

Abstract
A method by which to accomplish formal threonine ligation has been developed. The method accomplishes ligations of two peptide domains. We have also demonstrated the ability to successfully ligate two independent glycopeptide domains.
1. Introduction
Glycoproteins are a class of naturally occurring biomacromolecules, which are biosynthesized through post-translational protein glycosylation. A great deal of effort has been directed toward the examination of the role that glycosylation plays in various critical protein functions, such as protein folding, proteolytic stability, and intercellular communication.1 Of particular interest is the fact that many glycoproteins possess exploitable therapeutic activity, and may serve as promising candidates in the development of vaccines,2 diagnostic techniques,3 and therapeutic agents.4 Prominent examples of therapeutically valuable glycoproteins include the erythropoietic agent, erythropoietin (EPO)4a,5 and the fertility agent, human follicle stimulating hormone (hFSH).6 Despite considerable interest in this class of biomacromolecules, the field of glycobiology faces a significant obstacle to the rigorous evaluation of glycoproteins: the isolation of significant quantities of homogeneous glycoproteins from natural sources is often prohibitively difficult, due to the fact that most naturally occurring glycoproteins are biosynthesized as heterogeneous mixtures of glycoforms.
Given the great difficulties associated with the isolation of homogeneous glycoproteins, we recognized that an opportunity for chemical synthesis might lay in the challenge of using total chemical synthesis to gain access to homogeneous glycoprotein samples.7 The biology of such agents could then be studied in further detail. Moreover, through chemical synthesis, it would be possible to gain access to fully synthetic analog glycoproteins, possessing targeted modifications in the carbohydrate or protein domains. Of course, such targets could not necessarily be obtained through purely biologic means, since they would lack the viable biosynthetic pathways for reaching the desired structural types. In the context of our glycoprotein synthesis program, we have targeted for total synthesis the clinically relevant glycoproteins, EPO and hFSH.8 Several years ago, at the outset of our EPO total synthesis endeavor, we identified a number of significant methodological shortcomings that would need to be addressed before a viable total synthesis effort could be launched.
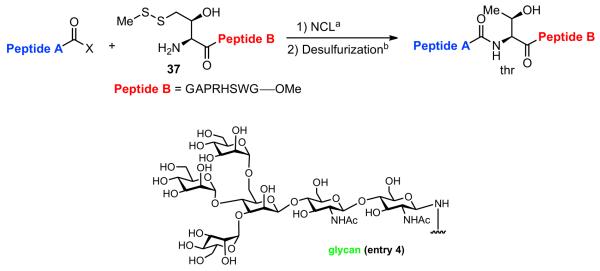
In particular, we were concerned with the dearth of general techniques and protocols available for the merging of two glycopeptide fragments. To address this issue, we developed an efficient method for the coupling of two differentially glycosylated peptide fragments. This advance was predicated on the landmark break-through of Kent and co-workers in the field of peptide synthesis, termed native chemical ligation (NCL).9 NCL is a widely used technique that allows for the merger of large peptide fragments, one of which presents a C-terminal thioester, and the other an N-terminal cysteine residue (Fig. 1a). Direct extension of the NCL method to the realm of glycopeptide synthesis is complicated by the difficulties associated with synthesizing pre-formed glycopeptide thioester. The solution developed in our laboratories (outlined in Fig. 1b) involves a relatively stable C-terminal ortho-thiophenolic ester, presented on one of the glycopeptides, and a protected N-terminal cysteine residue incorporated on the other fragment.10 Upon simultaneous reduction of the two disulfides, the phenol moiety undergoes intramolecular O→S migration, providing an intermediate thioester, which undergoes thioester exchange with the free cysteine of the second glycopeptide. The intermediate then suffers spontaneous intramolecular transfer to yield the bidomainal glycopeptide adduct, incorporating two differentiated sites of glycosylation. The general logic of this NCL method has more recently been extended to a direct oxo-ester variant, in which a phenolic ester equipped with para-NO2 or para-CN substitution, is sufficiently activated to undergo direct cysteine ligation without the need for the intermediacy of a thioester species.11
Figure 1.

(a) Native chemical ligation; (b) Glycopeptide NCL; (c) Free-radical-mediated reduction of Cys/Ala; (d) Proposed native chemical ligation at threonine.
These glycopeptide ligation protocols do suffer from an important limitation in terms of generalizability, in that they require the presence of a cysteine residue at the ligation site. In fact, there is often a paucity of cysteine sites in naturally occurring proteins and glycoproteins. The need for a menu of efficient cysteine-free ligation methods thus remains quite high.
A number of useful solutions to the cysteine ligation problem have been developed, many of which depend on the use of cysteine or a thiol-containing amino acid surrogate in the ligation step. Following thiol-mediated ligation, the cysteine or surrogate residue is converted to the desired amino acid. In this way, methionine ligation has been achieved through homo-cysteine coupling, followed by post-ligational methylation.12 Similarly, serine ligation has been accomplished through NCL followed by conversion of cysteine to serine.13 A number of cysteine-free ligation methods make use of a two-step ligation–desulfurization sequence. For instance, metal-based post-ligational thiol reduction has been used to accomplish formal alanine14 and phenylalanine15 ligations. Despite the appeal of such a strategy, we noted that traditional desulfurization methods suffer from a lack of substrate generality, due to the susceptibility of many common functional groups (particularly thiol moieties) to the standard harsh reduction conditions. Based on the disclosure by Hoffmann and Walling in the 1950s,16 we recently developed a very mild and chemoselective free-radical-mediated desulfurization strategy, which allows for the post-translational conversion of cysteine to alanine in complex glycopeptide and peptide settings (Fig. 1c).17 This method has proven to be tolerant of a wide range of functionalities, including sulfur-containing groups—such as Thz, Cys(Acm), biotin, and thioesters —as well as amino acids, including methionine, and even complex carbohydrate moieties. This overall alanine ligation strategy has now been successfully applied in the context of the synthesis of a complex glycopeptide fragment of erythropoietin (Ala1-Gly28).8c In addition, our mild sulfur reduction protocol has been employed to accomplish a formal valine ligation, through the coupling and post-ligational reduction of unnatural amino acid surrogates (γ-thiol valine18 or β-thiol valine18,19) to valine residues. Moreover, a dual native chemical ligation at lysine has also been achieved recently. A γ-thiol group on the N-terminal lysine mediates double chemical ligation at both α and ε amines, followed by a free-radical desulfurization.20
The development of a variety of methods by which to formally accomplish NCL at a diverse range of amino acid residues could be of substantial value. As noted above, due to its low frequency in nature, direct ligation at cysteine itself is of limited practical value. Furthermore, in developing a synthetic strategy toward a glycoprotein or glycopeptide target, it is often desirable to merge two pieces of relatively equal size. As the menu of amino acid ligation options expands, greater flexibility may be brought to the design of synthetic routes. Finally, the two-step ligation/reduction strategy provides the opportunity to explore the consequences of protein engineering with site-specifically modified glycoproteins.
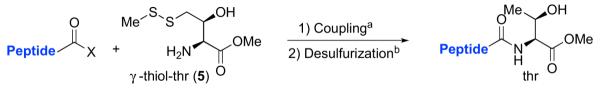
In the hopes of broadening the range of options for amino acid ligation, we sought to extend our two-step ligation/reduction protocol to the development of a formal threonine ligation. Threonine was selected for its relative abundance in nature, particularly in comparison with cysteine. We describe herein the discovery and formulation of a mild and efficient two-step formal threonine ligation protocol.
The central idea is outlined in Figure 1d. Thus, as in the case of the valine ligation,18 a thiol-containing threonine surrogate would be incorporated at the N-terminus of Peptide 2. One could envision two possible sites at which to install a thiol group onto the threonine residue: at the β position or the γ position. We expected that the γ-thiol threonine would serve as the more productive surrogate in establishing the initial acylation event. Peptide 2, incorporating the γ-thiol threonine, was expected to undergo trans-thioester-ification with Peptide 1 (presented as either a thioester or activated oxo-ester), to generate a thioester-linked intermediate, which would then undergo spontaneous intramolecular acyl transfer, generating the new amide bond. Subsequent radical-based desulfurization would serve to remove the thiol, providing the target peptide with threonine at the ligation site.
2. Results and discussion
Our first objective was to synthesize a γ-thiol threonine amino acid surrogate. To accomplish this, we drew on the earlier work of Rapoport and co-workers,21 in which allo-threonine was obtained from D-vinylglycine. Through modification of the Rapoport route, we sought to accomplish the diastereoselective syntheses of the γ-thiol threonine derivatives, 5 and 6 (Scheme 1). Compound 5 was to be used directly for single amino acid extension studies (see Table 1), while compound 6 would be elaborated to peptide 37, which would serve as the substrate in the threonine ligation studies at the peptide level (see Table 2). Thus, as shown in Scheme 1, vinylglycine 122 was epoxidized with an excess of mCPBA to afford a 5:1 ratio of syn and anti epoxides, 2a and 2b, which could be separated by chromatography. The major diastereomer, 2a, has the desired syn configuration, perhaps as a consequence of hydrogen bonding of mCPBA to the nitrogen functionality.23 Upon exposure to the sodium salt of thioacetic acid, epoxide 2a was opened to provide the acetylated thiol, 3. The latter was transformed, in a straightforward fashion, to the target compounds, 5 and 6.
Scheme 1.

Synthesis of g-thiol threonine.
Table 1.
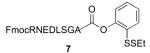
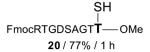
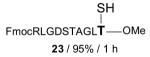
Threonine extension by coupling and subsequent free-radical desulfurization
 | |||
|---|---|---|---|
| Entry | Peptide | Coupling Product/Yield/Time | Desulfurization Product/Yield/Time |
| 1 |
|

|
FmocRNEDLSGAT—OMe 9 / 92 % / 2 h |
| 2 |
|

|
FmocRLGDSTAGQT—OMe 12 / 81% / 2 h |
| 3 |

|

|
FmocRLGDSTAGYT—OMe 15 / 89% / 2 h |
| 4 |

|

|
FmocRLGDSTAGWT—OMe 18 / 90% / 2 h |
| 5 |

|
|
FmocRTGDSAGTT—OMe 21 / 84% / 2 h |
| 6 |

|
|
FmocRLGDSTAGLT—OMe 24 / 80% / 2 h |
| 7 |
|
|
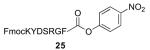
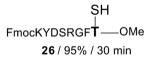
FmocKYDSRGFT—OMe 27 / 80% / 2 h |
| 8 |
|

|
FmocRTGDSAGVT—OMe 30 / 95% / 2 h |
| 9 |
|

|
FmocRTGDSAGIT—OMe 33 / 82% / 2 h |
| 10 |
|
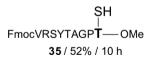
|
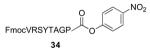
FmocVRSYTAGPT—OMe 36 / 83% / 2 h |
Key: (a) buffer at pH 6.5 (6.0 M Gn·HCl, 188.8 mM Na2HPO4), TCEP, rt (b) TCEP, VA-044, tBuSH, 37 °C. VA-044=2,2′-azobis-[2-(2-imidazolini-2-yl)propane] dihydrochloride.
TCEP=tris(2-carboxyethyl)phosphine.
Table 2.
NCL at threonine through ligation and subsequent free-radical desulfurization
 | |||
|---|---|---|---|
| Entry | Peptide A | Ligation Product/Yield/Time | Desulfurization Product/Yield/Time |
| 1 |

|
|
FmocRLGDSTAGYTGAPRHSWG—OMe 39 / 98% / 2 h |
| 2 |

|
|
FmocRLGDSTAGWTGAPRHSWG—OMe 41 / 85% / 2 h |
| 3 |
|
|
FmocRTGDSAGITGAPRHSWG—OMe 43 / 96% / 2 h |
| 4 |
|
|
|
Key: (a) buffer at pH 6.5 (6.0 M Gn·HCl, 188.8 mM Na2HPO4), TCEP, RT; (b) TCEP, VA-044, tBuSH, 37 °C.
With γ-thiol threonine derivative 5 in hand, we next sought to evaluate the feasibility of the proposed threonine ligation method. We first examined the protocol in the context of a single amino acid extension of a variety of peptide substrates. As shown in Table 1, when a relatively sterically less demanding amino acid (Ala, Gln, Tyr, Trp, Phe) was presented at the C-terminus of the peptide, the amino acid extension generally proceeded very quickly and with good yield (see entries 1–4 and 7). Even when the β-branched amino acid, threonine, was incorporated at the C-terminus, the coupling was complete within 1 h (entry 5). As expected, as the C-terminus became more sterically hindered (Leu, Val, Ile, Pro), the reaction rate suffered. However, coupling could still be accomplished within a reasonable time frame, and in moderate to good yields (entries 6, 8–10). As shown in Table 1, a variety of different C-terminal esters participated successfully in the extension protocol, including thiophenyl ester, ortho-thiophenolic ester, and para-nitrophenyl ester. The diversity of C-terminal esters amenable to this protocol is of significance, as the ortho-thiophenolic ester is compatible with glycopeptide ligation, while para-nitrophenyl ester is particularly efficient at promoting coupling at sterically hindered ligation sites. We also note that the threonine ligation protocol is able to accommodate the presence of an unprotected lysine in the substrate peptide (25, entry 7). All of the coupling products obtained in Table 1 were subsequently subjected to our standard radical-based desulfurization conditions to provide the desired threonine extension products in very good yields.
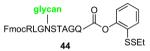
We now sought to examine the two-step ligation/reduction protocol in the context of a peptide-peptide coupling. Peptide 37, possessing the γ-thiol threonine surrogate at its N-terminus, was prepared from compound 6 (see Supplementary data for details). According to our general procedure, 37 and the peptide coupling partner were dissolved in a guanidine buffer solution. Upon addition of TCEP, the disulfide moieties were cleaved to presumably give rise to the free thiol functionalities, which then underwent the anticipated ligation reaction. As shown in Table 2, we found the ligation rate to be dependent on the nature of the C-terminal amino acid. Thus, when a less sterically demanding amino acid—such as Tyr (13) or Trp (16)—was present at the C-terminus, ligation was complete within 1 h (entries 1 and 2). However, in the case of the more hindered amino acid, Ile (31), the reaction took up to 7 h to reach completion (entry 3). Once more, in each case, subsequent free-radical-based desulfurization was readily achieved in high yields through use of our mild reduction method. Finally, as shown in Table 2, entry 4, our new protocol could be readily extended to a glycopeptide ligation setting. Thus, peptide 44, presenting an N-linked hexasaccharide domain, underwent ligation with peptide 37 to provide glycopeptide 45 in 82% yield. Upon exposure to our previously described reduction conditions, glycopeptide 46, incorporating threonine at the ligation site, was obtained in 96% yield.
3. Conclusion
In conclusion, we have described the development of a useful new entry in the field of native chemical ligation. Through an efficient two-step ligation/reduction protocol, it is now possible to formally achieve NCL at threonine sites, in both peptide and gly-copeptide settings. This methodological advance, taken in concert with the previous entries of our group and others, has served to significantly expand the NCL menu, which was originally restricted to cysteine-based ligations. Further applications and extensions of this method to the synthesis of important biologic level agents are underway in our laboratory.
4. Experimental section
4.1. General
Anhydrous THF, diethyl ether, CH2Cl2, toluene, and benzene were obtained from a dry solvent system (passed through column of alumina) and used without further drying. NMR spectra (1H and 13C) were recorded on a Bruker Advance DRX-500 MHz, or a Bruker DRX-600 MHz. Low-resolution mass spectral analyses were performed with a JOEL JMS-DX-303-HF mass spectrometer or Waters Micromass ZQ mass spectrometer.
HPLC: All separations of peptides and glycopeptides involved a mobile phase of 0.05% TFA (v/v) in water (solvent A)/0.04% TFA in acetonitrile (solvent B). Preparative and analytical HPLC separations were performed using a Rainin HPXL solvent delivery system equipped with a Rainin UV-1 detector. LC–MS chromatographic separations were performed using a Waters 2695 Separations Module and a Waters 996 Photodiode Array Detector equipped with X-Bridge™ C18 column (5.0 μm, 2.1×150 mm), X-Terra™ MS C18 column (3.5 μm, 2.1× 100.0 mm) or Varian Microsorb C18 column (2×150mm) at a flow rate of 0.2 mL/min. HPLC separations were performed using: X-Bridge™ Prep C18 column OBD™ (5.0 mm, 19×150 mm), a flow rate of 16 mL/min. Microsorb 100-5 C18 column at a flow rate of 16.0 mL/min or Microsorb 300-5 C4 column at a flow rate of 16.0 mL/min.
Automated peptide synthesis was performed on an Applied Biosystems Pioneer continuous flow peptide synthesizer. Peptides were synthesized under standard automated Fmoc protocols. The deblock mixture was a mixture of 100/5/5 of DMF/piperidine/DBU. Upon completion of automated synthesis on a 0.05 mmol scale, the peptide resin was washed into a peptide synthesis vessel with CH2Cl2. The resin cleavage was effected by treatment with AcOH/ TFE/CH2Cl2 (2:2:6) for 2 ×1 h to yield peptidyl acids in good yield. The peptidyl acids were modified on C-terminus and/or N-terminus. The resulting peptides were subjected to a deprotection cocktail (60.0 mg of phenol, 0.2 ml of water, 0.15 ml of triisopro-pylsilane, and 3.0 ml TFA) for 2.0 h. TFA was removed by N2. The oily residue was triturated with diethyl ether to give a white suspension, which was centrifuged and the ether subsequently decanted. The resulting solid was ready for HPLC purification.
4.2. Preparation and characterization of compounds 2a–6
4.2.1. (S)-Methyl 2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-2-((S)-oxiran-2-yl) acetate (2a)
To a stirred solution of 1 (200 mg, 0.593 mmol) in CH2Cl2 (6 mL) at 0 °C was added m-chloroperbenzoic acid (1.022 g, 5.93 mmol) and the reaction was warmed to rt. After 24 h, the mixture was filtered through a glass filter, the solids were extracted with CH2Cl2, and the combined organic phase was washed with 10% NaHCO3, water, dried, and evaporated. The two diastereomers were then separated by chromatography (CH2Cl2/ MeOH=300:1) to give 2a (138 mg, 67% yield) as a white solid. 1HNMR (500 MHz, CDCl3) δ 2.59 (m,1H), 2.76 (m,1H), 3.46 (s, 1H), 3.81 (s, 3H), 4.20 (t, J=6.8 Hz, 1H), 4.40 (d, J=6.9 Hz, 2H), 4.70 (dd, J= 1.5 Hz, 8.9 Hz, 1H), 5.23 (d, J=8.8 Hz, 1H), 7.29 (m, 2H), 7.32 (t, J=6.3 Hz, 2H), 7.56 (t, J=6.0 Hz, 2H), 7.76 (d, J=6.3 Hz, 2H); 13C NMR (125 MHz, CDCl3) δ 43.9, 47.1, 51.2, 53.0, 53.1, 67.2, 120.1, 125.1, 127.1, 127.1, 127.8, 141.3, 141.4, 143.6, 143.8, 156.2, 170.2; −5.21 (c 1.08, CHCl3); IR (liquid film) (vmax/cm−1): 3342, 3066, 3019, 2953, 2848, 1724; HRMS: m/e calcd for C20H19NO5Na+: 376.1161, found: 376.1153.
4.2.2. (2S,3S)-Methyl 2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-4-(acetylthio)-3-hydroxybutanoate (3)
Compound 2a was dissolved in toluene (3.2 mL) and treated with a solution of sodium acetate (88 mg, 1.077 mmol) and thioacetic acid (80 μL, 1.077 mmol) in DMF (1.6 mL). The reaction mixture was stirred at rt for 2 h. The mixture was concentrated under N2. The concentrate was dissolved in EtOAc and washed with NH4Cl, water, and brine. The organic layer was dried, concentrated, and purified by chromatography (Hexane/EtOAc=2:1) to give 3 (72 mg, 80%) as a light yellow foam. 1H NMR (500 MHz, CDCl3) δ 2.35 (s, 3H), 3.01 (m, 3H), 3.75 (s, 3H), 4.23 (m, 2H), 4.41 (d, J=7.0 Hz, 2H), 4.51 (d, J=9.3 Hz, 1H), 5.69 (d, J9.4 Hz, 1H), 7.31 (m, 2H), 7.39 (m, 2H), 7.61 (m, 2H), 7.75 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 30.6, 32.8, 47.2, 52.8, 57.3, 67.3, 71.5, 120.0, 120.0, 125.1, 125.2, 127.1, 127.8, 141.3, 141.4, 143.6, 143.8, 156.7, 170.9, 196.6; 11.69 (c 0.68, CHCl3); IR (liquid film) (vmax/cm−1): 3373, 3065, 3019, 2952, 2847, 1747, 1722, 1697; HRMS: m/e calcd for C22H23NO6SNa+: 452.1144, found: 452.1155.
4.2.3. (2S,3S)-Methyl 2-(((9H-fluoren-9-yl)methoxy)carbonylamino)-3-hydroxy-4-(methyldisulfanyl) butanoate (4)
Compound 3 (47 mg, 0.11 mmol) was dissolved in MeOH (1.87 mL) and treated with 0.2 N NaOH solution (1.87 mL) at 0 °C for 20 min. The reaction mixture was carefully neutralized by the addition of 1 N HCl at 0 °C, diluted with EtOAc and washed with water and brine. The organic layer was concentrated and dried in vacuo, generating 43 mg of crude compound (2S,3S)-methyl 2-(((9H-fluoren-9-yl)methoxy)-carbonylamino)-3-hydroxy-4-mercaptobutanoate, which was directly used in the next step.
S-Methyl methanethiolsulfonate (36 μL, 0.38 mmol) and DIEA (12 mL, 0.11 mmol) were added to CH2Cl2 (0.5 mL). The crude residue of (2S,3S)-methyl 2-(((9H-fluoren-9-yl)methoxy)carbonyl-amino)-3-hydroxy-4-mercaptobutanoate in CH2Cl2 (0.8 mL) was added dropwise to the above solution and stirred at rt for 2 h. The reaction mixture was concentrated and purified by chromatography (Hexane/EtOAc=2:1), to provide 4 (33 mg, 70% in two steps) as a light yellow foam. 1H NMR (500 MHz, CDCl3) δ 2.42 (s, 3H), 2.65 (dd, J=9.2 Hz 14.2 Hz, 1H), 2.71 (d, J=2.5 Hz, 1H), 2.85 (dd, J=3.8 Hz, 14.0 Hz, 1H), 3.79 (s, 3H), 4.22 (t, J=6.8 Hz, 1H), 4.43 (m, 3H), 4.51 (d, J=9.4 Hz, 1H), 5.58 (d, J=9.4 1H), 7.29 (m, 2H), 7.37 (t, J=7.1 Hz, 2H), 7.60 (t, J=7.1 Hz, 2H), 7.75 (m, 2H); 13C NMR (125 MHz, CDCl3) δ 22.9, 41.4, 47.2, 52.9, 57.1, 67.2, 69.9, 120.0, 120.1, 125.1, 127.1, 127.8, 141.3, 141.4, 143.6, 143.8, 156.7, 170.9; 48.90 (c 0.27, CHCl3); IR (liquid film) (vmax/cm−1): 3406, 3019, 2952, 2918, 2850, 1724; HRMS: m/e calcd for C21H23NO5S2Na+: 456.0915, found: 456.0909.
4.2.4. (2S,3S)-Methyl 2-amino-3-hydroxy-4-(methyldisulfanyl)butanoate (5)
To a stirred solution of 4 (57 mg, 0.131 mmol) in DMF (4.3 mL) was added diethylamine (1.4 mL). The reaction mixture was stirred at rt for 2 h and the solvent was evaporated. The residue was purified by chromatography (CH2Cl2/MeOH 40:1) to give 5 (25 mg, 89% yield) as a clear oil. 1H NMR (500 MHz, CDCl3) δ 2.43 (s, 3H), 2.84 (dd, J=6.0 Hz, 13.9 Hz, 1H), 2.92 (dd, J=7.0 Hz, 13.9 Hz, 1H), 3.66 (d, J=3.1 Hz, 1H), 3.76 (s, 3H), 4.17 (m, 1H); 13C NMR (125 MHz, CDCl3) d 23.1, 41.7, 52.5, 56.2, 70.6, 174.3; 76.7 (c 0.3, CHCl3); IR (liquid film) (vmax/cm−1): 3366, 3303, 2952, 2918, 1738, 1437, 1245, 1174, 1022; HRMS: m/e calcd for C6H13NO3S2H+: 212.0415, found: 212.0413.
4.2.5. (2S,3S)-2-(tert-Butoxycarbonylamino)-3-hydroxy-4-(methyldisulfanyl) butanoic acid (6)
To a solution of 5 (28 mg, 0.133 mmol) and Boc2O (57.8 mg, 0.265 mmol) in THF (0.7 mL) and MeOH (0.5 mL) was added TEA (0.056 mL, 0.399 mmol). The mixture was stirred for 2 h at room temperature. The reaction mixture was extracted with EtOAc three times. Combined organic layers were dried and purified by chromatography (Hex/EtOAc 2:1) to give (2S,3S)-methyl 2-(tert-butoxycarbonylamino)-3-hydroxy-4-(methyl-disulfanyl) butanoate (30 mg, 75% yield) as a clear oil. 1H NMR (500 MHz, CDCl3) d 1.43 (s, 9H), 2.42 (s, 3H), 2.71 (m, 1H), 2.88 (m, 1H), 3.78 (s, 3H), 4.42 (d, J=8.8 Hz, 2H), 5.32 (d, J=8.4 Hz, 1H); 13C NMR (125 MHz, CDCl3) δ 22.9, 28.3, 41.6, 52.8, 56.7, 70.0, 80.4, 156.1, 171.2; 59.84 (c 0.37, CHCl3); IR (liquid film) (vmax/cm−1): 3407, 2979, 2918, 1752, 1716; HRMS: m/e calcd for C11H21NO5S2Na+: 334.0759, found: 334.0749.
(2S,3S)-methyl 2-(tert-butoxycarbonylamino)-3-hydroxy-4-(methyldisulfanyl) butanoate (20 mg, 0.064 mmol) was dissolved in THF (1 mL) and treated with 1 N NaOH solution (1 mL). The mixture was stirred for 2 h at rt. The reaction mixture was washed with Et2O and the aqueous layer was adjusted to pH 3 using 1 N HCl, and extracted with Et2O three times. The combined organic layer was dried and purified by chromatography (CH2Cl2/ MeOH=20:1 to 15:1) to give 6 (14 mg, 74% yield).*1H NMR (500 MHz, CDCl3) δd 1.39 (s, 9H), 2.38 (s, 3H), 2,70 (m, 1H), 2.84 (m, 1H), 4.44 (m, 2H), 5.45 (d, J=8.8 Hz, 1H); 13C NMR (125 MHz, CDCl3) d 23.0, 28.3, 41.4, 56.6, 70.0, 80.7, 156.4, 174.7; 37.56 (c 0.28, CHCl3); IR (liquid film) (vmax/cm−1): 3393, 2979, 2929, 2853, 1710, 1691; HRMS: m/e calcd for C10H19NO5S2Na+: 320.0602, found: 320.0602.
4.3. General procedure for native chemical ligation at threonine site
To a solution of Peptide A (1.0 equiv, 4 mmol) and Peptide B (1.5 equiv, 6 μmol) in 0.5 mL of Guanidine buffer‡ was added 0.5 M bond-breaker® TCEP solution (Pierce) (5.0 equiv, 20 mmol). The reaction mixture was stirred at room temperature. The reactions were monitored by LC–MS and purified directly by HPLC upon consumption of the starting material.
4.4. General procedure for desulfurization
To a solution of the peptide (0.6 mM) in 200.0 mL of water (or buffer) and 100.0 mLofCH3CN were added 200.0 μL of 0.5 M bond-breaker® TCEP solution (Pierce), 20.0 μL of 2-methyl-2-propanethiol and 10.0 mL of radical initiator (0.1 M in water). The reaction mixture was stirred at 37 °C. The reactions were monitored by LC– MS and purified directly by HPLC upon consumption of the starting material.
4.4.1. MS characterization of peptides 7–46
Compound 7. ESIMS calcd for C56H74N12O17S2 [M+H]+ m/z=1251.48, found: 1251.60.
Compound 8. ESIMS calcd for C53H75N13O19S [M+H]+ m/z=1230.51, found: 1231.12.
Compound 9. ESIMS calcd for C53H75N13O19 [M+H]+ m/z=1198.54, found: 1198.88.
Compound 10. ESIMS calcd for C58H79N13O17S2 [M+H]+ m/z=1294.53, found: 1294.79.
Compound 11. ESIMS calcd for C55H79N14O19S [M+H]+ m/z=1273.55, found: 1274.08.
Compound 12. ESIMS calcd for C55H80N14O19 [M+H]+ m/z=1241.58, found: 1241.91.
Compound 13. ESIMS calcd for C60H76N12O16S [M+H]+ m/z=1253.53, found: 1254.20.
Compound 14. ESIMS calcd for C59H81N13O19S [M+H]+ m/z=1308.56, found: 1308.92.
Compound 15. ESIMS calcd for C59H81N13O19 [M+H]+ m/z=1276.59, found: 1277.07.
Compound 16. ESIMS calcd for C62H77N13O15S [M+H]+ m/z=1276.55, found: 1276.81.
Compound 17. ESIMS calcd for C61H82N14O18S [M+H]+ m/z=1331.58, found: 1331.93.
Compound 18. ESIMS calcd for C61H82N14O18 [M+H]+ m/z=1299.60, found: 1300.02.
Compound 19. ESIMS calcd for C49H63N11O15S [M+H]+ m/z=1078.43 [M+2H]2+ m/z=539.72, found: 1078.58, 540.12.
Compound 20. ESIMS calcd for C48H68N12O18S [M+H]+ m/z=1133.46, found: 1134.01.
Compound 21. ESIMS calcd for C48H68N12O18 [M+H]+ m/z=1101.49, found: 1101.83.
Compound 22. ESIMS calcd for C57H78N12O15S [M+H]+ m/z=1203.55, found: 1204.02.
Compound 23. ESIMS calcd for C56H83N13O18S [M+H]+ m/z=1258.58, found: 1259.00.
Compound 24. ESIMS calcd for C56H83N13O18 [M+H]+ m/z=1226.61, found: 1227.09.
Compound 25. ESIMS calcd for C60H70N12O16 [M+H]+ m/z=1215.51, [M+2H]2+ m/z=608.26; found: 1216.04, 608.71.
Compound 26. ESIMS calcd for C59H76N12O16S [M+H]+ m/z=1241.52, [M+2H]2+ m/z=621.27; found: 1241.94, 621.63.
Compound 27. ESIMS calcd for C59H76N12O16 [M+H]+ m/z=1209.56, [M+2H]2+ m/z=605.29; found: 1210.14, 605.69.
Compound 28. ESIMS calcd for C50H64N12O17 [M+H]+ m/z=1105.46, found: 1105.80.
Compound 29. ESIMS calcd for C49H70N12O17S [M+H]+ m/z=1131.48, [M+2H]2+ m/z=566.25; found: 1131.87, 566.81.
Compound 30. ESIMS calcd for C49H70N12O17 [M+H]+ m/z=1099.51, found: 1099.86.
Compound 31. ESIMS calcd for C51H66N12O17 [M+H]+ m/z=1119.48, found: 1120.10.
Compound 32. ESIMS calcd for C50H72N12O17S [M+H]+ m/z=1145.50, [M+2H]2+ m/z=573.26; found: 1145.93, 573.68.
Compound 33. ESIMS calcd for C50H72N12O17 [M+H]+ m/z=1113.52, found: 1113.87.
Compound 34. ESIMS calcd for C58H72N12O16 [M+H]+m/z=1193.53, found: 1193.59.
Compound 35. ESIMS calcd for C57H78N12O16S [M+H]+ m/z=1219.55, found: 1220.01.
Compound 36. ESIMS calcd for C57H78N12O16 [M+H]+ m/z=1187.57, found: 1188.02.
Compound 37. ESIMS calcd for C44H65N15O12S2 [M+H]+ m/z=1060.45, found: 1060.44.
Compound 38. ESIMS calcd for C97H133N27O28S [M+2H]2+ m/z=1078.99, [M+3H]3+ m/z=719.66, found: 1079.34, 719.96.
Compound 39. ESIMS calcd for C97H133N27O28 [M+2H]2+ m/z=1063.00, [M+3H]3+ m/z=709.00, found: 1063.35, 709.23.
Compound 40. ESIMS calcd for C99H134N28O27S[M+2H]2+ m/z=1090.50, [M+3H]3+ m/z=727.33, found: 1090.91, 727.57.
Compound 41. ESIMS calcd for C99H134N28O27 [M+2H]2+ m/z=1074.50, [M+3H]3+ m/z=716.68, found: 1074.79, 716.97.
Compound 42. ESIMS calcd for C88H124N26O26S [M+2H]2+ m/z=997.46, [M+3H]3+ m/z=665.31, found: 997.82, 665.60.
Compound 43. ESIMS calcd for C88H124N26O26 [M+2H]2+ m/z=981.47, [M+3H]3+ m/z=654.65, found: 981.72, 654.97.
Compound 44. ESIMS calcd for C98H146N16O46S2 [M+2H] + m/z=1174.46, found: 1175.42.
Compound 45. ESIMS calcd for C133H199N31O57S [M+2H]2+ m/z=1588.18, [M+3H]3+ m/z=1059.12, found: 1588.27, 1059.25.
Compound 46. ESIMS calcd for C133H199N31O57 [M+2H]2+ m/z=1572.19, [M+3H]3+ m/z=1048.46, found: 1572.66, 1048.74.
Supplementary Material
Acknowledgements
Support for this research was provided by the National Institutes of Health (CA28824 to SJD). We thank Prof. W.F. Berkowitz for helpful discussions. Special thanks go to Ms. Rebecca Wilson for editorial advice and consultation and to Ms. Dana Ryan for assistance with the preparation of the manuscript. We thank Dr. George Sukenick, Ms. Hui Fang, and Ms. Sylvi Rusli of the Sloan-Kettering Institute’s NMR core facility for mass spectral and NMR spectroscopic analysis.
Footnotes
Guanidine buffer: To 1.0 mL of 6.0 M guanidine buffer, were added 26.8 mg of Na2HPO4. The pH value of resulting solution was nearly 6.5.
Supplementary data Experimental procedures, NMR spectra, LC–MS spectra, compound characterization. Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.tet.2010.01.067.
References and notes
- 1(a).Imperiali B, O’Connor SE, Hendrickson T, Kellenberger C. Pure Appl. Chem. 1999;71:777–787. [Google Scholar]; (b) Lis H, Sharon N. Eur. J. Biochem. 1993;218:1–27. doi: 10.1111/j.1432-1033.1993.tb18347.x. [DOI] [PubMed] [Google Scholar]; (c) Rudd PM, Elliott T, Cresswell P, Wilson IA, Dwek RA. Science. 2001;291:2370–2376. doi: 10.1126/science.291.5512.2370. [DOI] [PubMed] [Google Scholar]; (d) Bertozzi CR, Kiessling LL. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 2.For selected examples, see Calarese DA, Scanlan CN, Zwick MB, Deechongkit S, Mimura Y, Kunert R, Zhu P, Wormald MR, Stanfield RL, Roux KH, Kelly JW, Rudd PM, Dwek RA, Katinger H, Burton DR, Wilson IA. Science. 2003;300:2065–2071. doi: 10.1126/science.1083182. Mensdorff-Pouilly S. von, Snijdewint FG, Verstraeten AA, Verheijen RH, Kenemans P. Int. J. Biol. Markers. 2000;15:343–356. doi: 10.1177/172460080001500413. Buskas T, Ingale S, Boons G. J. Angew. Chem., Int. Ed. 2005;44:5985–5988. doi: 10.1002/anie.200501818. Keding SJ, Danishefsky SJ. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11937–11942. doi: 10.1073/pnas.0401894101. Zhu JL, Wan Q, Ragupathi G, George CM, Livingston PO, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:4151–4158. doi: 10.1021/ja810147j.
- 3.For selected examples, see Dudkin VY, Miller JS, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:736–738. doi: 10.1021/ja037988s. Peracaula R, Tabares G, Royle L, Harvey DJ, Dwek RA, Rudd PM, de Llorens R. Glycobiology. 2003;13:457–470. doi: 10.1093/glycob/cwg041.
- 4.For selected examples, see Ridley DM, Dawkins F, Perlin E. J. Natl. Med. Assoc. 1994;86:129–135. Durand G, Seta N. Clin. Chem. 2000;46:795–805. Koeller KM, Wong CH. Nat. Biotechnol. 2000;18:835–841. doi: 10.1038/78435. Armitage JO. Blood. 1998;92:4491–4508.
- 5(a).Szymkowski DE. Curr. Opin. Drug Discov. Devel. 2005;8:590–600. [PubMed] [Google Scholar]; (b) Pavlou AK, Rechert JM. Nat. Biotechnol. 2004;22:1513–1519. doi: 10.1038/nbt1204-1513. [DOI] [PubMed] [Google Scholar]; (c) Jelkmann W, Wagner K. Ann. Hematol. 2004;83:673–686. doi: 10.1007/s00277-004-0911-6. [DOI] [PubMed] [Google Scholar]
- 6(a).Pang SC. Women’s Health. 2005;1:87–95. doi: 10.2217/17455057.1.1.87. [DOI] [PubMed] [Google Scholar]; (b) Herbert DC. Am. J. Anat. 1975;144:379–385. doi: 10.1002/aja.1001440311. [DOI] [PubMed] [Google Scholar]; (c) Dada MO, Campbell GT, Blake CA. Endocrinology. 1983;113:970–984. doi: 10.1210/endo-113-3-970. [DOI] [PubMed] [Google Scholar]; (d) Rathnam P, Saxena BB. J. Biol. Chem. 1975;250:6735–6746. [PubMed] [Google Scholar]; (e) Saxena BB, Rathnam P. J. Biol. Chem. 1976;251:993–1005. [PubMed] [Google Scholar]
- 7.For selected reviews on peptide ligations and glycoprotein synthesis, see: Hackenberger CPR, Schwarzer D. Angew. Chem., Int. Ed. 2008;47:10030–10074. doi: 10.1002/anie.200801313. Gamblin DP, Scanlan EM, Davis BG. Chem. Rev. 2009;109:131–163. doi: 10.1021/cr078291i. Pratt MR, Bertozzi CR. Chem. Soc. Rev. 2005;34:58–68. doi: 10.1039/b400593g. Brik A, Wong C-H. Chem.d Eur. J. 2007;13:5670–5675. doi: 10.1002/chem.200700330. For selected examples of recent syntheses or semisyntheses of homogeneous protein and glycoproteins, see:Piontek C, Silva DV, Heinlein C, Pohner C, Mezzato S, Ring P, Martin A, Schmid FX, Unverzagt C. Angew. Chem., Int. Ed. 2009;48:1941–1945. doi: 10.1002/anie.200804735. Becker CFW, Liu X, Olschewski D, Castelli R, Seidel R, Seeberger PH. Angew. Chem., Int. Ed. 2008;47:8215–8219. doi: 10.1002/anie.200802161. Pentelute BL, Gates ZP, Dashnau JL, Vanderkooi JM, Kent SBH. J. Am. Chem. Soc. 2008;130:9702–9707. doi: 10.1021/ja801352j. Yamamoto N, Tanabe Y, Okamoto R, Dawson PE, Kajihara Y. J. Am. Chem. Soc. 2008;130:501–510. doi: 10.1021/ja072543f. Kochendoerfer GG, Chen S-Y, Mao F, Cressman S, Traviglia S, Shao H, Hunter CL, Low DW, Cagle EN, Carnevali M, Gueriguian V, Keogh PJ, Porter H, Stratton SM, Wiedeke MC, Wilken J, Tang J, Levy JJ, Miranda LP, Crnogorac MM, Kalbag S, Botti P, Schindler-Horvat J, Savatski L, Adamson JW, Kung A, Kent SBH, Bradburne JA. Science. 2003;299:884–887. doi: 10.1126/science.1079085.
- 8.Significant progress has been made toward the syntheses of both of these complex glycoproteins.For recent progress toward EPO, see: Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424–5431. doi: 10.1021/ja808704m. Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5432–5437. doi: 10.1021/ja808705v. Kan C, Trzupek JD, Wu B, Wan Q, Chen G, Tan Z, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5438–5443. doi: 10.1021/ja808707w. For recent progress toward hFSH, see:Nagorny P, Fasching B, Li X, Chen G, Fasching B, Aussedat B, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5792–5799. doi: 10.1021/ja809554x.
- 9.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 10.Warren JD, Miller JS, Keding SJ, Danishefsky SJ. J. Am. Chem. Soc. 2004;126:6576–6578. doi: 10.1021/ja0491836. [DOI] [PubMed] [Google Scholar]
- 11.Wan Q, Chen J, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2008;130:15814–15816. doi: 10.1021/ja804993y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12(a).Tam JP, Yu Q. Biopolymers. 1998;46:319–327. doi: 10.1002/(SICI)1097-0282(19981015)46:5<319::AID-BIP3>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]; (b) Pachamuthu K, Schmidt RR. Synlett. 2003:659–662. [Google Scholar]; (c) Saporito A, Marasco D, Chambery A, Botti P, Pedone C, Ruvo M. Biopolymers. 2006;83:508–518. doi: 10.1002/bip.20582. [DOI] [PubMed] [Google Scholar]
- 13.Okamoto R, Kajihara Y. Angew. Chem., Int. Ed. 2008;47:5402–5406. doi: 10.1002/anie.200801097. [DOI] [PubMed] [Google Scholar]
- 14.Yan LZ, Dawson PE. J. Am. Chem. Soc. 2001;123:526–533. doi: 10.1021/ja003265m. [DOI] [PubMed] [Google Scholar]
- 15.Crich D, Banerjee A. J. Am. Chem. Soc. 2007;129:10064–10065. doi: 10.1021/ja072804l. [DOI] [PubMed] [Google Scholar]
- 16(a).Hoffmann FW, Ess RJ, Simmons TC, Hanzel RS. J. Am. Chem. Soc. 1956;78:6414. [Google Scholar]; (b) Walling C, Rabinowitz R. J. Am. Chem. Soc. 1957;79:5326. [Google Scholar]; (c) Walling C, Basedow OH, Savas ES. J. Am. Chem. Soc. 1960;82:2181–2184. [Google Scholar]
- 17.Wan Q, Danishefsky SJ. Angew. Chem., Int. Ed. 2007;46:9248–9252. doi: 10.1002/anie.200704195. [DOI] [PubMed] [Google Scholar]
- 18.Chen J, Wan Q, Yuan Y, Zhu JL, Danishefsky SJ. Angew. Chem., Int. Ed. 2008;47:8521–8524. doi: 10.1002/anie.200803523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haase C, Rohde H, Seitz O. Angew. Chem., Int. Ed. 2008;47:6807–6810. doi: 10.1002/anie.200801590. [DOI] [PubMed] [Google Scholar]
- 20.Yang R, Pasunooti KK, Li F, Liu X-W, Liu C-F. J. Am. Chem. Soc. 2009;131:13592–13593. doi: 10.1021/ja905491p. [DOI] [PubMed] [Google Scholar]
- 21.Shaw KJ, Luly JR, Rapoport HJ. Org. Chem. 1985;50:4515–4523. [Google Scholar]
- 22(a).Organ MG, Xu J, N’Zemba B. Tetrahedron Lett. 2002;43:8177–8180. [Google Scholar]; (b) Afzali-Ardakani A, Rapoport H. J. Org. Chem. 1980;45:4817–4820. [Google Scholar]; (c) Itaya T, Shimizu S, Nakagawa S, Morisue M. Chem. Pharm. Bull. 1994;42:1927–1930. [Google Scholar]
- 23(a).Narula AS. Tetrahedron Lett. 1983;24:5421–5424. [Google Scholar]; (b) Wade PA, Singh SM, Pillay MK. Tetrahedron. 1984;40:601–611. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.