50 for inhibiting phosphorylation of Akt was between 0.05 and 0.1 μM (Figures S4A and S4B), consistent with the range of concentration required to inhibit cell proliferation (Figures S4C and S4D), and did not differ dramatically between cells wild-type or mutant at PTEN.
We next tested PI-103 against a larger panel of glioma cell lines that differed in mutational status of PTEN and p53. Cell lines were treated with PI-103 (0.5 μM) or LY294002 (10 μM) and analyzed at 24 hr by immunoblot and flow cytometry. Regardless of PTEN or p53 status, phosphorylation of Akt and S6 were both decreased in response to PI-103 (Figure S4E). PI-103 (0.5 μM) was more active than LY294002 (10 μM) in inducing arrest at G0G1 in all cell lines (Table S1), although one cell line (LN-Z308) was relatively resistant to both compounds. Cell cycle arrest induced by PI-103 was not accompanied by apoptosis in any lines tested, as indicated by measurement of the sub-G1 fraction TUNEL and cleaved caspase 3 assays (data not shown). These data demonstrate that PI-103 blocks proliferation in a broad range of glioma cell lines and argue that combined inhibition of p110α and mTOR represents a promising therapeutic strategy in glioma, irrespective of PTEN or p53 status.
Amplification and mutation of EGFR occurs commonly in glioma and is associated with high-grade glioblastoma multiforme tumors (Agosti et al., 1992; Ekstrand et al., 1992; Schlegel et al., 1994). Because amplification of EGFR leads to activation of PI3 kinase, we also tested whether the efficacy of PI-103 was impacted by activation of EGFR. Since amplification of EGFR is not maintained in cultured glioma cell lines, we transduced U8MG human glioma cells with vector, EGFR, or ΔEGFR—a tumor-derived allele that signals constitutively in the absence of ligand (Ekstrand et al., 1992; Wong et al., 1992). Each line was treated with LY294002 (10 μM) or PI-103 (0.5 μM) in the presence or absence of EGF (for EGFR-transduced cells). Again, PI-103 was more effective than LY294002 in blocking phosphorylation of Akt (Figure 4A) and did not affect phosphorylation of the receptors. PI-103 paralleled LY294002 in blocking phosphorylation of the mTOR target S6 protein in U87MG and U87MG:ΔEGFR cells. Neither agent impacted levels of phosphorylated S6 in U87MG:EGFR cells. Although wild-type EGFR differed subtly from ΔEGFR in response to PI-103, these data clearly demonstrated equivalent biochemical activity of PI-103 in glioma cell lines with either low or high levels of EGFR.
Figure 4. Inhibition of p110α and of mTOR represents a safe and effective strategy in EGFR-driven glioma in vitro and in vivo.

A: U87MG, U87MG:ΔEGFR, and U87MG:EGFR cells were treated with inhibitors shown. EGF (50 ng/ml) was added 30 min prior to harvest, where indicated. Phosphorylation status of EGFR/ΔEGFR was probed with anti-phosphotyrosine antibody 4G10. Although U87MG:ΔEGFR cells show increased basal activation of both Akt and mTOR signaling in comparison with U87MG cells, treatment with PI-103 inhibited activation of Akt and of mTOR pathways in both cell types.
B: U87MG:ΔEGFR cells were injected subcutaneously in BALB/c nu/nu animals, five mice in each group, and allowed to establish for 12 days. Animals were treated with daily PI-103 or DMSO vehicle for 18 days. Each point represents mean tumor volume ± SE obtained from five mice.
C: Representative tumors after 18 days of treatment.
D: Immunoblot of tumor lysates demonstrates blockade of p-Akt and p-rpS6 after treatment with PI-103 (T3, T4), compared with control (T1, T2).
E: Five animals treated as in B were injected with BrdU 2 hr prior to sacrifice. Tumors were analyzed by immunofluorescence with antibody to BrdU (red) or to cleaved caspase 3 (green) to measure proliferation and apoptosis, respectively. Quantification of five high-power microscopic fields from five animals (in each group) demonstrated a decrease in BrdU levels from 19.1% to 11.4% (p < 0.0001, Student’s t test). Levels of apoptosis were unchanged, 1.2% control versus 1.3% treated (p = 0.13, Student’s t test), suggesting that PI-103 acts in a tumor-static manner. Panel shows representative images. Scale bar, 100 μm.
Inhibition of p110α and of mTOR represents a safe and effective strategy in EGFR-driven glioma in vivo
We next asked whether PI-103 could be both safe and effective in treating established U87:ΔEGFR xenografts in vivo. U87: ΔEGFR cells were implanted subcutaneously into nude mice. When tumors reached 50–100 mm3, animals were randomized and treated with vehicle or PI-103. PI-103 showed significant activity in vivo (Figures 4B and 4C), reducing average tumor size by 4-fold after 18 days (p = 0.0002, Student’s t test). Mice treated with PI-103 had no obvious signs of toxicity premorbidly (based on body weight, food and water intake, activity, and general exam) or at necropsy. Treated tumors showed decreased levels of phosphorylated Akt and S6, consistent with blockade of p110α and mTOR in vivo (Figure 4D).
To assess whether treatment with PI-103 was cytotoxic or cytostatic for tumor cells, we treated xenografted animals with PI-103 or DMSO control and assessed tumors from five mice for proliferation-BrdU incorporation or apoptosis-caspase 3 activation. Levels of BrdU were decreased in treated tumors in comparison with control (Figure 4E). Although it has been suggested that inhibition of mTORC2 may regulate Akt-dependent survival processes (Guertin and Sabatini, 2005), preclinical treatment of glioma xenografts with PI-103 (which inhibits PI3 kinase α, mTORC1, and mTORC2) blocked proliferation without inducing apoptosis (Figure 4E). These experiments suggest that PI-103 treatment is cytostatic to glioma xenografts in vivo.
Discussion
The dramatic clinical efficacy of imatinib combined with the discovery of frequent mutations in protein and lipid kinases that promote oncogenesis has led to the hope that targeted inhibition of specific signaling proteins might revolutionize cancer therapy (Druker, 2004). It is increasingly clear, however, that many highly selective kinase inhibitors are displaying limited efficacy in clinical trials, despite the fact that their targets have been extensively validated to promote tumor progression (Krause and Van Etten, 2005). Conversely, several of the most effective kinase inhibitors in clinical development have been shown to act through complex inhibition of multiple targets (Fabian et al., 2005). Yet this multitargeted activity was not intentionally designed into these compounds based on predictive models of signal transduction.
These observations present a paradox for target-based cancer therapy, since this paradigm depends on target validation prior to compound optimization. We believe that the approach described in this study illustrates one path toward resolving this problem, based on the unbiased parallel evaluation of diverse chemotypes of potent and selective inhibitors of a single enzyme family. Through careful correlation of the unique biochemical selectivity profiles of these compounds with their efficacy in cellular and animal model systems, it is possible to pharmacologically validate specific combinations of targets that are likely to have efficacy in a particular cancer. In work reported here, this approach led us to identify a compound that uniquely inhibits two targets important for glioma proliferation (p110α and mTOR), despite the fact that many other chemotypes were able to potently block PI3-K signaling in these cells. Cooperativity between blockade of PI3 kinase and mTOR signaling also impacts autophagy in glioma cells (Takeuchi et al., 2005) and may contribute to the efficacy observed in our studies.
The malignant gliomas as a group are rapidly lethal tumors. In this context, the possibility of later malignancies, secondary to inhibition of a DNA-PK or other DNA repair checkpoint by PI-103, presents a relatively minor concern and is not supported by data included to address this point (Figure S1). As therapy for these tumors improves, however, it will be important to identify selective inhibitors of the PI3 kinase family of lipid kinases that do not inhibit DNA-PK, or other protein kinases critical for regulation of DNA repair checkpoints. In addition, the family of PI3 kinase inhibitors could impact such general processes as insulin signaling, diabetes, obesity, and aging. In regards to such potential toxicities, we believe the key advantage of model compounds such as PI-103 is to identify potential novel target combinations that display efficacy in cellular and animal systems. Actual drugs derived from these lead compounds must then be evaluated extensively for both efficacy and safety.
Inhibitors of mTOR are currently being tested clinically and have been somewhat disappointing as monotherapy in cancer. Although these compounds are cytostatic in many preclinical studies (Easton and Houghton, 2004; Takeuchi et al., 2005), mTOR inhibitors have failed to achieve similar cytostasis in clinical trials (Margolin et al., 2005), suggesting that the utility of these agents may be improved through combination therapy (Choo and Blenis, 2006). It has recently been proposed that selective mTOR inhibition may lead to PI3 kinase activation, thereby limiting the effectiveness of these agents (Hay, 2005). Our findings in glioma support this model, provide pharmacological evidence that this feedback can be overcome by dual inhibition of mTOR and PI3 kinase α, and suggest that inhibition of PI3 kinase and mTOR in combination should more effectively achieve cytostasis than inhibition of mTOR alone.
These data support a mechanistic rationale by which inhibition of a class 1 PI3 kinase can augment the efficacy of mTOR inhibitors, and in which mTOR inhibition reciprocally augments the efficacy of class 1 PI3 kinase inhibitors. Activation of mTOR is influenced by both growth factor- and nutrient-sensing pathways. While growth factor-sensing pathways signal through class 1 PI3 kinases, nutrients activate mTOR through a class 3 PI3 kinase (Byfield et al., 2005; Nobukuni et al., 2005). Blockade of the class 1 PI3 kinase α thus impacts only one of two inputs driving activation of mTOR. Combining a class 1 PI3 kinase inhibitor with an mTOR inhibitor effectively blocks two separate inputs promoting activation of mTOR, while also preventing the activation of class 1 PI3 kinases observed in response to monotherapy with mTOR inhibitors.
In contrast to results using inhibitors of PI3 kinase α, multiple small molecule inhibitors directed against PI3 kinase β showed little activity in blocking the proliferation of glioma cells in vitro. In addition, blockade of PI3 kinase β did not cooperate with blockade of mTOR, did not affect the activation of p-Akt observed in response to inhibiting mTOR, and did not augment the efficacy of PI3 kinase α inhibitors. These data support the importance of PI3 kinase α rather than β as the major isoform driving malignant progression in glioma, a conclusion further supported by siRNA directed against PI3 kinase α. Interestingly, small molecule and siRNA approaches were inconsistent regarding a role for PI3 kinase β in this tumor. While we provide a number of experiments suggesting that the efficacy of siRNA directed against PI3 kinase β may be attributable to decreased levels of p85, it is alternatively possible that the biology of perturbations in the PI3 kinase signaling pathway may differ depending on approach (kinase knockdown versus kinase inhibition). Our experiments are motivated by translational research and the discovery of small molecule drug targets for treating human cancers. In this context, the data presented support further development of PI3 kinase α-targeting small molecules that also inhibit mTOR and argue that targeting PI3 kinase β will not be efficacious in this disease setting.
We have reported here an unbiased screen of diverse small molecule PI3 kinase inhibitors against malignant glioma. This screen identified a single agent, PI-103, that induced proliferative arrest in vitro and inhibited growth of established human tumor xenografts in vivo. Significantly, this high degree of efficacy was achieved at a low dose of PI-103 with no compound-related side effects. We were able to trace the unique activity of PI-103 to combinatorial inhibition of mTOR and p110α, by showing that selective inhibitors of mTOR and p110α have limited efficacy in isolation but phenocopy PI-103 when combined. This suggests a potentially effective strategy for cancer therapy based on dual inhibition of these two PI3-K family members.
Experimental procedures
Cell lines, flow cytometry, and cell staining
Human glioma cell lines were obtained from the Brain Tumor Research Center at UCSF. Cells were harvested and fixed, treated with RNAase and propidium iodide, and filtered through 95 mM nylon mesh (Small Parts, Inc.). Ten thousand stained nuclei were analyzed in a FACS Calibur flow cytometer (Becton Dickinson). DNA histograms were modeled offline using Modifit-LT software (Verity Software, Topsham, ME). For crystal violet staining, 105 cells were seeded in 12-well plates in the presence or absence of PI-103.
Immunoblotting, siRNAs, and assessment of cell death
Membranes were blotted with antisera to p-Akt (Ser473), Akt, p-p70 S6 kinase (Thr389), p-70 S6 kinase, p-S6 ribosomal protein (Ser235/236), S6 ribosomal protein, p-4E-BP1 (Thr37/46), p-Erk (Thr202/204), p-p53 (Ser15), p53, p-γH2A.X (Ser139), H2A.X, and PI3 kinase p110α (all from Cell Signaling); Erk, PI3 kinase p110β, and PI3 kinase p85α (from Santa Cruz); 4G10 and β-tubulin (Upstate Biotechnology); or EGFR/ΔEGFR (Ab5, NeoMarkers). Immunoblotting and detection were as previously described (Fan et al., 2002). siRNA against p110β was from Santa Cruz Biotechnology. Control siRNA directed against luciferase GL3 (Elbashir et al., 2001) and p110α siRNA (Czauderna et al., 2003) were from Dharmacon. Cells were transfected with siRNA using Lipofectamine 2000 (Invitrogen). For assessment of cell death, U87MG cells were treated with PI-103 or LY294002 for 24 hr. Cell death was quantified by colorimetric determination of LDH activity using a cytotoxicity detection kit (Roche). Percentage of cell death (mean of three 12-well plates per experimental point) was calculated [(experimental value − low control)/(high control − low control) × 100], where the low-control cells were DMSO treated and high-control cells were Triton treated (1% Triton X-100, 30 min, 37°C).
Immunofluorescence
Immunofluorescence staining and visualization of BrdU, cleaved caspase-3, and p-γH2AX were performed as described previously (Fan et al., 2002). Sections were incubated overnight at 4°C with rat monoclonal anti-BrdU (1:200, Accurate Chemicals) followed by incubation at room temperature for 1 hr with both anti-rat Alexa Fluor 555 (1:200 Molecular Probes, OR) and anti-cleaved caspase-3 (Asp175 FITC conjugate, Cell Signaling). For p-γH2AX staining, cells were permeabilized and incubated with anti-p-γH2AX (Ser139, Cell Signaling), washed, and incubated for 1 hr in anti-rabbit Alexa Fluor 555 secondary antibody (Molecular Probes). Nuclei were labeled with To-Pro-3 iodide (Molecular Probes) for 30 min at RT. Sections and cells were mounted with Vectashield mounting media (Vector Laboratories) and analyzed with a Zeiss 510 LSM confocal microscope.
Xenografts
U87MG:ΔEGFR cells (106) were injected subcutaneously just caudal to the left forelimb in 6- to 12-week-old Balbc nu/nu mice (Harlan Sprague-Dawley). Mice with established tumors (50–100 mm3) were randomly allocated to IP treatment with 5 mg/kg PI-103 in 50% DMSO, or 50% DMSO alone (control). Tumor diameters were measured with calipers at 4 day intervals, and volumes were calculated from five mice per data point (mm3 = width2 × length/2). At necropsy, gross and microscopic analyses of all organs were performed to assess toxicity. The UCSF Institutional Animal Care and Use Committee approved all experiments.
SIGNIFICANCE.
PI3 kinases have elicited intense interest as drug targets because primary tumors show frequent inactivation of the tumor suppressor PTEN (a negative regulator of PI3 kinase) or activating mutations in PI3 kinase α. Yet it remains unclear which kinases in this family should be targeted in cancer. We report a strategy for pharmacological target validation, based on screening an array of chemically diverse inhibitors that potently target the PI3 kinase family. We demonstrate that dual blockade of a growth factor-dependent pathway (PI3 kinase) and a nutrient-sensing pathway (mTOR) provides an unusually effective therapeutic window. Moreover, the lead inhibitor that we report highlights how the conserved nature of the PI3 kinase catalytic domain can be exploited to discover multitargeted inhibitors with unexpected efficacy.
Supplementary Material
Acknowledgments
We are grateful to Jay Debnath, Morri Feldman, Frank McCormick, David Morgan, Greg Plowman, Pablo Rodriguez-Viciana, and Eli Zunder for useful discussions; Russ Pieper and Cynthia Cowdry for cell lines; Pablo Rodriguez-Viciana for viral constructs; Jack Chen for assistance with UV irradiation; and Gabriele Bergers and Christopher Hackett for critical review of this manuscript. Z.A.K. is an HHMI predoctoral fellow. This work was supported by the Brain Tumor Society, the Goldhirsh and Samuel G. Waxman Foundations, the Sandler Family, and the Brain Tumor SPORE Program.
Footnotes
The Supplemental Data include five supplemental figures and one supplemental table and can be found with this article online at http://www.cancercell.org/cgi/content/full/9/5/341/DC1/.
References
- Agosti RM, Leuthold M, Gullick WJ, Yasargil MG, Wiestler OD. Expression of the epidermal growth factor receptor in astrocytic tumours is specifically associated with glioblastoma multiforme. Virchows Arch A Pathol Anat Histopathol. 1992;420:321–325. doi: 10.1007/BF01600211. [DOI] [PubMed] [Google Scholar]
- Aguirre V, Uchida T, Yenush L, Davis R, White MF. The c-Jun NH(2)-terminal kinase promotes insulin resistance during association with insulin receptor substrate-1 and phosphorylation of Ser(307) J Biol Chem. 2000;275:9047–9054. doi: 10.1074/jbc.275.12.9047. [DOI] [PubMed] [Google Scholar]
- Aoki M, Vogt PK. Retroviral oncogenes and TOR. Curr Top Microbiol Immunol. 2004;279:321–338. doi: 10.1007/978-3-642-18930-2_19. [DOI] [PubMed] [Google Scholar]
- Bachman KE, Argani P, Samuels Y, Silliman N, Ptak J, Szabo S, Konishi H, Karakas B, Blair BG, Lin C, et al. The PIK3CA gene is mutated with high frequency in human breast cancers. Cancer Biol Ther. 2004;3:772–775. doi: 10.4161/cbt.3.8.994. [DOI] [PubMed] [Google Scholar]
- Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC. Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Mol Cell Biol. 2005;25:1596–1607. doi: 10.1128/MCB.25.5.1596-1607.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broderick DK, Di C, Parrett TJ, Samuels YR, Cummins JM, McLendon RE, Fults DW, Velculescu VE, Bigner DD, Yan H. Mutations of PIK3CA in anaplastic oligodendrogliomas, high-grade astrocytomas, and medulloblastomas. Cancer Res. 2004;64:5048–5050. doi: 10.1158/0008-5472.CAN-04-1170. [DOI] [PubMed] [Google Scholar]
- Byfield MP, Murray JT, Backer JM. hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem. 2005;280:33076–33082. doi: 10.1074/jbc.M507201200. [DOI] [PubMed] [Google Scholar]
- Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo AY, Blenis J. TORgeting oncogene addiction for cancer therapy. Cancer Cell. 2006;9:77–79. doi: 10.1016/j.ccr.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Czauderna F, Fechtner M, Aygun H, Arnold W, Klippel A, Giese K, Kaufmann J. Functional studies of the PI(3)-kinase signalling pathway employing synthetic and expressed siRNA. Nucleic Acids Res. 2003;31:670–682. doi: 10.1093/nar/gkg141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druker BJ. Imatinib as a paradigm of targeted therapies. Adv Cancer Res. 2004;91:1–30. doi: 10.1016/S0065-230X(04)91001-9. [DOI] [PubMed] [Google Scholar]
- Easton JB, Houghton PJ. Therapeutic potential of target of rapamycin inhibitors. Expert Opin Ther Targets. 2004;8:551–564. doi: 10.1517/14728222.8.6.551. [DOI] [PubMed] [Google Scholar]
- Ekstrand AJ, Sugawa N, James CD, Collins VP. Amplified and rearranged epidermal growth factor receptor genes in human glioblastomas reveal deletions of sequences encoding portions of the N- and/or C-terminal tails. Proc Natl Acad Sci USA. 1992;89:4309–4313. doi: 10.1073/pnas.89.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Fabian MA, Biggs WH, III, Treiber DK, Atteridge CE, Azimioara MD, Benedetti MG, Carter TA, Ciceri P, Edeen PT, Floyd M, et al. A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol. 2005;23:329–336. doi: 10.1038/nbt1068. [DOI] [PubMed] [Google Scholar]
- Fan QW, Zhang C, Shokat KM, Weiss WA. Chemical genetic blockade of transformation reveals dependence on aberrant oncogenic signaling. Curr Biol. 2002;12:1386–1394. doi: 10.1016/s0960-9822(02)01070-9. [DOI] [PubMed] [Google Scholar]
- Guertin DA, Sabatini DM. An expanding role for mTOR in cancer. Trends Mol Med. 2005;11:353–361. doi: 10.1016/j.molmed.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Gupta N, Scharenberg AM, Fruman DA, Cantley LC, Kinet JP, Long EO. The SH2 domain-containing inositol 5′-phosphatase (SHIP) recruits the p85 subunit of phosphoinositide 3-kinase during FcγRIIb1-mediated inhibition of B cell receptor signaling. J Biol Chem. 1999;274:7489–7494. doi: 10.1074/jbc.274.11.7489. [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110:177–189. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–183. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- Jackson SP, Schoenwaelder SM, Goncalves I, Nesbitt WS, Yap CL, Wright CE, Kenche V, Anderson KE, Dopheide SM, Yuan Y, et al. PI 3-kinase p110β: a new target for antithrombotic therapy. Nat Med. 2005;11:507–514. doi: 10.1038/nm1232. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110:163–175. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Shokat KM. Features of selective kinase inhibitors. Chem Biol. 2005;12:621–637. doi: 10.1016/j.chembiol.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Chiang GG, Alaimo PJ, Kenski DM, Ho CB, Coan K, Abraham RT, Shokat KM. Isoform-specific phosphoinositide 3-kinase inhibitors from an arylmorpholine scaffold. Bioorg Med Chem. 2004;12:4749–4759. doi: 10.1016/j.bmc.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Knight ZA, Gonzalez B, Feldman ME, Zunder ER, Goldenberg DD, Williams O, Loewith R, Stokoe D, Balla A, Toth B, et al. A pharmacological map of the PI3-K family defines a role for p110a in insulin signaling. Cell. 2006;125 doi: 10.1016/j.cell.2006.03.035. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzeniewski C, Callewaert DM. An enzyme-release assay for natural cytotoxicity. J Immunol Methods. 1983;64:313–320. doi: 10.1016/0022-1759(83)90438-6. [DOI] [PubMed] [Google Scholar]
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. doi: 10.1056/NEJMra044389. [DOI] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10:457–468. doi: 10.1016/s1097-2765(02)00636-6. [DOI] [PubMed] [Google Scholar]
- Lowndes NF, Toh GW. DNA repair: The importance of phosphorylating histone H2AX. Curr Biol. 2005;15:R99–R102. doi: 10.1016/j.cub.2005.01.029. [DOI] [PubMed] [Google Scholar]
- Margolin K, Longmate J, Baratta T, Synold T, Christensen S, Weber J, Gajewski T, Quirt I, Doroshow JH. CCI-779 in metastatic melanoma: a phase II trial of the California Cancer Consortium. Cancer. 2005;104:1045–1048. doi: 10.1002/cncr.21265. [DOI] [PubMed] [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, et al. Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA. 2005;102:14238–14243. doi: 10.1073/pnas.0506925102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RD, James CD. Altered molecular pathways in gliomas: an overview of clinically relevant issues. Semin Oncol. 2004;31:595–604. doi: 10.1053/j.seminoncol.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Sadhu C, Dick K, Tino WT, Staunton DE. Selective role of PI3Kδ in neutrophil inflammatory responses. Biochem Biophys Res Commun. 2003;308:764–769. doi: 10.1016/s0006-291x(03)01480-3. [DOI] [PubMed] [Google Scholar]
- Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- Schlegel J, Merdes A, Stumm G, Albert FK, Forsting M, Hynes N, Kiessling M. Amplification of the epidermal-growth-factor-receptor gene correlates with different growth behaviour in human glioblastoma. Int J Cancer. 1994;56:72–77. doi: 10.1002/ijc.2910560114. [DOI] [PubMed] [Google Scholar]
- Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, Khuri FR. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–7058. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Kondo Y, Fujiwara K, Kanzawa T, Aoki H, Mills GB, Kondo S. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336–3346. doi: 10.1158/0008-5472.CAN-04-3640. [DOI] [PubMed] [Google Scholar]
- Ueki K, Fruman DA, Brachmann SM, Tseng YH, Cantley LC, Kahn CR. Molecular balance between the regulatory and catalytic subunits of phosphoinositide 3-kinase regulates cell signaling and survival. Mol Cell Biol. 2002;22:965–977. doi: 10.1128/MCB.22.3.965-977.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Wong AJ, Ruppert JM, Bigner SH, Grzeschik CH, Humphrey PA, Bigner DS, Vogelstein B. Structural alterations of the epidermal growth factor receptor gene in human gliomas. Proc Natl Acad Sci USA. 1992;89:2965–2969. doi: 10.1073/pnas.89.7.2965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S, McLaughlin J, Cheng D, Zhang C, Shokat KM, Witte ON. Sole BCR-ABL inhibition is insufficient to eliminate all myeloproliferative disorder cell populations. Proc Natl Acad Sci USA. 2004;101:17456–17461. doi: 10.1073/pnas.0407061101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wymann MP, Zvelebil M, Laffargue M. Phosphoinositide 3-kinase signalling–which way to target? Trends Pharmacol Sci. 2003;24:366–376. doi: 10.1016/S0165-6147(03)00163-9. [DOI] [PubMed] [Google Scholar]
- Zheng XF, Florentino D, Chen J, Crabtree GR, Schreiber SL. TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin. Cell. 1995;82:121–130. doi: 10.1016/0092-8674(95)90058-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.