

Abstract
The union of functionalized pyrroloindolizines for the synthesis of heterodimeric products relevant to myrmicarin alkaloids is described. Design and synthesis of tricyclic substrates and new methods for their union enable the investigation of late-stage cyclopentannulation strategies. The rapid assembly of dimeric structures using unique modes of pyrroloindolizine reactivity presents a concise approach to the dimeric myrmicarins and relevant derivatives.
1. Introduction*
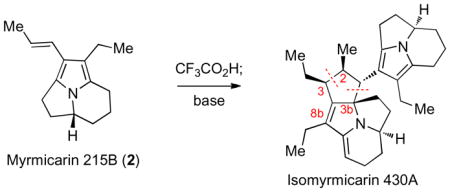
The myrmicarins are a family of exceedingly air-sensitive alkaloids isolated from the poison gland of the African ant species Myrmicaria opaciventris (Figure 1).i While these alkaloids are responsible for the paralytic activity of the secretion, their mechanism of action remains unknown.ii The relative stereochemistries of myrmicarin 430A (4)ib and myrmicarin 663 (6)ic have been elucidated through a series of spectroscopic studies by Schröder and coworkers, whereas the extreme fragility and limited quantities of myrmicarin 645 (5)ic have precluded stereochemical assignment. The fascinating structures and marked sensitivity of these alkaloids prompted us to develop methods for concise assembly of the complex members. We envisioned that a biogenetically inspired dimerization of activated pyrroloindolizine derivatives may provide rapid access to the highly sensitive complex structures.iii
Figure 1.

Representative myrmicarin alkaloids.
Pyrroloindolizine structures with a Lewis base substituent at C8 are predisposed to generate azafulvenium ions upon electrophilic activation (Scheme 1). This reactivity enables a range of activated derivatives to undergo addition by neutral vinyl pyrroloindolizines to afford corresponding dimeric or heterodimeric compounds.iv Use of C8-heteroatom substituted vinyl pyrroloindolizine nucleophiles provides hexacyclic derivatives bearing a C1 functional group relevant to late-stage cyclopentannulation chemistries. We envisioned that strategic design of the dimerization partners would enable us to access air and acid stable hexacyclic structures with enhanced capacity for functional group manipulation at C1. With this in mind, we embarked on the development of versatile methods for fragment assembly to allow rapid investigation of alternative radical,v metal-mediated,vi and electrophilic cyclization methods. Herein we describe the synthesis of functionalized pyrroloindolizine dimers and our studies on the unique reactivity of these structures for the synthesis of complex myrmicarins.
Scheme 1.

Retrosynthetic analysis of myrmicarin 430A (4).
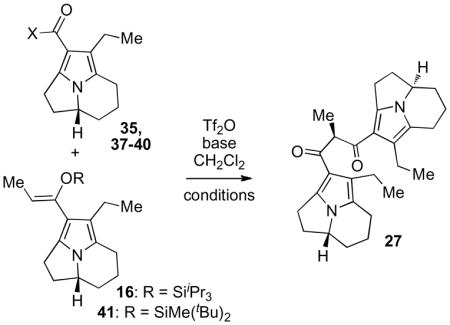
2. Heterodimerization of strategically functionalized pyrroloindolizines
To facilitate various prospective modes of cyclization we sought tricyclic subunits that would stabilize a radical or transition metal substituent at C8a (Scheme 2). We reasoned that conjugation to an alkene bearing an appropriate substituent at C7 would confer enhanced stability. Use of a corresponding functionalized tricyclic ketone as an electrophile (11, Scheme 1) would introduce this substituted alkene in a dimeric structure. With these considerations, we identified readily available tricycle 15vii (Scheme 3) as an appropriate heterodimerization substrate to introduce the desired alkene and vinyl bromide substituent as a versatile handle for modification.
Scheme 2.

Possible modes of cyclization of heterodimer 14.
Scheme 3.

Synthesis of hexacycle 18 and dehydration product 21. Conditions: a) Tf2O, 2,6-di-tert-butyl-4-methylpyridine, CH2Cl2, −78 °C, 2 h, 78%. b) DIBAL-H, CH2Cl2, −78 °C, 2 h, 100%. c) AcOH, C6D6, 23 °C, 1.8 h, 68%.
To effect heterodimerization employing 15 as an electrophile we focused on application of our previously reported trifluoromethanesulfonic anhydride-mediated activation of 15 and π-nucleophilic addition of silyl enol ether 16.iiia At the outset, we noted that appendage of the electron-withdrawing alkene to the pyrrole π system may reduce the capacity of 15 to undergo electrophilic activation, potentially diminishing the efficiency of the reaction. Gratifyingly, portion wise addition of trifluoromethanesulfonic anhydride to a dichloromethane solution of ketone 15 and silyl enol ether 16 (1:1) in the presence of 2,6-di-tert-butyl-4-methylpyridine smoothly provided heterodimer 17. As noted with previous substrates, completely regioselective elimination of trifluoromethanesulfonic acid proceeded via deprotonation at the less hindered site to provide the C3-C9 alkene. Neither the yield nor the rate of this reaction was significantly influenced by the presence of the bromoalkene in 15, underscoring the versatility of this protocol with regard to the structure of the ketone electrophile.iiia To investigate formation of the C1-C8b bond we targeted derivatives that would provide a pseudobenzylic C1 radical upon activation.v Examination of number of hydride reducing agents revealed that efficient reduction of the vinylogous amide carbonyl at C1 in the presence of the C7 bromoalkene in 17 was achieved using diisobutylaluminium hydride in dichloromethane at −78 °C (Scheme 3). As noted in structures lacking the C7–C8 bromoalkene, 18 exhibited marked sensitivity to conditions involving activation of the C1 alcohol. Treatment of 18 with Lewis or Brønsted acids in order to promote substitution of the C1 alcohol resulted in facile elimination to yield the exceedingly air-sensitive diene 21. Protocols to introduce C1 halide or selenide derivatives aimed at avoiding electrophilic ionization of the C1 alcohol resulted in decomposition of 18.
Interestingly, no cyclopentannulation byproducts arising from intramolecular alkylation at C3b were observed in these studies. By contrast, corresponding structures lacking a C7–C8 bromoalkene undergo rapid alkylation at C3b without visible accumulation of hexacyclic alkene intermediates.viii Pursuing this observation, we found that treatment of a benzene solution of 21 containing trace amounts of water with one equivalent of trichloroacetic acid effected conversion to heptacycle 24 (Scheme 4). Full structural characterization of this air-sensitive compound using a combination of two-dimensional NMR techniques revealed that the vinyl bromide 21 had undergone conversion to the corresponding C7 ketone 24. The absence of heptacyclic products possessing the C7-C8 bromoalkene and failure of 18 to cyclize upon activation of the C1 alcohol with acetic acid suggest that a stronger acid was needed for the hydrolysis of the bromoalkene to enable C3b alkylation. Importantly, the suppression of this alkylation by the presence of the C7–C8 bromoalkene demonstrated that strategic structural modification of this pyrroloindolizine subunit could be used to prevent undesired alkylation. While these studies demonstrated the applicability of our trifluoromethanesulfonic anhydride activation protocol to structurally modified pyrroloindolizine electrophiles, manipulation of the resulting heterodimers remained challenging. The sensitivity of hexacyclic compounds bearing ionizable C1 substituents motivated us to consider hexacyclic structures that would show enhanced stability and propensity for functionalization to alternative cyclization substrates.
Scheme 4.

Acid-promoted hydrolysis and cyclization of 21. Conditions: a) Cl3CCO2H, C6H6, 23 °C, 1.8 h, 48%.
3. Preparation and functionalization of stable heterodimers
To address the sensitivity of these heterodimers we considered symmetrical diketone 27 as a versatile hexacyclic structure (Scheme 5). We anticipated that conjugation of an electron withdrawing carbonyl substituent to each of the pyrrole subunits would enhance the air stability of 27 and suppress undesired C3b alkylation. Importantly, preparation of β-substituted enone derivatives would be facilitated by enhanced acidity of the C2 methine in this structure, providing versatile substrates for radical, metal-mediated, and alternative electrophilic cyclization chemistries.ix Finally, the symmetry of this structure provided a powerful simplifying element, and enabled us to consider stereoselective introduction of the C3 ethyl substituent after heptacycle formation.
Scheme 5.

Retrosynthesis of myrmicarin 430A (4) to heterodimer 27.
We envisioned that decarbonylation of readily available ketone 28iiib and introduction of an appropriate acyl substituent via Friedel-Crafts reaction would provide the requisite truncated tricycle for preparation of 27 (Scheme 6). Accordingly, removal of the propanoyl group from 28 could be effected in high yield by heating a 1,2-dichloroethane solution of ketone 28 to 65 °C in the presence of trifluoromethanesulfonic acid and ethylene glycol. Treatment of pyrrole 29 with trichloroacetyl chloride in 1,2-dichloroethane at 65 °C in the absence of Lewis acid provided trichloroacetyl ketone 30 in 65% yield.x Methanolysis of 30 proceeded efficiently to provide methyl ester 35. Alternatively, hydrolysis of 30 and derivatization of acid 36 enabled preparation of a range of tricyclic acid, ester, and amides as electrophiles for heterodimerization with ketone 28.
Scheme 6.

Synthesis of acid, ester, and amide derivatives. Conditions: a) TfOH, ethylene glycol, 1,2-dichloroethane, 65 °C, 1 h, 100%. b) Cl3CCOCl, 1,2-dichloroethane, 65 °C, 2 h, 74%.
Having established the efficiency of our trifluoromethanesulfonic anhydride mediated heterodimerization with ketone-substituted pyrroloindolizine electrophiles, we anticipated that this method would be applicable to the corresponding ester electrophiles. Unexpectedly, heterodimerization between methyl ester 35 and silyl enol ether 16 under previously optimized conditions provided diketone 27 in low yield. Raising the reaction temperature (Table 1, entries 1 and 2) decreased the yield of 27. Use of an alternative base additive (Table 1, entry 3) resulted in decomposition of the silyl enol ether nucleophile. Modification of the silyl group of the nucleophile had little effect on the efficiency of the reaction (Table 1, entries 1 and 4). Use of electrophilic ester derivatives 37 (X = OC6F5), 38 (X = OCH2CF3), and 39 (X = OPh) and more Lewis basic morpholine amide 40 provided none of the desired product (Table 1, entries 5–8). Failure of these substrates to undergo heterodimerization may be a result of the reduced electrophilicity of the activated esters and amides relative to the corresponding ketone derivatives, which prevents addition of the silyl enol ether. The ability to access a range of activated acid and ester derivatives from pyrrolyl carboxylic acid 36 (Scheme 6) motivated us to investigate their direct reaction with a metalloenolate of ketone 28. Surprisingly, the only products of attempted addition of the lithium enolate of 28 to the acid chloride 31, acid fluoride 32, or anhydride 33 were recovered 28 and tricyclic acid 36. Efforts to add the potassium or lithium enolates of 28 to the activated ester derivatives 37, 38, or 39 in the presence of various solvents and additives at different reaction temperatures failed to provide the desired heterodimer. The conversion of hydrolytically robust ester derivatives to the corresponding acid suggested that the metalloenolates of 28 may be undergoing O-acylation in preference to C-acylation and hydrolysis upon aqueous work up. In line with this hypothesis, we envisioned that the phosphate ester 34 (Scheme 6)xi may provide strong coordination to the lithium cation of the corresponding enolate of 28 and promote a closed transition state to favor C-acylation. Gratifyingly, treatment of a toluene solution of ketone 28 with lithium bis(trimethylsilyl)amide at −78 °C followed by addition of phosphate ester 34 and slow warming to −40 °C furnished diketone 27 in 70% yield. No product was observed under identical conditions when tetrahydrofuran, a coordinating solvent, was employed as solvent. As anticipated, hexacyclic diketone 27 demonstrated greatly enhanced stability relative to previous hexacyclic dimeric structures and could be stored for extended periods as a white solid. Interestingly, 1H and 13C spectra of diketone 27 in benzene-d6 show two distinct sets of signals, which may reflect restricted rotation in the C1–C2 linkage and a preference for each pyrrole unit to maintain conjugation with its carbonyl substituent.
Table 1.
Trifluoromethanesulfonic anhydride-promoted fragment coupling
 | ||||||
|---|---|---|---|---|---|---|
| entry | X (35, 37–40) | nucleophile | base | temp (°C) | time (h) | yield (%) |
| 1 | OMe | 16 | DTBMP | −78→0 | 7.5 | 30 |
| 2 | OMe | 16 | DTBMP | −30→40 | 1.5 | 18 |
| 3 | OMe | 16 | 2-ClPyr | −78 | 0.5 | 0a |
| 4 | OMe | 41 | DTBMP | −78→0 | 4 | 20 |
| 5 | OC6F5 | 41 | DTBMP | −78→0 | 3 | 0 |
| 6 | OCH2CF3 | 16 | DTBMP | −78→23 | 3 | 0 |
| 7 | OPh | 41 | DTBMP | −78→0 | 5 | 0 |
| 8 | N(CH2CH2)2O | 41 | DTBMP | −78→0 | 2 | 0 |
Immediate decomposition of 16. DTMBMP = 2,6-di-tert-butyl-4-methylpyridine; 2-ClPyr = 2-chloropyridine.
An efficient synthesis of diketone 27 enabled us to explore preparation of β-functionalized enone substrates for alternative cyclization strategies. We anticipated that deprotonation of the C2 methine and trapping of the resulting enolate could provide vinyl trifluoromethanesulfonate, p-toluenesulfonate, or phosphate derivatives.vi Importantly, these conditions avoided electrophilic activation at C1 and provided useful substrates for a range of cyclization protocols. Quantitative formation of the symmetrical enol 42xii upon treatment of a tetrahydrofuran solution of diketone 27 with potassium bis(trimethylsilyl)amide followed by water quench verified the propensity of 27 to undergo enolization (Scheme 8). Attempts to trap the corresponding potassium or lithium enolate as a vinyl trifluoromethanesulfonate or p-toluenesulfonate, or its conversion to a vinyl selenide were unsuccessful. However, sequential treatment of diketone 27 in tetrahydrofuran with lithium bis(trimethylsilyl)amide and diethyl cyanophosphonate at −78 °C efficiently provided the vinyl phosphate 43. Interestingly, heating a dimethylformamide solution of phosphate 43 saturated with lithium chloride at 75 °C for 4 hours provided the vinyl chloride 44 as an alternative cyclization substrate.xiii
Scheme 8.

Synthesis of enol 42 and β-substituted enone derivatives. Conditions: a) KHMDS, THF, −78→0 °C, 15 min; H2O, 100%. b) LHMDS, (EtO)2POCN, THF, −78 °C, 1.2 h, 67%. c) LiCl (sat.), DMF, 75 °C, 4 h, 43%.
The ability to prepare 44 via substitution of phosphate from 43 suggested that we may be able to improve the yield of 44 and apply this method to the synthesis of more reactive cyclization substrates. However, attempts to prepare 44 and the corresponding vinyl bromide derivative by activation of the C1 carbonyl and addition of halide typically resulted in hydrolysis to diketone 27 with poor mass recovery. Close monitoring of the reaction mixture upon treatment of 43 with triphenylphosphine dichloride in acetonitrile at 0 °C revealed that diketone 27 was produced concomitantly with another highly fragile compound, which likewise underwent conversion to 27. Careful purification of this acid, base, and air-sensitive compound and full structural characterization using two-dimensional NMR analysis revealed it to be the heptacyclic alcohol 46 (Scheme 9). Formation of 46 is consistent with enone activation followed by intramolecular pyrrole alkylation at C3b. Interestingly, hydrative trapping occurred exclusively at C3a, potentially as a consequence of strain relief upon sp2 to sp3 rehybridzation at the junction between the two five-membered rings.
Scheme 9.

Acid-promoted cyclization of 43. Conditions: a) Ph3PCl2, CH3CN, 0 °C, 2 h, 36%.
Successful preparation of the air stable diketone 27 and the corresponding β-substituted enone derivatives significantly expanded our knowledge regarding these dimeric pyrroloindolizine structures. These observations, combined with our expanded arsenal of methods to prepare heterodimeric structures, inspired strategies based on alternative electrophilic cyclization reactions.
4. Design and synthesis of heterodimers for alternative electrophilic cyclization reactions
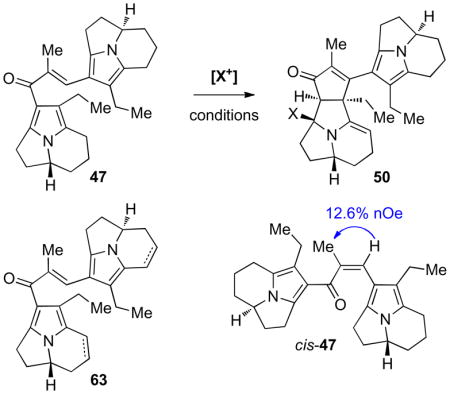
The propensity of the pyrroloindolizine subunit to undergo electrophilic trapping at C3b in dimeric structures suggested that we may be able to circumvent undesired alkylation by engaging this pyrrole nucleus in a reversible interaction with an alternative electrophile. Careful design of the dimeric structure may enable cyclization pathways in activated pyrrole substrates where C1-C3b bond formation could not occur. Specifically, we envisioned that reversible protonation, halogenation, or coordination of a metal salt at C3b would generate a dienone substructure such as 48, which may be susceptible to a Nazarov cyclization to give heptacycle 50 (Scheme 10).xiv
Scheme 10.

Potential activation and Nazarov cyclization of 47.
To examine the acid-promoted reactivity of a tricyclic substrate related to our proposed Nazarov cyclization we used in situ 1H NMR monitoring to investigate protonation and deuterium incorporation in ketone 28 upon treatment with excess trifluoroacetic acid-d1 (TFA-d1).xv 1H NMR analysis of the reaction mixture showed that addition of TFA-d1 to a solution of 28 in benzene-d6 (50% v/v) produced a 9:2 mixture of two protonated compounds. Heating this solution to 70 °C for 24 hours resulted in deuterium incorporation at C9, but also at C7 and C11 (Scheme 11). Deuterium incorporation at C9 is consistent with tautomerization of the C8 carbonyl to the transient C8–C9 enol 51 and reversible protonation at C9 by TFA-d1. Additionally, H/D exchange at C7 and C11 suggests an equilibrium involving protonation of the pyrrole ring. Protonation at C7a followed by deprotonation at C11 may generate the vinyl enamine 52, whereupon protonation by TFA-d1 would result in deuterium incorporation at C11. Protonation at C2a and/or C1 would result in D incorporation at C7 via 53 and/or 54. The inability to detect the proposed enamine intermediates by 1H NMR indicates that they are present in exceedingly low concentrations relative to related ring protonated pyrrolinium derivatives.
Scheme 11.

Deuterium incorporation in 28 in the presence of excess TFA-d1.
With evidence for reversible ring protonation of ketone 28 we aimed to exploit this reactivity to affect Nazarov cyclization in a dimeric structure. We envisioned that an aldol addition reaction between ketone 28 and tricyclic aldehyde 58xvi followed by elimination of the C1 alcohol could provide the enone substrate 47 (Scheme 10). Surprisingly, attempts to add lithium enolate 55 to aldehyde 58 failed to provide β-hydroxy ketone 59, returning both starting materials (Equation 1). Likewise, treatment of a mixture of 28 and 58 with titanium tetrachloride and N,N-diisopropylethylamine to effect in situ formation of the titanium enolate 56 and addition to 58 yielded no dimeric products. Use of Mukaiyama conditions for addition of triethylsilyl enol ether 57 to aldehyde 58 upon activation by titanium tetrachloride was also unsuccessful.
Equation 1.
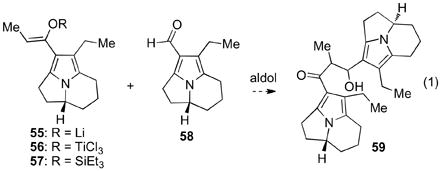
Attempted aldol addition reaction for the synthesis of ketone 59.
The low electrophilicity of aldehyde 58 and the potential for the dimeric β-alkoxy ketone 59 to undergo retro-aldol reaction suggested that successful conditions may entail activation of 58 with concomitant generation of a silylated alcohol in the product. The unique susceptibility of carbonyl substituted pyrroloindolizine structures to activation by trialkylsilyl trifluoromethanesulfonates and accessibility of silyl enol ether derivatives of ketone 28 indicated that a one-pot Mukaiyama aldol conditions may be optimal for our system.xvii In the event, addition of trimethylsilyl trifluoromethane sulfonate to a solution of ketone 28 and aldehyde 58 in the presence of N,N-diisopropylethylamine efficiently generated the desired β-silyloxy ketone 62 in 92% yield (Scheme 12). Attempts to remove the trimethylsilyl group by treatment of 62 with tetra-n-butylammonium fluoride resulted in fragmentation to starting components 28 and 58, consistent with our hypothesis that the β-alkoxy ketone is subject to a retro-aldol fragmentation. Fortunately, direct treatment of a dichloromethane solution of β-silyloxy ketone 62 with titanium tetrachloride at −78 °C afforded the desired enone 47 in 84% yield, precluding the need for desilylation. This two-step sequence provided efficient access to our requisite Nazarov substrate.
Scheme 12.

Synthesis of enone 47. Conditions: a) TMSOTf, CH2Cl2, 0 °C, 30 min, 92%, 20: 13: 5: 5, mixture of diastereomers. b) TiCl4, CH2Cl2, −78 °C, 25 min, 84%.
We considered a variety of electrophiles that may reversibly add to the pyrrole nucleus to generate a dienone substructure upon activation of 47. In addition to use of acids to protonate at C3b we envisioned that mild electrophilic halogenating agents or mercury (II) salts may reversibly add to C3b to generate 48 (Scheme 10). Interestingly, in situ monitoring showed that treatment of a benzene-d6 solution of enone 47 with excess TFA-d1 (50% v/v) and heating to 65 °C afforded three major ring protonated species; however, no cyclization products could be detected, and basic quench returned starting enone 47 (Table 2, entry 1). By contrast, treatment of an acetonitrile-benzene (4:1) solution of 47 with the mild chlorinating agent 2,4,6,6-tetrachloro-2,4-cyclohexadienone (TCCHD) slowly generated a mixture of mono- and bis-alkene products 63 (Table 2, entry 2). This result is consistent with chlorination at C2a to form the desired intermediate for Nazarov cyclization (48, X = Cl, Scheme 10) followed by net elimination of hydrochloric acid to give the alkene(s) 63. However, increased reaction temperatures resulted only in further oxidation and decomposition. Likewise, treatment of an acetonitrile-d3 solution of enone 47 with mercury (II) trifluoroacetate resulted in rapid oxidation to 63 accompanied by isomerization to the corresponding enone cis-47 (Table 2, entry 3). Exposure of 47 to mercury (II) acetate or mercury (II) chloride and heating to 65 °C resulted only in partial isomerization (Table 2, entries 4 and 5).
Table 2.
Treatment of 47 with Brønsted and Lewis acids
 | ||||||
|---|---|---|---|---|---|---|
| entry | [X+] | additive | solvent | temp (°C) | time (h) | product(s) (% yield) |
| 1 | TFA | - | C6D6 | 65 | 6 | 47 (67) |
| 2 | TCCHD | - | CH3CN | 23 | 4 | 63 (59) |
| 3 | Hg(O2CCF3)2 | - | CD3CN | 23 | 3 | 63 (57) |
| 4 | Hg(OAc)2 | - | CD3CN | 65 | 7 | 47 (88), cis-47 (4) |
| 5 | HgCl2 | - | CD3CN | 65 | 12 | 47 (47), cis-47 (5) |
| 6 | TFA | LiCl | THF | 65 | 4 | 47 (37), cis-47 (8) |
| 7 | TFA | LiClO4 | THF | 85 | 13 | 2 (20), 47 (30), cis-47 (30) |
TFA = trifluoroacetic acid; TCCHD = 2,4,6,6-tetrachloro-2,4-cyclohexadienone.
We reasoned that nucleophilic additives might efficiently trap an initial cationic species 48 at C8a (Scheme 10) and increase the lifetime of the dienone substructure. Addition of saturating lithium chloride to an acetonitrile-d3-benzene-d6 (4:1) solution of enone 47 in the presence of TFA (3.00 equiv) and heating to 65 °C resulted in partial isomerization to the Z-isomer cis-47; however, no cyclization products were observed (Table 2, entry 6). Notably, no net isomerization occurred under these conditions in the absence of lithium chloride (Table 2, entry 1). Heating a solution of enone 47 and TFA (3.00 equiv) in tetrahydrofuran saturated with lithium perchlorate at 85 °C gradually produced a 1:1 mixture of 47 and cis-47 and resulted in fragmentation to myrmicarin 215B (2) (Table 2, entry 7). Use of alternative acids such as formic acid, trifluoromethanesulfonic acid, and perchloric acid resulted in varying degrees of fragmentation and isomerization, however, none of these conditions generated any observable cyclized products.
To investigate the feasibility of the Nazarov pathway in a simplified system containing only one pyrrole nucleus susceptible to activation we prepared the tricyclic enone substrate 64. In contrast to our results with 47, treatment of a dichloromethane solution of 64 with trifluoromethanesulfonic acid for 6 hours followed by treatment 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diaza-phosphorine (BEMP) yielded the tetracyclic product 67 (Scheme 13). Formation of this compound is consistent with protonation of the enone carbonyl followed by bond-formation at C2a. Deprotonation at C7 would generate the enamine 66, which may be susceptible to air oxidation upon exposure to atmosphere. In our earlier studies,iiic we observed that heptacyclic enamine intermediates similar to 66 were extremely sensitive to isolation due to rapid decomposition/oxidation pathways, rarely allowing isolation of identifiable oxidation products. In the present case, we were able to observe compound 66 by 1H NMR and isolate the oxidation product 67 albeit in low yield.
Scheme 13.

Acid-induced cyclization of 64. Conditions: a) TfOH, CH2Cl2, 23 °C, 6 h; BEMP, 23 °C, 45 min, 16%.
Interestingly, while both tricyclic enone 64 and the β-functionalized enone 43 (Scheme 9) undergo C3b alkylation upon activation by Brønsted acid, enone 47 failed to show any evidence of analogous cyclization. The observation of ring-protonated derivatives of 47 by 1H NMR and oxidation byproducts potentially arising via activation of the pyrrole ring provide evidence for formation of the desired Nazarov cyclization intermediate 48 (Scheme 10). Additionally, the failure of 47 to undergo C3b alkylation under these conditions provides validation for strategies that block undesired reactivity at this site similar to those described in the vinyl pyrrole series (Scheme 3). These studies on unique modes of pyrroloindolizine reactivity in dimeric structures inform the design of strategically functionalized heterodimers that are predisposed to differing reaction manifolds. Currently, we are pursuing the development of pyrroloindolizine derivatives that capitalize on these observations and are incorporating these derivatives into dimeric structures for the desired cyclization using the directed heterodimerization methods described herein.
5. Conclusion
Pyrroloindolizine structures undergo unique modes of electrophilic reactivity. Activation of C8-heteroatom substituted pyrroloindolizines and addition of neutral derivatives provides the corresponding dimeric structures. In this manner, we have employed trifluoromethanesulfonic anhydride activation of functionalized pyrroloindolizine substrates to prepare a range of hexacyclic structures. Studies on cyclopentannulation to form heptacyclic derivatives motivated the design of stable dimeric structures susceptible to further functionalization and derivatization. Accordingly, condensation between functional dimerization partners enabled us to prepare an air stable symmetrical diketone. Synthesis of a hexacyclic enone derivative through a uniquely efficient Mukaiyama aldol reaction allowed us to explore potential Nazarov cyclization in an activated dimeric structure. Our protocols for directed heterodimerization in these systems provide exciting opportunities to rapidly introduce the requisite functional groups in dimeric structures. In addition, fascinating observations on the reversible dimerization of myrmicarin 215B introduce the possibility of strategically modifying monomeric and dimeric pyrroloindolizine derivativesiiid to influence the structure of heptacyclic products formed under equilibrium conditions.xviii The merger between strategic design of pyrroloindolizine structures and powerful methods of fragment assembly continues to guide our design of synthetic strategies relevant to these intriguing alkaloids and their derivatives.
6. Experimental section
6.1. General procedures
All reactions were performed in oven-dried or flame-dried round-bottomed flasks, modified Schlenk (Kjeldahl shape) flasks, or glass pressure vessels. The flasks were fitted with rubber septa and reactions were conducted under a positive pressure of argon. Stainless steel syringes or cannulae were used to transfer air- and moisture-sensitive liquids. Flash column chromatography was performed as described by Still et al. using silica gel (60 Å pore size, 40–63 μm, 4–6% H2O content, Zeochem).xix Where necessary (so noted), silica gel was neutralized by treatment with the eluent containing 2.5% triethylamine. Analytical thin-layer chromatography was performed using glass plates pre-coated with 0.25 mm 230–400 mesh silica gel impregnated with a fluorescent indicator (254 nm). Where necessary (so noted), silica gel plates were neutralized by treatment with a solution of 2.5% triethylamine in ethyl acetate–hexanes followed by heating on a hot plate (~250 °C). Thin layer chromatography plates were visualized by exposure to ultraviolet light and/or by exposure to an aqueous solution of ceric ammonium molybdate (CAM) followed by heating (<1 min) on a hot plate (~250 °C). Organic solutions were concentrated on Büchi R-200 rotary evaporators at ~20 Torr at 25–35 °C unless otherwise indicated.
6.2. Materials
Commercial reagents and solvents were used as received with the following exceptions: dichloromethane, diethyl ether, tetrahydrofuran, acetonitrile, and toluene were purchased from J.T. Baker (Cycletainer™) and were purified by the method of Grubbs et al. under positive argon pressure.xx Triethylamine, diisopropylethylamine, benzene, and 1,2-dichloroethane were distilled over calcium hydride immediately before use. Ethylene glycol was distilled over magnesium sulfate at reduced pressure. Trichloroacetyl chloride was distilled at reduced pressure immediately before use. Trimethylsilyl trifluoromethanesulfonate was distilled at reduced pressure immediately before use.
6.3. Instrumentation
Proton nuclear magnetic resonance (1H NMR) spectra were recorded with a Varian 300 Mercury or a Varian inverse probe 500 INOVA spectrometer or a Bruker 400 spectrometer or a Bruker inverse probe 600 Avance spectrometer. Chemical shifts are recorded in parts per million on the δ scale and are referenced from the residual protium in the NMR solvent (C6D5H: δ7.16). Data is reported as follows: chemical shift [multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, app = apparent, br = broad), coupling constant(s) in Hertz, integration, assignment]. Carbon-13 nuclear magnetic resonance (13C NMR) spectra were recorded with a Varian 500 INOVA spectrometer and are recorded in parts per million on the δ scale and are referenced from the carbon resonances of the solvent (benzened6: δ 128.4). Phosphorus-31 nuclear magnetic resonance (31P NMR) spectra were recorded with a Varian 500 INOVA spectrometer and are referenced to phosphoric acid (H3PO4: δ0.00) as an external standard. Infrared data were obtained with a Perkin-Elmer 2000 FT-IR and are reported as follows: [frequency of absorption (cm−1), intensity of absorption (s = strong, m = medium, w = weak, br = broad), assignment]. We are grateful to Dr. Li Li for obtaining the mass spectrometric data at the Department of Chemistry’s Instrumentation Facility, Massachusetts Institute of Technology. High-resolution mass spectra (HRMS) were recorded on a Bruker APEX 4.7 Tesler FTMS spectrometer using electronspray ion source (ESI) or electronspray (ES).
6.4. Positional numbering system
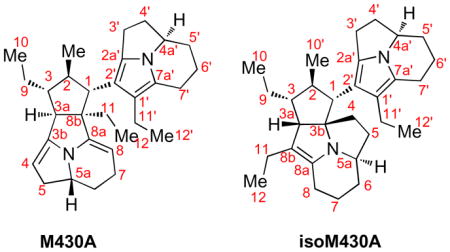
The numbering system for proton and carbon assignments for all pyrroloindolizine structures is consistent with the isolation reports for the naturally occurring tricyclic myrmicarins.ia For direct comparison, the numbering system for proton and carbon assignments for all heterodimeric structures is consistent with the isolation reports for myrmicarin 430Aib and the numbering system of isomyrmicarin 430A.iiic
6.5. Experimental procedures
6.5.1. Vinyl Bromide 15
A solution of 2,4,4,6-tetrabromo-2,5-cyclohexadienone (TBCHD, 222 mg, 542 μmol, 2.00 equiv) in tetrahydrofuran (5.00 mL) was added to solution of tricyclic ketone 28iiib (62.6 mg, 271 μmol, 1 equiv) in tetrahydrofuran (5.00 mL) at 0 °C over 10 min. After an additional 10 min the reaction mixture was concentrated under reduced pressure at 30 °C. The resulting green-brown oil was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 2.5 cm, ht. 15 cm, eluent: 1% Et3N and 14% EtOAc in hexanes) to afford vinyl bromide 15 (56.4 mg, 68%) as a yellow solid. 1H and 13C signals were assigned with the aid of gCOSY, HSQC, gHMBC, and nOe analysis. 1H NMR (500 MHz, C6D6, 20 °C): 6.68 (d, J = 2.9 Hz, 1H, C7–H), 3.34-3.27 (m, 1H, C4a –H), 2.96 (q, J = 7.4 Hz, 2H, C11–H, C11–H′), 2.45–2.34 (m, 2H, C9–H, C9–H′), 2.32 (dd, J = 16.0, 8.3 Hz, 1H C3–Ht), 2.26 (dd, J = 16.4, 5.5 Hz, 1H, C5–Hc), 2.16 (ddd, J = 16.3, 9.9, 6.8 Hz, 1H, C3–Hc), 2.02 (ddd, J = 16.1, 12.6, 3.2 Hz, 1H, C5–Ht), 1.62 (dt, J = 12.4, 6.2 Hz, 1H, C4–Hc), 1.39 (t, J = 7.4 Hz, 3H, C12–H), 1.27 (t, J = 7.3 Hz, 3H, C10–H), 1.18–1.10 (m, 1H, C4–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 194.5 (C8), 139.8 (C2a), 128.8 (C1), 121.2 (C7a), 121.0 (C7), 117.8 (C2), 113.8 (C6), 53.3 (C4a), 41.4 (C5), 36.0 (C4), 34.7 (C9), 29.0 (C3), 20.0 (C11), 16.7 (C12), 9.1 (C10). FTIR (thin film) cm−1: 2922 (m), 1650 (s), 1488 (m), 1401 (w), 1264 (w), 1097 (m). HRMS (ESI): calc’d for C15H19BrNO [M+H]+: 308.0645, found: 308.0657. TLC (silica gel, 20% EtOAc in hexanes), Rf: 0.50 (UV, CAM).
6.5.2. Hexacyclic Ketone 17
Trifluoromethanesulfonic anhydride (Tf2O, 51.0 μL, 228 μmol, 2.00 equiv) was added portion-wise (3 × 20 μL, 30 min intervals) to a solution of vinyl bromide 15 (47.0 mg, 152 μmol, 1 equiv), silyl enol ether 16iiia (70.9 mg, 183 μmol, 1.20 equiv) (dried as a mixture by concentration from benzene under reduced pressure, 3 × 1.00 mL), and 2,6-di-tert-butyl-4-methylpyridine (DTBMP, 157 mg, 570 μmol, 5.00 equiv) in dichloromethane (2.20 mL) at −78 °C. After 30 min, saturated aqueous sodium bicarbonate solution (2 mL) was added and the mixture was diluted with dichloromethane (5 mL) and allowed to warm to 23 °C. The aqueous layer was extracted with dichloromethane (4 × 10 mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting oil was purified by flash column chromatography (neutral alumina gel: diam. 3.0 cm, ht. 21 cm; eluent: 10% EtOAc in hexanes) to afford hexacyclic ketone 17 (61.6 mg, 78%, 9:2 mixture of diastereomers) as a yellow foam. 1H and 13C signals for the major diastereomer were assigned with the aid of gCOSY, HSQC, gHMBC, and gNOESY analysis. 1H NMR (500 MHz, C6D6, 20 °C): 6.82 (d, J = 2.6 Hz, 1H, C8–H), 6.02–5.96 (m, 1H, C9–H), 3.99 (q, J = 7.0 Hz, 1H, C2–H), 3.59–3.45 (m, 1H, C5a–H), 3.12 (dq, J = 13.6, 7.0 Hz, 1H, C11′–Hx), 3.07–2.82 (m, 2H, C11′–Hy, C4a′–H), 2.72–2.52 (m, 1H, C3′–Hc), 2.58 (dd, J = 15.4, 8.1 Hz, 1H, C4–Hx), 2.51–2.31 (m, 5H, C3′–Ht, C4–Hy, C11–Hx, C11–Hy, C7′–Hc), 2.29–2.20 (m, 1H, C6–Hy), 2.17–2.04 (m, 2H, C7′–Ht, C6–Hx), 1.90–1.66 (m, 5H, C10-H, C5–Hx, C4′–Hc), 1.60 (d, J = 7.1 Hz, 3H, C10′–H), 1.54–1.47 (m, 1H, C6′–Ht), 1.43 (t, J = 7.4 Hz, 3H, C12′–H), 1.40–1.27 (m, 3H, C5–Hy, C4′–Ht, C5′–Hc), 1.17–1.08 (m, 4H, C12–H, C6′–Hc), 0.66 (tdd, J = 13.1, 10.8, 2.3 Hz, 1H, C5′–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 196.3 (C1), 137.0 (C2a′), 136.8 (C3), 132.4 (C3b), 126.4 (C1′), 124.7 (C9), 122.1 (C8), 120.9 (C7a′), 119.6 (C8b), 117.9 (C2′), 115.6 (C3a), 111.4 (C8a), 111.2 (C7), 56.0 (C4a′), 53.3 (C5a), 51.9 (C2), 41.8 (C6), 37.4 (C5), 36.2 (C4′), 29.7 (C5′), 28.4 (C3′), 26.9 (C4), 22.7 (C6′), 20.2 (C11), 20.0 (C7′), 19.7 (C11′), 18.1 (C10′), 17.0 (C12), 16.3 (C12′), 15.7 (C10). HRMS (ESI): calc’d for C30H38BrN2O [M+H]+: 521.2162, found: 521.2164. TLC (silica gel, 20% EtOAc in hexanes), Rf: 0.39 (UV, CAM).
6.5.3. Diene 21
To a solution of hexacyclic alcohol 18 (17.3 mg, 33.0 μmol, 1 equiv) in benzene (1.30 mL) at 23 °C was added acetic acid (25.0 μL, 437 μmol, 13.0 equiv). After 1.8 h, saturated aqueous sodium bicarbonate solution (2 mL) was added and the mixture was diluted with dichloromethane (5 mL). The layers were separated and the aqueous layer was extracted with dichloromethane (4 × 5 mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (neutral alumina gel: diam. 2 cm, ht. 15 cm; eluent: 12.5% EtOAc in hexanes) to afford diene 21 (11.3 mg, 68%) as a yellow oil. Due to the air- and acid-sensitivity of 21, best spectra were obtained by immediate spectroscopic analysis. 1H and 13C signals were assigned with the aid of gCOSY, HSQC, and gHMBC analysis. 1H NMR (500 MHz, C6D6, 20 °C): 6.88 (d, J = 2.8 Hz, 1H, C8–H), 6.76 (s, 1H, C1–H), 6.04 (m, 1H, C9–H), 3.68–3.57 (m, 1H, C5a–H), 3.30 (tdd, J = 10.6, 5.1, 3.8 Hz, 1H, C4a′–H), 2.64 (ddd, J =14.9, 10.9, 6.5 Hz, 1H, C3′–Hc), 2.62–2.39 (m, 7H, C7′–Hc, C11–Hx, C11–Hy, C11′–Hx, C11′–Hy, C4–Hx, C4–Hy), 2.46 (dd, J = 15.0, 8.2 Hz, 1H, C3′–Ht), 2.40–2.29 (m, 2H, C7′–Ht, C6–Hx), 2.25 (s, 3H, C10′–H), 2.26–2.21 (m, 1H, C6–Hy), 1.94 (dt, J = 11.4, 5.8 Hz, 1H, C4′–Hc), 1.86 (d, J = 6.8 Hz, 3H, C10–H), 1.75–1.67 (m, 1H, C5–Hx), 1.67–1.60 (m, 1H, C6′–Ht), 1.50 (dq, J = 12.4, 3.3 Hz, 1H, C5′–Hc), 1.48–1.40 (m, 2H, C5–Hy, C4′–Ht), 1.37–1.26 (m, 1H, C6′–Hc), 1.22–1.17 (m, 6H, C12′–H, C12–H), 0.77 (tdd, J = 13.0, 10.9, 2.2 Hz, 1H, C5′–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 140.7 (C2), 132.9 (C3a), 132.8 (C3b), 129.4 (C2a′), 127.9 (C1′), 123.3 (C1), 122.8 (C8b), 122.2 (C8) 121.5 (C9), 120.0 (C8a), 119.5 (C7a′), 114.5 (C3), 114.1 (C2′), 111.2 (C7), 55.6 (C4a′), 53.3 (C5a), 42.0 (C6), 37.8 (C4′), 37.8 (C5), 30.4 (C5′), 27.8 (C3′), 26.5 (C4), 22.8 (C6′), 20.8 (C7′) 19.8 (C11′), 19.4 (C11), 17.1 (C12′), 17.0 (C12), 16.8 (C10′), 16.4 (C10). HRMS (ESI): calc’d for C30H38BrN2 [M+H]+: 505.2213, found: 505.2233. TLC (silica gel, 20% EtOAc in hexanes), Rf: 0.57 (UV, CAM).
6.5.4. Heptacyclic Ketone 24
Trichloroacetic acid solution (0.16 M in benzene, 3.95 mg, 24.2 μmol, 1.05 equiv) was added portion-wise (3 × 50 μL, 30 min intervals) to a solution of diene 21 (12.0 mg, 23.0 μmol, 1 equiv) in benzene (1.50 mL) at 23 °C. After 1.8 h, saturated aqueous sodium bicarbonate solution (2 mL) was added and the mixture was diluted with dichloromethane (3 mL). The layers were separated and the aqueous layer was extracted with dichloromethane (4 × 4 mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting brown oil was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 1.0 cm, ht. 15 cm; eluent: 1% Et3N and 14→99% EtOAc in hexanes) to afford heptacyclic ketone 24 (5.00 mg, 48%) as a yellow oil. Due to the fragility and air-sensitivity of 24, best spectra were obtained by immediate spectroscopic analysis. 1H and 13C signals were assigned with the aid of gCOSY, gHSQC, gHMBC, and gNOESY analysis. 1H NMR (500 MHz, C6D6, 20 °C): 5.60 (s, 1H, C8–H), 3.52–3.48 (m, 1H, C5a–H), 3.34 (s, 1H, C1–H), 3.08–3.02 (m, 1H, C4a′–H), 2.65 (dd, J = 17.7, 7.5 Hz, 1H, C6–Hy), 2.63–2.54 (m, 4H, C7′–Hc, C11′–H, C11′–H′, C3′–Hc), 2.41–2.33 (m, 1H, C7′–Hx), 2.37 (d, J = 17.7 Hz, 1H, C6–Hx), 2.29–2.12 (m, 5H, C3′–Ht, C11–H, C11–H′, C9–H, C9–H′), 2.05 (ddd, J = 12.9, 10.7, 2.1 Hz, 1H, C4–Hx), 1.98–1.88 (m, 2H, C4′–Hc, C5–Hx), 1.70 (dt, J = 11.6, 8.8 Hz, 1H, C4–Hy), 1.60 (s, 3H, C10′–H), 1.58–1.48 (m, 2H, C4′–Ht, C6′–Ht), 1.42–1.36 (m, 2H, C5′–Hc, C5–Hy), 1.31 (t, J = 7.5 Hz, 3H, C12′–H), 1.21–1.18 (m, 1H, C6′–Hc), 1.08 (t, J = 7.6 Hz, 3H, C12–H), 0.99 (t, J = 7.6 Hz, 3H, C10–H), 0.80-0.72 (m, 1H, C5′–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 189.6 (C7), 177.0 (C8a), 164.2 (C3a), 152.4 (C2), 134.1 (C3), 127.5 (C2a′), 124.5 (C8b), 121.8 (C1′), 118.3 (C7a′), 111.4 (C2′), 97.9 (C8), 88.0 (C3b), 58.5 (C5a), 55.8 (C1), 55.0 (C4a′), 41.1 (C4), 38.4 (C6), 37.0 (C4′), 31.2 (C5), 29.9 (C5′), 26.7 (C3′), 22.8 (C6′), 21.1 (C7′), 19.6 (C11′), 19.1 (C11), 18.4 (C9), 17.6 (C12), 15.1 (C10′), 14.6 (C10), 14.4 (C12). FTIR (thin film) cm−1: 2856 (s), 1704 (m), 1643 (m), 1453 (m), 1206 (m), 1061 (m). HRMS (ESI): calc’d for C30H39N2O [M+H]+: 443.3057, found: 443.3076. TLC (1.0% Et3N in EtOAc, Et3N neutralized silica gel), Rf: 0.55 (UV, CAM).
6.5.5. Pyrrole 29
To a vigorously stirred solution of ketone 28iiib (124.0 mg, 536 μmol, 1 equiv) and ethylene glycol (600 μL, 10.7 mmol, 20.0 equiv) in 1,2-dichloroethane (5.25 mL) at 23 °C was added triflurormethanesulfonic acid (142 μL, 1.61 mmol, 3.00 equiv) and the resulting yellow solution was heated to 65 °C. After 1 h, the tan coloured reaction mixture was allowed to cool to 23 °C, was poured into 3N KOH (17.5 mL), and was diluted with diethyl ether (20 mL). The layers were separated and the aqueous layer was extracted with diethyl ether (3 × 20 mL). The combined organic layers were washed with brine (20 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting material was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 2.5 cm, ht. 15 cm; eluent: 2% Et3N and 3% EtOAc in hexanes) to afford pyrrole 28 (93.9 mg, 100%) as white needles. The spectroscopic data was consistent with the data reported in the literature.xxi 1H NMR (500 MHz, C6D6, 20 °C): 5.95 (s, 1H, C2–H), 3.26 (tdd, J = 10.7, 4.8, 3.8 Hz, 1H, C4a–H), 2.65–2.55 (m, 4H, C3–Hc, C3–Ht, C11–H, C11′–H), 2.52 (dd, J = 14.5, 8.1 Hz, C7–Hc), 2.38 (ddd, J = 16.8, 11.3, 6.1 Hz, 1H, C7–Ht), 1.94 (dt, J = 11.5, 5.8 Hz, 1H, C4–Hc), 1.69–1.64 (m, 1H, 6–Ht), 1.58–1.48 (m, 2H, C4–Ht, C5–Hc), 1.40–1.30 (m, 1H, C6–Hc), 1.32 (t, J = 7.5 Hz, 3H, C12–H), 0.82 (tdd, J = 13.0, 10.6, 2.5 Hz, 1H, C5–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 130.7, 123.5, 118.3, 100.3, 55.5, 37.8, 30.3, 25.9, 23.1, 20.9, 20.9, 16.8. FTIR (thin film) cm−1: 2958 (s), 1425 (m), 1322 (m), 1168 (w), 767 (m). HRMS (ESI): calc’d for C12H18N [M+H]+: 176.1434, found: 176.1443. TLC (silica gel pretreated with Et3N, 2.5% Et3N in [17.5% EtOAc in hexanes]), Rf: 0.69 (UV, CAM).
6.5.6. Trichloroacetylpyrrole 30
To a solution of pyrrole 29 (68.8 mg, 393 μmol, 1 equiv) in 1,2-dichloroethane (4.00 mL) at 23 °C was added trichloroacetyl chloride (65.9 μL, 590 μmol, 1.50 equiv) and the solution was heated to 65 °C. After 2 h, the resulting deep red reaction mixture was allowed to cool to 23 °C, was poured into saturated aqueous sodium bicarbonate solution (10 mL), and was diluted with ethyl acetate (10 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (3 × 12.5 mL). The combined organic layers were washed with brine (10 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting material was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 2.0 cm, ht. 23 cm; eluent: 2% Et3N and 3% EtOAc in hexanes) to afford trichloroacetylpyrrole 30 (93.5 mg, 74%) as a pale yellow solid. 1H NMR (500 MHz, C6D6, 20 °C): 3.03 (dd, J = 16.2, 7.8 Hz, 1H, C3–Ht), 2.95–2.80 (m, 3H, C11-H, C11-H′, C4a–H), 2.49 (ddd, J = 16.5, 10.8, 6.1 Hz, 1H, C3–Hc), 2.26 (dd, J =16.6, 6.3 Hz, 1H, C7–Hc), 1.99 (ddd, J = 16.7, 12.0, 6.7 Hz, 1H, C7–Ht), 1.68 (dt, J = 11.7, 5.9, 5.9 Hz, 1H, C4–Hc), 1.48–1.41 (m, 1H, C6–Ht), 1.35 (t, J = 7.4 Hz, 3H, C12–H), 1.35–1.25 (m, 2H, C4–Ht, C5–Hc), 1.10–1.00 (m, 1H, C6–Hc), 0.57 (tdd, J =13.2, 11.0, 2.3 Hz, 1H, C5–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 177.2, 138.3, 129.8, 122.0, 107.2, 99.1, 56.5, 36.0, 31.6, 29.2, 22.2, 20.5, 19.7, 15.8. FTIR (thin film) cm−1: 2958 (s), 1666 (s), 1456 (m), 1319 (m), 813 (m). HRMS (ESI): calc’d for C14H17Cl3NO [M+H]+: 320.0370, found: 320.0358. TLC (silica gel pretreated with Et3N, 2.5% Et3N in [2.5% EtOAc in hexanes]), Rf: 0.22 (UV, CAM).
6.5.7. Methyl Ester 35
To a solution of trichloroacetylpyrrole 30 (40.0 mg, 125 μmol, 1 equiv) in methanol–dichloromethane (15:1, 3.20 mL) at 23 °C was added sodium methoxide (135.1 mg, 2.50 mmol, 20.0 equiv) in a single portion. After 1.5 h, aqueous ammonium chloride solution (10 mL) was added and the reaction mixture was diluted with ethyl acetate (12.5 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (4 × 7.5 mL). The combined organic layers were washed with brine (10 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting material was purified by flash column chromatography (silica gel: diam. 1.5 cm, ht. 18 cm; eluent: 7.5→15% EtOAc in hexanes) to afford methyl ester 35 (25.2 mg, 87%) as a white solid. 1H NMR (500 MHz, C6D6, 20 °C): 3.65 (s, 3H, OCH3), 3.11–2.92 (m, 4H, C11–H, C11′–H, C3–Ht, C4a–H), 2.70 (ddd, J = 16.2, 10.4, 6.3 Hz, 1H, C3–Hc), 2.40 (ddd, J = 16.3, 6.5, 1.1, 1H, C7–Hc), 2.18 (ddd, J = 16.4, 11.9, 6.8 Hz, 1H, C7–Ht), 1.78 (ddd, J = 11.2, 6.2, 5.6 Hz, 1H, C4–Hc), 1.56–1.50 (m, 1H, C6–Ht), 1.46 (t, J = 7.4 Hz, 3H, C12–H), 1.41–1.32 (m, 2H, C4–Ht, C–5Hc), 1.21–1.11 (m, 1H, C6–Hc), 0.66 (tdd, J = 13.1, 11.0, 2.3 Hz, 1H, C5–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 166.2, 138.8, 125.6, 120.3, 106.8, 56.1, 50.4, 36.4, 29.8, 27.5, 22.7, 20.2, 19.8, 16.6. FTIR (thin film) cm−1: 2965 (s), 1702 (s), 1514 (m), 1435 (m), 1275 (m), 1110 (m). HRMS (ESI calc’d for C14H20NO2 [M+H]+: 234.1489, found: 234.1496. TLC (silica gel, 10% EtOAc in hexanes), Rf: 0.25 (UV, CAM).
6.5.8. Acid Phosphate 34
To a vigorously stirred suspension of acid 36 (40.3 mg, 183 μmol, 1 equiv) and diethyl cyanophosphonate (39.7 μL, 265 μmol, 1.45 equiv) in ethyl acetate (1.83 mL) at 23 °C was added triethylamine (34.4 μL, 247 μmol, 1.35 equiv). After 1.5 h, the reaction mixture was concentrated under reduced pressure to a volume of approximately 100 μL and the resulting material was immediately purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 1.5 cm, ht. 15 cm; eluent: 2.5% Et3N and 67.5% EtOAc in hexanes) to afford acid phosphate 34 (55.7 mg, 85%) as a pale yellow oil. Acid phosphate 34 was sensitive to hydrolysis and was used immediately after preparation. 1H NMR (500 MHz, C6D6, 20 °C): 4.30–4.20 (m, 4H, OCH2CH3), 2.95–2.78 (m, 4H, C11–H, C11′–H, C4a–H, C3–Ht), 2.57 (ddd, J = 16.3, 10.4, 6.2 Hz, 1H, C3–Hc), 2.27 (dd, J = 16.6, 3.8 Hz, 1H, C7–Hc), 2.04 (ddd, J = 16.5, 11.8, 6.7 Hz, 1H, C7–Ht), 1.69 (dt, J = 11.7, 6.0 Hz, 1H, C4–Hc), 1.50–1.44 (m, 1H, C6–Ht), 1.36 (t, J = 7.5 Hz, 3H, C12–H), 1.30 (dq, J = 12.5, 3.4 Hz, 1H, C5–Hc), 1.27–1.19 (m, 1H, C4–Ht), 1.10–1.02 (m, 7H, OCH2CH3, C6–Hc), 0.58 (tdd, J = 13.1, 11.0, 2.3 Hz, 1H, C5–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 158.9 (d, J = 7.7 Hz), 141.5, 126.7 (d, J = 1.4 Hz), 121.3, 105.1 (d, J = 9 Hz), 64.6 (d, J = 5.4 Hz), 64.6 (d, J = 5.4 Hz), 56.4, 35.9, 29.5, 27.8, 22.5, 20.0, 19.5, 16.6 (d, J = 6.8 Hz), 16.6 (d, J = 6.8 Hz), 16.2. 31P NMR (200 MHz, C6D6, 20 °C): −6.14. FTIR (thin film) cm−1: 2965 (m), 1726 (s), 1513 (m), 1273 (s), 1031 (s), 1004 (s). HRMS (ESI): calc’d for C17H26NNaO5P [M+Na]+: 378.1441, found: 378.1443. TLC (silica gel pretreated with Et3N, 2.5% Et3N in [47.5% EtOAc in hexanes]), Rf: 0.37 (UV, CAM).
6.5.9. Diketone 27
A solution of ketone 28iiib (63.2 mg, 273 μmol, 1 .76 equiv) in toluene (300 μL) was transferred via cannula to a solution of lithium hexamethyldisilazide (LHMDS, 97% w/w, 46.2 mg, 268 μmol, 1.60 equiv) in toluene (550 μL) at −78 °C and the transfer was completed with a toluene rinse (2 × 100 μL). The reaction flask was warmed to 0 °C for 15 minutes then cooled to −78 °C for 5 min. A solution of acid phosphate 34 (55.7 mg, 155 μmol, 1 equiv) in toluene (300 μL) was added via cannula and transfer was complete with a toluene rinse (2 × 100 μL). The reaction flask was placed on a dry- ice–acetone bath at −40 °C and allowed to warm slowly to 0 °C. After 1 h, aqueous ammonium chloride solution (7.5 mL) was added and the reaction mixture was diluted with ethyl acetate (12.5 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (4 × 10 mL). The combined organic layers were washed with brine (10 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting material was purified by flash column chromatography (silica gel: diam. 2.0 cm, ht. 17 cm; eluent: 40→55% EtOAc in hexanes, followed by 1% AcOH in 99% EtOAc for elution of acid 36) to afford diketone 27 (47.0 mg, 70%) as a white solid. In addition, ketone 28 (28.3 mg, 38% recovered) and acid 36 (8.1 mg, 16% recovered based on acid phosphate 34) were separated during chromatography and isolated. 1H and 13C signals were assigned with the aid of gCOSY, HSQC, and gHMBC analysis. 1H NMR (500 MHz, C6D6, 20 °C): 4.45 (m, 1H, C2–H), 3.18–2.98 (m, 3H, C11–Hx, C11–Hy, C4a′–H), 3.05 (q, J = 7.4 Hz, 2H, C11′–Hx, C11′-Hy), 2.97–2.89 (m, 1H, C5a–H), 2.94 (dd, J = 15.4, 8.2 Hz, 1H, C4–Ht), 2.78–2.66 (m, 2H, C3′–Hc, C3′–Ht), 2.58 (ddd, J = 15.5, 10.6, 6.5 Hz, C4–Hc), 2.40–2.29 (m, 2H, C8–Hc, C7′–Hc), 2.17–2.07 (m, 2H, C8–Ht C7′–Ht), 1.81–1.75 (m, 2H, C5–Hc, C4′–Hc), 1.78 (d, J = 7.2 Hz, 3H, C10′–H), 1.53–1.36 (m, 4H, C7-Hc, C6′-Hc, C5–Ht, C4′–Ht), 1.51 (t, J = 7.4 Hz, 3H, C12–H), 1.45 (t, J = 7.4 Hz, 3H, C12′–H), 1.36–1.29 (m, 2H, C6–Hc, C5′–Hc), 1.20–1.10 (m, 1H, C6′–Ht), 1.12–1.02 (m, 1H, C7–Ht), 0.68 (tdd, J = 14.8, 13.2, 2.3 Hz, 1H, C6–Ht), 0.59 (tdd, J = 14.8, 13.3, 2.3 Hz, 1H, C5′–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 193.3 (C1), 193.0 (C3), 136.2 (C3b), 136.2 (C2a′), 126.7 (C8b), 126.5 (C1′), 121.1 (C7a′), 120.9 (C8a), 117.6 (C3a), 117.6 (C2′), 56.6 (C2), 56.0 (C4a′), 55.9 (C5a), 36.5 (C5), 36.3 (C4′), 29.7 (C6), 29.4 (C5′), 28.6 (C3′), 28.1 (C4), 22.7 (C6′), 22.5 (C7), 20.4 (C8), 20.3 (C7′), 20.0 (C11), 19.9 (C11′), 16.4 (C12), 16.3 (C12′), 15.8 (C10′). FTIR (thin film) cm−1: 2928 (s), 1650 (s), 1494 (m), 1428 (m), 1320 (w), 972 (s). HRMS (ESI): calc’d for C28H36N2NaO2 [M+Na]+: 455.2669, found: 455.2664. TLC (silica gel, 50% EtOAc in hexanes), Rf: 0.35 (UV, CAM).
6.5.10. Vinyl Phosphate 43
A solution of diketone 27 (17.1 mg, 39.5 μmol, 1 equiv) in tetrahydrofuran (200 μL) was added drop wise to a solution of potassium hexamethyldisilazide (KHMDS, 16.6 mg, 79.1 μmol, 2.00 equiv) in tetrahydrofuran (550 μL) at −78 °C. The reaction flask was warmed to 0 °C for 15 minutes then cooled to −78 °C for 5 min and diethyl cyanophosphonate (13.1 μL, 79.1 μmol, 2.00 equiv) was added. After 1.2 h, water was added (6 mL) and the reaction mixture was diluted with ethyl acetate (10 mL) and brine (1 mL) and allowed to warm to 23 °C. The layers were separated and the aqueous layer was extracted with ethyl acetate (4 × 7.5 mL). The combined organic layers were washed with brine (7 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting material was purified by flash column chromatography (silica gel: diam. 1.5 cm, ht. 20 cm; eluent: 50→85% EtOAc in hexanes) to afford vinyl phosphate 43 (15.0 mg, 67%) as a pale yellow oil. 1H and 13C signals were assigned with the aid of gCOSY and gHMBC analysis. 1H NMR (500 MHz, C6D6, 20 °C): 3.90–3.65 (m, 4H, OCH2CH3), 3.57–3.30 (m, 3H, C4–Hx, C4–Hy, C3′–Hx), 3.30–3.20 (m, 2H, C4a′–H, C5a–H), 3.22–3.12 (m, 1H, C11′–Hx), 3.00 (dq, J = 13.7, 7.1 Hz, 1H, C11′–Hy), 2.83–2.71 (m, 2H, C3′–Hy, C11–Hx), 2.66 (dq, J = 14.6, 7.4 Hz, 1H, C11–Hy), 2.50 (dd, J = 16.2, 6.1 Hz, 1H, C8–Hc), 2.43 (dd, J = 16.5, 5.6 Hz, 1H, C7′–Hc), 2.27 (ddd, J = 16.8, 11.3, 6.1 Hz, 1H, C8–Ht), 2.22–2.09 (m, 2H, C7′–Ht, C4′–Hc), 2.06 (d, J = 2.6 Hz, 3H, C10′–H), 2.06–2.00 (m, 1H, C5–Hc), 1.83–1.65 (m, 2H, C4′–Ht, C5–Ht), 1.65–1.48 (m, 3H, C7–Ht, C6′–Ht, C6–Hc), 1.52 (t J = 7.4 Hz, 3H, C12′–H), 1.45-1.40 (m, 1H, C5′–Hc), 1.37 (t, J = 7.6 Hz, 3H, C12–H), 1.30 (m, 1H, C7–Hc), 1.20 (m, 1H, C6′–Hc), 0.93 (td, J = 7.1, 0.8 Hz, 3H, OCH2CH3), 0.86 (td, J = 7.1, 0.9 Hz, 3H, OCH2CH3), 0.70–0.62 (m, 2H, C5′–Ht, C6–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 191.9 (d, J = 0.9 Hz, C3), 140.8 (C2a′), 139.1 (d, J = 6.9 Hz, C1), 132.2 (C3b), 127.2 (d, J = 9.6 Hz, C2), 125.7 (C1′), 123.5 (C8b), 121.1 (C7a′), 119.1 (C8a), 116.7 (C2′), 108.6 (C3a), 63.7 (d, J = 5.5 Hz, OCH2CH3), 63.4 (d, J = 5.5 Hz, OCH2CH3), 56.4 (C4a′), 55.9 (C5a), 37.5 (C5), 36.6 (C4′), 30.4 (C6), 29.9 (C5′), 27.9 (C3′), 26.3 (C4), 23.0 (C6′), 22.8 (C7), 21.0 (C11′), 20.2 (C8), 20.2 (C7′), 19.8 (C11), 17.1 (C10′), 16.5 (d, J = 2.3 Hz, OCH2CH3), 16.4 (d, J = 2.3 Hz, OCH2CH3), 16.4 (C12′), 15.9 (C12). 31P NMR (200 MHz, C6D6, 20 °C): –4.33. FTIR (thin film) cm−1: 2931 (s), 1704 (s), 1621 (s), 1503 (m), 1267 (m), 1091 (m), 1047 (m). HRMS (ESI): calc’d for C32H46N2O5P [M+H]+: 569.3139, found: 569.3137. TLC (silica gel, 65% EtOAc in hexanes), Rf: 0.15 (UV, CAM).
6.5.11. Heptacyclic Alcohol 46
Triphenylphosphine dichloride (PPh3Cl2, 6.5 mg, 20.0 μmol, 1.50 equiv) was added as solid in one portion to a solution of vinyl phosphate 43 (7.4 mg, 13.0 μmol, 1 equiv) in acetonitrile (280 μL) at 0 °C. After 2 h, saturated aqueous sodium bicarbonate solution (3 mL) was added and the mixture was diluted with ethyl acetate (5 mL). The layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 5 mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 1.0 cm, ht. 15 cm; eluent: 2.5% Et3N and 7.5% EtOAc in hexanes) to afford heptacyclic alcohol 46 (2.0 mg, 36%) as a pale yellow oil. Due to the fragility and air-, acid-, and base-sensitivity of 46, best spectra were obtained by immediate spectroscopic analysis. 1H and 13C signals were assigned with the aid of gCOSY, gHSQC, gHMBC, and gNOESY analysis. 1H NMR (500 MHz, C6D6, 20 °C): 3.51–3.45 (m, 1H, C4a′–H), 3.16–3.11 (m, 1H, C5a–H), 3.02 (br s, 1H, OH), 2.89 (ddd, J = 15.5, 10.9. 6.2 Hz, 1H, C3′–Hc), 2.73 (dd, J = 15.2, 7.9 Hz, 1H, C3′–Ht), 2.57 (dd, J = 16.5, 6.8 Hz, 1H, C7′–Hc), 2.45–2.32 (m, 7H, C4–Hx, C11–H, C11–H′, C11′–H, C11′–H′, C7′–Ht, C8–Hx), 2.06–1.99 (m, 2H, C4′–Hc, C8–Hy), 1.95 (s, 3H, C10′–H), 1.93–1.86 (m, 1H, C4–Hy), 1.72–1.62 (m, 2H, C6′–Ht, C5–Hx), 1.60–1.28 (m, 7H, C5′–Hc, C6–Hx, C6–Hy, C4′–Ht, C6′–Hc, C7–Hc, C7–Ht), 1.25–1.19 (m, 1H, C5–Hy), 1.21 (t, J = 7.5 Hz, 3H, C12–H), 1.16 (t, J = 7.5 Hz, 3H, C12′–H), 0.82 (m, 1H, C5′–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 206.8 (C3), 165.3 (C1), 145.4 (C8a), 136.0 (C2), 129.8 (C2a′), 122.9 (C1′), 120.2 (C7a′), 115.1 (C8b), 109.3 (C2′), 89.3 (C3a), 83.2 (C3b), 62.5 (C5a), 56.1 (C4a′), 37.8 (C4′), 33.0 (C6), 30.1 (C5′), 29.5 (C4), 29.0 (C5), 27.5 (C3′), 24.5 (C8), 22.7 (C6′), 21.2 (C7), 21.1 (C7′), 19.8 (C11′), 17.9 (C11), 16.6 (C12), 16.3 (C12′), 10.9 (C10′). FTIR (thin film) cm−1: 3445 (br m), 2936 (s), 1687 (s), 1628 (s), 1452 (m), 1315 (m), 1046(m). HRMS (ESI): calc’d for C28H37N2O2 [M+H]+: 433.2850, found: 433.2857. TLC (2.5% Et3N in [12.5% EtOAc in hexanes], Et3N neutralized silica gel), Rf: 0.20 (UV, CAM).
6.5.12. Hexacyclic Enone 47
To a solution of β-trimethylsilyloxy ketone 62 (32.6 mg, 64.3 μmol, 1 equiv) in dichloromethane (1.25 mL) at −78 °C was added titanium tetrachloride (1.0 M in dichloromethane, 96.5 μL, 96.5 μmol, 1.50 equiv). After 30 min, brine was added (4 mL) and the reaction mixture was diluted with ethyl acetate (6 mL) and allowed to warm to 23 °C. The layers were separated and the aqueous layer was extracted with ethyl acetate (3 × 5 mL). The combined organic layers were washed sequentially with saturated aqueous sodium bicarbonate solution (4 mL) and brine (4 mL), were dried over anhydrous sodium sulfate, were filtered, and were concentrated under reduced pressure. The resulting material was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 2.0 cm, ht. 14 cm; eluent: 2.5% Et3N and 20→30% EtOAc in hexanes) to afford enone 47 (22.4 mg, 84%) and cis-enone cis-47 (1.4 mg, 5%) as yellow solids. 1H and 13C signals for enone 47 were assigned with the aid of gCOSY, HSQC, and gHMBC analysis. 1H signals for cis-enone cis-47 were assigned with the aid of gCOSY analysis. Data for 47: 1H NMR (500 MHz, C6D6, 20 °C): 7.64 (q, J = 1.4 Hz, 1H, C1–H), 3.30 (dq, J = 13.9, 7.1 Hz, 1H, C11–Hx), 3.26–3.20 (m, 1H, C5a–H), 3.16–3.08 (m, 1H, C4a′–H), 3.00 (dq, J = 13.6, 7.4 Hz, C11–Hy), 2.75–2.52 (m, 3H, C3′–Hc, C3′–Ht, C4–Hc), 2.50 (d, J = 1.3 Hz, 3H, C10′–H), 2.50–2.35 (m, 5H, C4–Ht, C8–Hc, C11′–Hx, C11′–Hy, C7′–Hc), 2.32–2.19 (m, 2H, C8–Ht, C7′–Ht), 1.92 (dt, J = 11.7, 5.9 Hz, 1H, C5–Hc), 1.72 (dt, J = 11.7, 5.9 Hz, 1H, C4′–Hc), 1.63–1.51 (m, 2H, C7–Ht, C6′–Ht), 1.53 (t, J = 7.4 Hz, 3H, C12–H), 1.52–1.32 (m, 4H, C5–Ht, C6–Hc, C5′–Hc, C4′–Ht), 1.32–1.12 (m, 2H, C7–Hc, C6′–Hc), 1.09 (t, J = 7.6 Hz, 3H, C12′–H), 0.79–0.65 (m, 2H, C5′–Ht, C6–Ht). 13C NMR (125 MHz, C6D6, 20 °C): 194.1 (C3), 137.6 (C2a′), 134.3 (C2), 132.6 (C1), 131.0 (C3b), 126.0 (C8b), 123.5 (C1′), 121.1 (C8a), 120.2 (C7a′), 117.7 (C3a), 113.6 (C2′), 56.0 (C4a′), 55.8 (C5a), 37.8 (C5), 36.9 (C4′), 30.1 (C6), 30.0 (C5′), 28.9 (C3′), 28.5 (C4), 22.7 (C6′), 22.6 (C7), 20.7 (C7′), 20.3 (C8), 20.1 (C11), 19.1 (C11′), 16.9 (C12′), 16.6 (C12), 16.0 (C10′). FTIR (thin film) cm−1: 2928 (s), 1607 (s), 1426 (m), 1320 (m), 1035 (w), 734 (w). HRMS (ESI): calc’d for C28H37N2O [M+H]+: 417.2900, found: 417.2913. TLC (silica gel pretreated with Et3N, 2.5% Et3N in [25% EtOAc in hexanes]), Rf: 0.25 (UV, CAM). Data for cis-47: 1H NMR (500 MHz, C6D6, 20 °C): 6.54 (q, J = 1.4 Hz, 1H, C1–H), 3.33–3.16 (m, 2H, C11–Hx, C11–Hy), 3.11 (dd, J = 15.8, 8.2 Hz, 1H, C3′–Ht), 3.00–2.89 (m, 2H, C5a–H, C3′–Hc), 2.81 (dd, J = 15.9, 8.1 Hz, 1H, C4–Ht), 2.77–2.61 (m, 4H, C4–Hc, C4a′–H, C11′–Hx, C11′–Hy), 2.47 (dd, J = 16.2, 6.4 Hz, 1H, C8–Hc), 2.39 (dd, J = 16.6, 6.1 Hz, 1H, C7′–Hc), 2.36–2.27 (m, 1H, C7′–Ht), 2.16 (ddd, J = 17.0, 11.5, 6.3 Hz, 1H, C8–Ht), 1.86 (dt, J = 11.4, 5.8 Hz, 1H, C5–Hc), 1.74 (td, J = 11.7, 5.9 Hz, 1H, C4′–Hc), 2.25 (d, J = 1.3 Hz, 1H, C10′–H), 1.62 (t, J = 7.4 Hz, 3H, C12–H), 1.50–1.39 (m, 2H, C6′–Ht, C7–Ht), 1.38–1.31 (m, 2H, C4′–Ht, C5–Ht), 1.35 (t, J = 7.5 Hz, 3H, C12′–H), 1.29–1.13 (m, 2H, C6–Hc, C5′–Hc), 1.05–0.89 (m, 2H, C6′–Hc, C7–Hc), 0.74–0.61 (m, 2H, C5′–Ht, C6–Ht). TLC (silica gel pretreated with Et3N, 2.5% Et3N in [25% EtOAc in hexanes]), Rf: 0.44 (UV, CAM).
6.5.13. Tetracycle 67
To a solution of tricyclic enone 64 (8.4 mg, 34.0 μmol, 1 equiv) in dichloromethane (800 μL) at 23 °C was added trifluoromethanesulfonic acid (6.1 μL, 69.0 mmol, 2.00 equiv). After 6 h, 2-tert-butylimino-2-diethylamino-1,3-dimethyl-perhydro-1,3,2-diazaphosphorine on polystyrene (BEMP, 165 mg, 340 mmol, 10.00 equiv) was added. After 45 min, the reaction mixture filtered through a cotton plug and the orange filtrate was concentrated under reduced pressure. The resulting residue was purified by flash column chromatography (silica gel pretreated with triethylamine: diam. 1.0 cm, ht. 12 cm; eluent: 2.5% Et3N and 12.5% EtOAc in hexanes) to afford tetracycle 67 (1.4 mg, 16%) as a yellow oil. 1H and 13C signals were assigned with the aid of gCOSY, HSQC, and gHMBC analysis. 1H NMR (500 MHz, C6D6, 20 °C): 3.29 (dqd, J = 14.3, 7.5, 0.8 Hz, 1H, C11–Hx), 3.02 (dq, J = 9.9, 6.4 Hz, 1H, C4a–H), 2.94(s, 1H, C2-H), 2.72 (dqd, J = 14.5, 7.4, 1.7 Hz, 1H, C11–Hy), 2.10–2.00 (m, 2H, C6–Hx, C9–H), 1.98 (td, J = 14.9, 4.2 Hz, 1H, C6–Hy), 1.64 (dd, J =13.4, 5.3 Hz, 1H, C10–Hx), 1.55–1.47 (m, 2H, C4–Hx, C10–Hy), 1.42 (ddt, J = 13.6, 5.9, 4.0 Hz, 1H, C5–Hx), 1.39–1.31 (m, 1H, C3–Hx), 1.19 (t, J = 7.5 Hz, 3H, C12–H), 1.19–1.12 (m, 1H, C3–Hy), 1.03 (d, J = 7.4 Hz, 3H, C10′–H), 0.99–0.80 (m, 2H, C4–Hy, C5–Hy). 13C NMR (125 MHz, C6D6, 20 °C): 216.4 (C8), 195.4 (C7), 138.2 (C7a), 126.2 (C1), 78.5 (C2a), 63.4 (C2), 58.1 (C4a), 44.4 (C9), 42.7 (C10′), 37.8 (C6), 36.4 (C3), 32.5 (C4), 27.1 (C5), 20.2(C11), 17.1 (C10), 13.4 (C12). FTIR (thin film) cm−1: 2962 (s), 1737 (s), 1692 (s), 1599 (m), 1453 (m), 1198 (m), 1032 (w). HRMS (ESI): calc’d for C16H22NO2 [M+H]+: 260.1645, found: 260.1646. TLC (silica gel pretreated with Et3N, 2.5% Et3N in [12.5% EtOAc in hexanes]), Rf: 0.18 (UV, CAM).
Scheme 7.

Synthesis of heterodimer 27. Conditions: a) 28, LHMDS, THF, −78 °C, 15 min; 34, −78→ −40 °C, 1 h, 70%.
Acknowledgments
We are grateful for financial support by NIH-NIGMS (GM074825) and the corresponding ARRA Supplement. M.M. is an Alfred P. Sloan Research Fellow and a Camille Dreyfus Teacher-Scholar. A.E.O. acknowledges a Novartis Graduate Fellowship. We thank Professor Robert G. Griffin and Dr. Tony Bielecki for use of a high-field instrument at the MIT-Harvard Center for Magnetic Resonance (EB002026). We thank Dr. Li Li for obtaining mass spectrometric data at the Department of Chemistry’s Instrumentation Facility (MIT).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- i.Myrmicarins 215A, 215B, and 217: Schröder F, Franke S, Francke W, Baumann H, Kaib M, Pasteels JM, Daloze D. Tetrahedron. 1996;52:13539–13546.Myrmicarin 430A: Schröder F, Sinnwell V, Baumann H, Kaib M. Chem Commun. 1996;18:2139–2140.Myrmicarins 663 and 645: Schröder F, Sinnwell V, Baumann H, Kaib M, Francke W. Angew Chem Int Ed Engl. 1997;36:77–80.
- ii.(a) Kaib M, Dittebrand H. Chemoecology. 1990;1:3–11. [Google Scholar]; (b) Ritter FJ, Rotgans IEM, Talman E, Verwiel PEJ, Stein F. Experientia. 1973;29:530–531. [Google Scholar]; (c) Attygalle AB, Morgan ED. Chem Soc Rev. 1984;13:245–278. [Google Scholar]; (d) Blum MS. J Toxicol. 1992;11:115–164. [Google Scholar]; (e) Jones T, Blum MS, Fales HM. Tetrahedron. 1982;38:1949–1958. [Google Scholar]
- iii.For our previous studies on the total synthesis of complex myrmicarin alkaloids, see: Movassaghi M, Ondrus AE, Chen B. J Org Chem. 2007;72:10065–10074. doi: 10.1021/jo701981q.For our enantioselective total synthesis of the tricyclic myrmicarins, see: Movassaghi M, Ondrus AE. Org Lett. 2005;7:4423–4426. doi: 10.1021/ol051629f.For our discovery of Brønsted acid-promoted dimerization of M215B, see: Ondrus AE, Movassaghi M. Tetrahedron. 2005;62:5287–5297. doi: 10.1016/j.tet.2006.01.108.Ondrus AE, Movassaghi M. Chem Commun. 2009:4151–4165. doi: 10.1039/b903995n.
-
iv.In the presence of Brønsted acid, myrmicarin 215B (2) undergoes highly efficient and diastereoselective dimerization via sequential formation of the C2-C3 bond and intramolecular alkylation at C3b to yield the heptacyclic structure of isomyrmicarin 430A. See refs. iiia and 3c for details.
- v.For examples of dearomatizing radical cyclizations onto pyrroles, see: Jones K, Ho TCT, Wilkinson J. Tetrahedron Lett. 1995;36:6743–6744.Escolano C, Jones K. Tetrahedron Lett. 2000;41:8951–8955.Escolano C, Jones K. Tetrahedron. 2002;58:1453–1464.Gross S, Reissig HU. Org Lett. 2003;5:4305–4307. doi: 10.1021/ol0355581.Guindeuil S, Zard SZ. Chem Commun. 2006;6:665–667. doi: 10.1039/b516020k.
- vi.For examples of palladium-mediated cyclizations onto pyrroles, see: Bowie AL, Chambers CH, Trauner D. Org Lett. 2005;7:5207–5209. doi: 10.1021/ol052033v.Garg KN, Caspi DD, Stoltz BM. J Am Chem Soc. 2004;126:9552–9553. doi: 10.1021/ja046695b.For reviews on transition metal-catalyzed reactions involving pyrroles, see: Seregin HV, Gevorgyan V. Chem Soc Rev. 2007;36:1173–1193. doi: 10.1039/b606984n.Banwell MG, Goodwen TE, Ng S, Smith JA, Wong D. Eur J Org Chem. 2006;14:3043–3060.
- vii.Prepared in one step by electrophilic bromination of the corresponding C6 debrominated, C6–C7 saturated derivative (28, Scheme 6).
- viii.Movassaghi M, Ondrus AE. unpublished results. MIT; [Google Scholar]
- ix.Attempts to deprotonate at C2 in previous heterodimers were unsuccessful.
- x.Use of Lewis acids or alternative acylating agents, solvents, or reaction temperatures afforded 30 in low yield.
- xi.For an example of formation of an imidazolyl acyl cyanide using diethylcyanophosphonate, see: Nagra BS, Shaw G, Robinson DH. Chem Commun. 1985;8:459–460.For the use of diethylcyanophosphonate for the formation of aromatic amines and esters from the corresponding acids, see: Shioiri T, Yokoyama Y, Kasai Y, Yamada S. Tetrahedron. 1976;32:2211–2217.
- xii.Only one set of signals corresponding to a hexahydropyrroloindolizine substructure is observed in the 1H NMR spectrum of 42 in benzene-d6. Interestingly, 42 is not obtained following an aqueous ammonium chloride quench of the Claisen condensation reaction. Subjection of 42 to silica gel chromatography results in complete tautomerization and quantitatively returns 27.
- xiii.Subjection of 43 to conditions described for Heck reaction of vinyl phosphates failed to effect cyclization, instead returning 43 or diketone 27. Attempts to achieve intermolecular Heck reaction also failed to provide the corresponding products, consistent with the expectation that 43 would not be an optimal substrate for these transformations. Likewise, subjection of 44 to conditions known to effect cross coupling with aryl chlorides did not induce Heck cyclization.
- xiv.For recent reviews, see: Frontier AJ, Collison C. Tetrahedron. 2005;61:7577–7606.Pellissier H. Tetrahedron. 2005;61:6479–6517.Harmata M. Chemtracts. 2004;17:416–435.
- xv.Our prior studies (see ref 3) demonstrated that treatment of a benzene solution of myrmicarin 215B (2) with excess trifluoroacetic acid (50% v/v) generated a 3:2 mixture of two monomeric pyrrole ring-protonated compounds and no dimeric products. Heating this sample for 13 hours at 80 °C followed by quench with aqueous sodium bicarbonate solution at 23 °C quantitatively returned 2 with complete deuterium incorporation at C9 exclusively.
- xvi.Aldehyde 58 was prepared via Vilsmeir-Haack formylation of the C2 unsubstituted pyrrole 29 (Scheme 6) under conditions reported by Valleé and coworkers: Sayah B, Pelloux-Léon N, Milet A, Pardillos-Guindet J, Vallée Y. J Org Chem. 2001;66:2522–2525. doi: 10.1021/jo001769x.
- xvii.Downey CW, Johnson MW. Tetrahedron Lett. 2007;48:3559–3562. [Google Scholar]
- xviii.Ondrus AE, Movassaghi M. Org Lett. 2009;11:2960–2963. doi: 10.1021/ol9008552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- xix.Still WC, Kahn M, Mitra A. J Org Chem. 1978;43:2923–2925. [Google Scholar]
- xx.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518–1520. [Google Scholar]
- xxi.Sayah B, Pelloux-Léon N, Valleé Y. J Org Chem. 2000;65:2824–2826. doi: 10.1021/jo9918697. [DOI] [PubMed] [Google Scholar]