

Abstract
Background and purpose:
Obovatol isolated from the medicinal herb Magnolia obovata exhibits a variety of biological activities. Here, the effect of obovatol and its mechanism of action on microglial activation, neuroinflammation and neurodegeneration were investigated.
Experimental approach:
In microglial BV-2 cells stimulated with lipopolysaccharide (LPS), we measured nitric oxide (NO) and cytokine production, and activation of intracellular signalling pathways by reverse transcription-polymerase chain reaction and Western blots. Cell death was assayed in co-cultures of activated microglia (with bacterial LPS) and neurons and in LPS-induced neuroinflammation in mice in vivo.
Key results:
Obovatol inhibited microglial NO production with an IC50 value of 10 µM. Obovatol also inhibited microglial expression of proinflammatory cytokines and inducible nitric-oxide synthase, which was accompanied by the inhibition of multiple signalling pathways such as nuclear factor kappa B, signal transducers and activators of transcription 1, and mitogen-activated protein kinases. In addition, obovatol protected cultured neurons from microglial toxicity and inhibited neuroinflammation in mice in vivo. One molecular target of obovatol in microglia was peroxiredoxin 2 (Prx2), identified by affinity chromatography and mass spectrometry. Obovatol enhanced the reactive oxygen species (ROS)-scavenging activity of Prx2 in vitro, thereby suppressing proinflammatory signalling pathways of microglia where ROS plays an important role.
Conclusions and implications:
Obovatol is not only a useful chemical tool that can be used to investigate microglial signalling, but also a promising drug candidate against neuroinflammatory diseases. Furthermore, our results indicate that Prx2 is a novel drug target that can be exploited for the therapeutic modulation of neuroinflammatory signalling.
Keywords: neuroinflammation, microglia, neuroprotection, chemical genetics, peroxiredoxin 2, obovatol
Introduction
Traditionally, the central nervous system (CNS) has been considered as an immunologically privileged region and inflammation was viewed only as a passive response to neuronal damage. However, an increasing number of reports suggest that the CNS is immunologically specialized and inflammation plays an active role in neurodegenerative disease progression (Block et al., 2007). Several cell types in the CNS have been shown to be contributors to inflammation-mediated neurodegeneration, but microglia, the resident innate immune cells in the brain, have been implicated as active contributors to neuronal damage in neurodegenerative diseases. In the mature brain, microglia exist in a resting state characterized by ramified morphology and are responsible for host defence and tissue repair (Nimmerjahn et al., 2005). However, once microglia are activated in response to brain injury or immunological stimuli, they undergo morphological changes into an amoeboid type. Overactivated microglia produce various proinflammatory cytokines and neurotoxic compounds such as tumour necrosis factor (TNF)α, interleukin (IL)-1β, IL-6, reactive oxygen and nitrogen species. These molecules are considered to contribute to neuronal damage and neurodegenerative processes (Block et al., 2007; McGeer and McGeer, 2003). The expression of these cytokines, nitric oxide (NO) and other inflammatory mediators is regulated at many levels. Protein kinases [such as protein kinase C, protein kinase A, protein tyrosine kinases and mitogen-activated protein kinases (MAPKs)] and transcription factors [such as nuclear factor κB (NF-κB), activator protein-1 (AP-1) and signal transducers and activators of transcription (STATs)] play important roles in the signalling pathways that lead to the inflammatory activation of microglia (Jongeneel, 1995; Hellendall and Ting, 1997; Bhat et al., 1998; Kleinert et al., 2004). Thus, based on the precise understanding of microglial signalling pathways, inhibition of microglial activation and the resulting neuroinflammation would be an effective therapeutic strategy to alleviate the progression of neurodegenerative diseases.
Obovatol is the main component of leaves of Magnolia obovata, a medicinal plant that has been used as a folk remedy in East Asia. Previous studies showed various properties of obovatol such as anti-bacterial, anti-tumour and anti-platelet activities (Ito et al., 1982; Pyo et al., 2002; Lee et al., 2008). The anti-inflammatory activity of obovatol was also reported in lipopolysaccharide (LPS)-stimulated mouse macrophage RAW264.7 cells (Choi et al., 2007). Little is known about its effects in the CNS. Recently, we reported that obovatol has anxiolytic effects mediated by GABA-benzodiazepine receptors (Seo et al., 2007). However, the effects of obovatol on brain inflammation or microglia have not been investigated.
In the present study, the effects of obovatol on microglial activation and neuroinflammation were examined using cultured cells and a mouse model of neuroinflammation. The results showed that obovatol inhibited inflammatory activation of microglia in vitro and neuroinflammation in vivo, and the compound exerted protective effects against microglia-mediated neurotoxicity. Affinity chromatography followed by mass spectrometric analysis identified peroxiredoxin 2 (Prx2) as one of the molecular targets of obovatol. The current study using obovatol as a chemical probe demonstrated the important role of redox regulation in microglial activation and neuroinflammation. Our results also suggest that Prx2 is a novel drug target that can be exploited for therapeutic modulation of microglial activation and neuroinflammation.
Methods
Isolation of obovatol
Obovatol (purity ≥ 95%) was isolated from the leaves of Magnolia obovata. The leaves of Magnolia obovata were harvested in Daejeon, Korea and identified by Dr Byung-Mog Kwon (Kwon et al., 1997). The dried leaves (1 kg) of Magnolia obovata were extracted with MeOH for 48 h at room temperature. After the combined extract was filtered and concentrated, the residue was partitioned between H2O and ethyl acetate (EtOAc; 1:1, v/v) to give a EtOAc-soluble fraction (15.2 g). This EtOAc-soluble fraction was concentrated, and then the residue was chromatographed on a silica gel (1 kg) column, eluted with a gradient of n-hexane–EtOAc to provide 10 fractions. The fractions containing obovatol were collected and concentrated to yield 0.8 g. The fraction was re-subjected to a C18 column and it was eluted with a gradient of MeOH–H2O from 60% MeOH/water to 80% MeOH/water to provide obovatol (220 mg). Analyis of this material by NMR provided the following results: 1H NMR (300 MHz, CDCl3) for obovatol: d 7.15 (2H, d, J = 9 Hz), 6.94 (2H, d, J = 9 Hz), 6.59 (1H, d, J = 1.5 Hz), 6.30 (1H, d, J = 1.5 Hz), 5.97 (2H, m), 5.06 (4H, m), 3.38 (2H, d, J = 6.6 Hz), 3.20 (2H, d, J = 6.6 Hz) ppm. These data were compatible with the structure of obovatol as shown in Figure 1. Obovatol was solubilized in dimethyl sulfoxide (DMSO) and added to cell cultures to the desired concentration.
Figure 1.
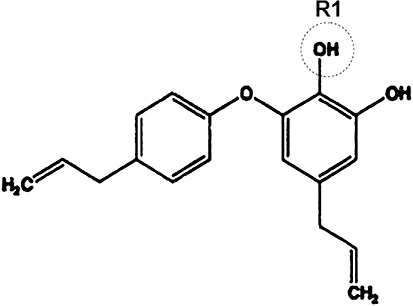
The chemical structure of obovatol. The structure of obovatol is shown. The R1 (circle) indicates the position that was conjugated with biotin moiety.
Cell culture
The immortalized BV-2 murine microglia cell line (Blasi et al., 1990) was maintained in Dulbecco's modified Eagle's medium (DMEM) containing 5% heat-inactivated FBS and 50 µg·mL−1 gentamicin at 37°C. Highly aggressively proliferating immortalized (HAPI) rat microglia cell line (Cheepsunthorn et al., 2001) and B35 rat neuroblastoma cell line (ATCC, CRL-2754) (Schubert et al., 1974) stably expressing enhanced green fluorescent protein (EGFP) were maintained in DMEM containing 10% heat-inactivated FBS, 2 mM glutamine, 10 U·mL−1 penicillin and 10 µg·mL−1 streptomycin (Gibco, Gaithersburg, MD, USA) at 37°C under a humidified atmosphere with 5% CO2.
All animal care and experimental procedures were approved by the Institutional Review Board of Kyungpook National University School of Medicine and were carried out in accordance with the guidelines in the NIH Guide for the Care and Use of Laboratory Animals. The animals were maintained under temperature- and humidity-controlled conditions with a 12-h light/12-h dark cycle. Mouse primary microglial cultures were prepared by mild trypsinization as previously described with minor modifications (Saura et al., 2003). In brief, forebrains of newborn Institute of Cancer Research (ICR) mice were chopped and dissociated by mechanical disruption using a nylon mesh. The cells were seeded into poly-L-lysine-coated flasks. After in vitro culture for 10–14 days, microglia cells were isolated from mixed glial cultures by mild trypsinization. Mixed glial cultures were incubated with a trypsin solution (0.25% trypsin, 1 mM EDTA in Hank's balanced salt solution) diluted 1:4 in phosphate-buffered saline (PBS; 150 mM NaCl, 5 mM phosphate, pH 7.4) containing 1 mM CaCl2 for 30–60 min. This resulted in the detachment of an upper layer of astrocytes in one piece, whereas microglia remained attached to the bottom of the culture flask. The detached layer of astrocytes was aspirated, and the remaining microglia were used for experiments. The purity of microglia cultures was greater than 95% as determined by isolectin B4 staining (data not shown). Primary cultures of dissociated cerebral cortical neurons were prepared from embryonic day 20 (E20) ICR mice as described previously (Araki et al., 2000). Briefly, mouse embryos were decapitated, and the brains were rapidly removed and placed in a culture dish with cold PBS. The cortices were isolated and then transferred to a culture dish containing 0.25% trypsin-EDTA (Gibco-BRL) in PBS for 30 min at 37°C. After two washes in serum-free neurobasal media (Gibco-BRL), the cortical tissues were mechanically dissociated with a gentle pipetting. Dissociated cortical cells were seeded onto poly D-lysine coated plates using neurobasal media containing 10% FBS, 0.5 mM glutamine, 100 U·mL−1 penicillin, 100 µg·mL−1 streptomycin, N2 supplement (Gibco-BRL) and B27 supplement (Gibco-BRL). Cells were maintained by changing the media every 2–3 days and grown at 37°C in a 5% CO2 humidified atmosphere. The purity of neuronal cultures was determined by immunocytochemical staining using an antibody against a neuron-specific marker, microtubule-associated protein 2 (Promega, Madison, WI, USA).
Nitrite quantification
The production of NO was estimated by measuring the amount of nitrite, a stable metabolite of NO, using the Griess reagent (Appendix S1).
Cell viability
Cell viability was assessed by a modified 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay as previously described (Ock et al., 2006). After treatment with LPS in the absence or presence of obovatol, diphenyliodonium (DPI; an NADPH oxidase inhibitor), or N-acetyl cysteine [NAC; a reactive oxygen species (ROS) scavenger] for 24 h, the culture media were aspirated and MTT (0.5 mg·mL−1 in PBS) was added to cells and then incubated at 37°C for 3 h. The resulting formazan crystals were dissolved in DMSO. Absorbance was determined at 570 nm using a microplate reader.
ELISA for TNFα
BV-2 cells were treated with LPS in the absence or presence of obovatol. After 24 h incubation, the levels of TNFα in culture media were measured with rat monoclonal anti-mouse TNFα antibody as the capture antibody and goat biotinylated polyclonal anti-mouse TNFα antibody as the detection antibody (ELISA development reagent; R&D systems) as previously described (Zheng et al., 2008).
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted using TRIZOL (Invitrogen, Carlsbad, CA, USA). Reverse transcription was carried out using M-MLV reverse transcriptase (Promega). PCR amplification was carried out in 50 µL PCR reaction mixture containing 10 mM Tris-HCl (pH 8.3), 50 mM KCl, 2 mM MgCl2, 20 pmol primer sets, two units of Taq DNA polymerase (CoreBio system Co., Seoul, Korea), 0.2 mM dNTPs and 2 µL of cDNA reaction. PCR condition was described in the Appendix S1.
Western blot analysis
BV-2 cells were pretreated with obovatol for 30 min and stimulated with LPS. After LPS stimulation for 30 min, cells lysates were subjected to Western blot analysis as previously described (Zheng et al., 2008) (see Appendix S1 for the commercial sources of antibodies used).
Nuclear extract preparation and electrophoretic mobility shift assay
BV-2 cells were pretreated with obovatol for 30 min and stimulated with LPS for 1 h. Nuclear extracts were then prepared from the cells as described previously (Schreiber et al., 1989). Synthetic double-stranded oligonucleotides of consensus NF-κB binding sequence, 5′-GAT CCC AAC GGC AGG GGA-3′ (Promega), were end-labelled with [γ-32P] ATP using T4 polynucleotide kinase. Labelled nucleic acids were purified using a mini Quick Spin Column (Roche, Indianapolis, IN, USA). Nuclear extracts were incubated with the labelled oligonucleotides in the presence of poly(dI-dC) in a binding buffer containing 20 mM HEPES at room temperature for 30 min. For supershift assays, 0.2 µg of antibodies against p65 subunit of NF-κB (Santa Cruz Biotechnology Inc.) were co-incubated with nuclear extracts in the reaction mixture for 30 min at 4°C before adding the radiolabelled probe. The specificity of binding was also examined by competition with an 80-fold molar excess of unlabelled oligonucleotide. DNA–protein complexes were resolved by electrophoresis on a 6% non-denaturing polyacrylamide gel at 180 V in 0.5× Tris boric acid EDTA (TBE). Gels were vacuum dried for 1 h at 80°C, and then exposed to X-ray film at −70°C for 12 h.
Microglia-neuron co-cultures
Microglia-neuron co-culture was performed using either HAPI rat microglia cells and B35 rat neuroblastoma cells or primary microglia and neurons. For the co-culture of microglia cell line and neuroblastoma cells, HAPI microglia cells were pretreated for 30 min with obovatol prior to the application of LPS. Then, the culture media were discarded and EGFP-expressing B35 (B35-EGFP) neuroblastoma cells and LPS were added. After additional incubation, B35-EGFP neuroblastoma cells were counted to assess cell viability. For the co-culture of primary microglia and primary cortical neurons, primary microglia cultures were pretreated with obovatol for 30 min and then washed to remove obovatol. Primary cortical neurons (labelled with 5-chloromethylfluorescein diacetate; CMFDA) and LPS were then added to the microglial culture. After chronic stimulation of microglia with LPS for 72 h, CMFDA-positive neuronal cells were counted (detailed methods were described in Appendix S1). Preparation of microglia-conditioned media was described in the Appendix S1.
Mouse model of neuroinflammation
Lipopolysaccharide was injected peripherally (i.p.) to evoke neuroinflammation in mice as previously described (Cunningham et al., 2005; Qin et al., 2007). All experiments were carried out on 11-week-old male C57BL/6 mice (25–30 g) supplied by Koatech (Pyongtaec City, Korea). Animals were divided into three experimental groups in each experiment: group 1 no reagents or treatment; group 2 treated with LPS and obovatol diluted in saline containing 5% propylene glycol; group 3) treated with LPS and 0.5% DMSO diluted in saline containing 5% propylene glycol. DMSO was included in the vehicle because obovatol was dissolved in DMSO. Obovatol (10 mg·kg−1) or vehicle (saline containing 0.5% DMSO and 5% propylene glycol) was administered i.p.once daily for 4 days. LPS (from E. coli 055:B5; Sigma) was administered i.p. at a dose of 5 mg·kg−1 on day 2 for a single challenge. For histochemical analysis, mice were anesthetized at 72 h after the LPS injection (day 5), and then transcardially perfused with saline and fixed in 4% paraformaldehyde for 72 h. Microglial activation was assessed by isolectin B4 staining (Appendix S1). For mRNA analysis, mice were anesthetized at 6 h after LPS injection, and transcardially perfused with saline. The brains were removed and stored at −80°C until analysis. At least three animals were used for each experimental group.
DNA microarray analysis
BV-2 cells were plated at a density of 1 × 106 cells per well in 6-well plates, allowed to adhere for 4 h, and then treated with LPS in the absence or presence of obovatol for 6 h. Total RNA was extracted using TRIZOL (Invitrogen) and analysed using mouse WG-6 v2.0 Expression BeadChip (Illumina Inc., San Diego, CA, USA) (Appendix S1). Microarray data were analysed by the standard procedure (Appendix S1).
Detection of obovatol-binding proteins
BV-2 cells were washed with PBS and then lysed in CSK buffer (50 mM NaCl, 10 mM PIPES, 3 mM MgCl2, 0.5% Triton X-100, 300 mM sucrose, 1 mM PMSF and 10 µg·mL−1 leupeptin; pH 6.8) for 20 min at 4°C. Cells were scraped from the plates and sedimented by centrifugation at 16 609×g for 10 min. The supernatant was collected and cleared by incubation with monomeric avidin (Pierce Chemical Co., Rockford, IL, USA) for 1 h at 4°C and then centrifuged at 500×g for 5 min. The cleared supernatant was incubated with biotin-obovatol (final concentration of 20 µM), equal volume of DMSO (vehicle as a negative control) or a mixture of biotin-obovatol (20 µM) and obovatol (20 µM) for the competition reaction. After incubation for 2 h at 4°C, proteins associated with biotin-obovatol were precipitated with avidin-agarose (Pierce Chemical Co.). Precipitated samples were applied to the column. Samples were washed with 10 bed volumes of washing buffer [50 mM HEPES (pH 7.5), 30 mM NaCl, 1 mM EDTA, 2.5 mM EGTA, 0.1% Tween 20, 10% glycerol, 1 mM NaF, 0.1 mM Na3VO4 and protease inhibitor mixture] and eluted from the column with 2 mL of 0.1 M glycine buffer (pH 2.8). Eluted fractions were concentrated with Centricon YM-3 (cut-off 3000 Da, Millipore, Bedford, MA, USA) to 100–150 µL. Protein concentration was determined using Bradford protein assay kit (Bio-Rad, Hercules, CA, USA).
Two-dimensional gel electrophoresis (2-DE) and protein identification
Proteins (40 µg) were solubilized in rehydration sample buffer (7 M urea, 2 M thiourea, 2% CHAPS, 100 mM dithiothreitol (DTT), 1% IPG buffer and 0.002% bromophenol blue) and applied onto 7 cm immobilized pH gradient (IPG) strips (Bio-Rad, pH 3–10, L). Isoelectric focusing (IEF) was performed for 30 min at 500 V, 30 min at 1000 V, 1 h 40 min at 5000 V and 3 h at 5000 V. Thereafter, the IPG strips were equilibrated for 15 min in SDS equilibration buffer A [50 mM Tris-HCl (pH 8.8), 6 M urea, 30% glycerol, 2% SDS, 1% DTT, 0.002% bromophenol blue] and then for 15 min in SDS equilibration buffer B [50 mM Tris-HCl (pH 8.8), 6 M urea, 30% glycerol, 2% SDS, 2.5% iodoacetamide, 0.002% bromophenol blue]. For the second dimension, vertical slab gels (10% SDS PAGE) were prepared. Then an equilibrated IPG gel strip was laid on top of the gel and sealed with 0.5% agarose solution. Gel electrophoresis was carried out at 16°C at 150 V for 1 h until the bromophenol blue reached the bottom of the gel. The gel was fixed in 50% methanol and 12% acetic acid overnight at 4°C. Proteins were detected by silver-staining as previously described (Shevchenko et al., 1996) and the protein spots were compared. Protein identification was performed by liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS) as previously described (Kim et al., 2007).
Flow cytometric measurement of ROS generation
Dichlorodihydrofluorescein diacetate (H2DCF-DA) enters cells passively and is deacetylated by esterases and then reacts with ROS to form fluorescent dichlorofluorescein (DCF). H2DCF-DA was dissolved in DMSO at 10 mM and was diluted to give H2DCF-DA at 10 µM. For intracellular ROS measurement, obovatol was added 30 min before treatment with LPS for various time periods. Then, cells were incubated with 10 µM H2DCF-DA in PBS for 30 min at 37°C. Cells were detached and fluorescence was measured by the flow cytometry.
Assay for Prx activity
Peroxiredoxin 2 activity was determined by measuring the remaining H2O2 after in vitro incubation of recombinant Prx2 protein with known concentrations of H2O2 (Thurman et al., 1972). In brief, for the measurement of Prx2 activity, DTT (2 mM), H2O2 (50 µM), recombinant Prx2 proteins and obovatol in a volume of 100 µL were mixed and incubated for 10 min at 37°C. Bovine serum albumin (BSA) or DMSO, instead of Prx2 or obovatol, were used as a control respectively. Then, 20 µL of 1 N HCl was added to stop the reaction, and 20 µL of 10 mM Fe(NH4)SO4 and 10 µL of 2.5 M potassium thiocyanate were added to visualize the remaining H2O2 as a red-colored complex. The mixture was centrifuged at 10 000×g for 30 s and 100 µL of supernatant was added to wells of a microtiter plate. The optical density was measured at 450 nm. Prx2 activity was estimated based on the amount of H2O2 remaining in the reaction.
Statistical analysis
All data are presented as mean ± SD from three or more independent experiments, unless stated otherwise. Statistical comparisons between different treatments were made either by a Student's t-test or by one-way anova with Bonferroni's post hoc tests, using the SPSS version 14.0K program (SPSS Inc., Chicago, IL, USA). Differences with a value of P < 0.05 were considered to be statistically significant.
Materials
Bacterial LPS from Escherichia coli 0111:B4 and ATP were obtained from Sigma (St Louis, MO, USA). Recombinant mouse interferon gamma (IFN-γ) was purchased from R&D Systems (Minneapolis, MN, USA) and recombinant human Prx2 was purchased from Abnova (Taipei, Taiwan) or generously provided by Dr Sang Won Kang (Ewha Womans University, Seoul, Korea). All nomenclature for the therapeutic agents, drugs, proteins and receptors used in the study followed the guidelines in Alexander et al. (2009).
Results
Obovatol inhibited microglial NO production in a dose-dependent manner
The effect of obovatol on NO production in LPS-stimulated BV-2 microglia cells was investigated. BV-2 cells were stimulated with LPS in the absence or presence of obovatol for 24 h. The accumulated nitrite in the culture media estimated by the Griess reaction was used as an index for NO synthesis. LPS-induced NO production was decreased by obovatol in a dose-dependent manner with an IC50 value of 10 µM (Figure 2A). Therefore, this concentration was used for subsequent experiments. In order to rule out the possibility that the reduction of NO production may be due to the cytotoxicity of obovatol, cell viability was assessed (Figure 2A). No significant cytotoxicity from obovatol was observed at the concentrations tested. The NO inhibitory effect of obovatol was further confirmed at several different concentrations using primary microglia cultures and HAPI rat microglia cells (Figure 2C). The inhibitory effect of obovatol on NO production was also tested in ATP- or IFN-γ-stimulated microglia cells. As shown in Figure 2B, obovatol inhibited NO production in ATP or IFN-γ-stimulated BV-2 microglia cells without significant cytotoxicity. These results indicate that the inhibitory effect of obovatol on microglial NO production is not limited to just LPS stimulation condition. The results also suggest that obovatol may modulate multiple inflammatory signalling pathways in microglia.
Figure 2.
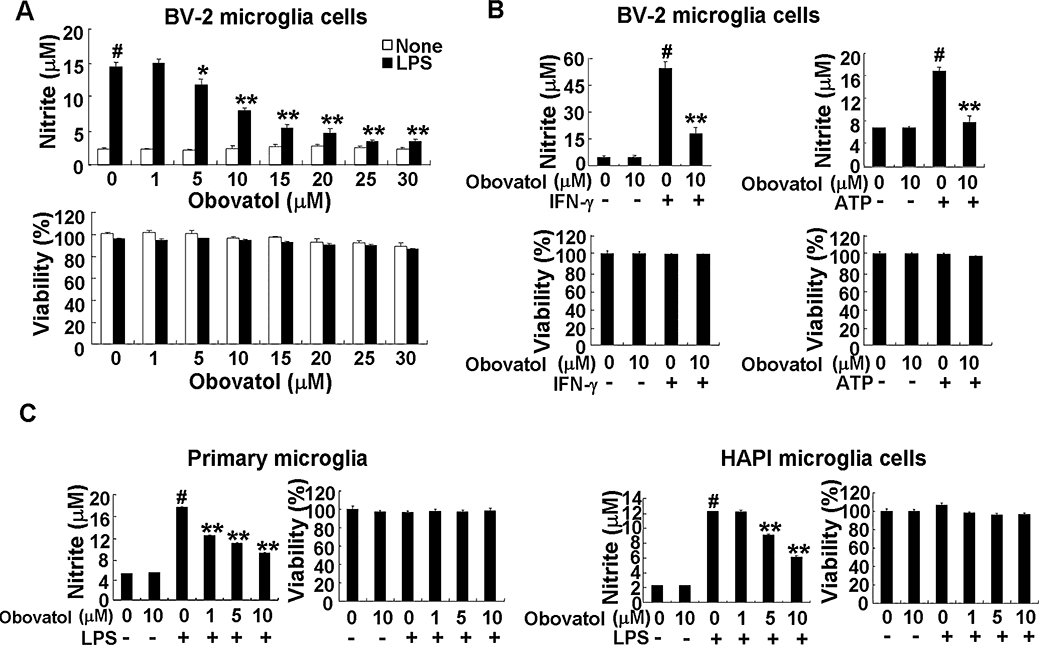
Obovatol suppressed NO production in BV-2 microglia cells, primary microglia cultures or HAPI microglia cells. Microglia cells (either microglia cell lines or primary cultures as indicated) were treated with LPS (100 ng·mL−1) or ATP (2 mM) or IFN-γ (50 U·mL−1) in the absence or presence of indicated concentration of obovatol (A, 1–30 µM in BV-2 cells; B, 10 µM in BV-2 cells; C, 1-10 µM in primary microglia culture and HAPI rat microglia cells) for 24 h. Nitrite content was measured using the Griess reaction, and cytotoxicity of obovatol was assessed by MTT assays (A, B and C). The data were expressed as the mean ± SD (n= 3). #P < 0.01 versus untreated control; *P < 0.05, **P < 0.01 versus LPS, IFN-γ, or ATP alone. NO, nitric oxide; HAPI, highly aggressively proliferating immortalized; LPS, lipopolysaccharide; IFN, interferon.
Obovatol inhibited the expression of IL-1β, iNOS and TNFα in microglia
Expression of iNOS is induced in activated microglia and mediates synthesis of large amounts of NO. Levels of proinflammatory cytokines such as TNFα and IL-1β also increase in activated microglia. Therefore, the effect of obovatol on IL-1β, iNOS and TNFα gene expression at the transcriptional level was determined by RT-PCR. Obovatol inhibited LPS-induced expression of IL-1β, iNOS and TNFα, whereas the compound alone did not induce any significant change in gene expression (Figure 3A). The effect of obovatol on iNOS gene expression was confirmed at the protein level by Western blot analysis (Figure 3B). TNFα production in the microglial culture media was analysed by ELISA (Figure 3C) and obovatol significantly inhibited LPS-induced TNFα production.
Figure 3.
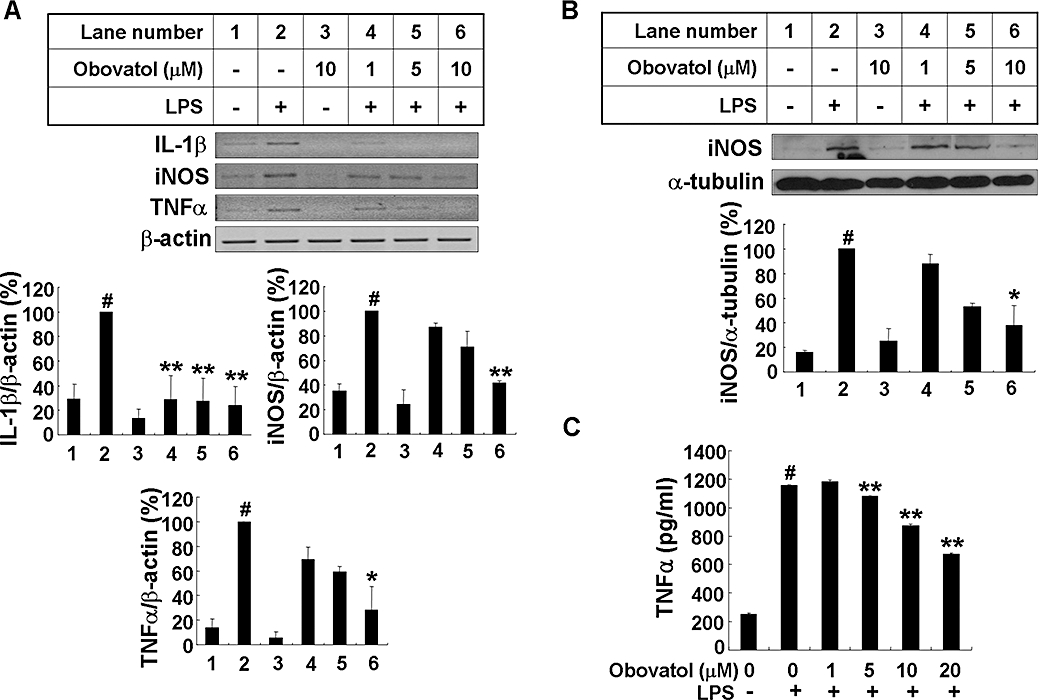
Obovatol suppressed expression of iNOS, IL-1β, and TNFα in LPS-stimulated BV-2 microglia cells. BV-2 microglia cells were treated with LPS (100 ng·mL−1) in the absence or presence of 1–10 µM obovatol for 6 h (RT-PCR), 14 h (Western blot) or 24 h (TNFα ELISA). After treatment, total RNA was isolated and specific mRNA levels were determined by RT-PCR analysis (A; upper), and then subjected to densitometric quantification (A; lower). Levels of IL-1β, iNOS and TNFα were normalized to β-actin levels and expressed as a relative change in comparison with the LPS treatment, which was set to 100% (lane 2). Alternatively, BV-2 microglia cell lysates were subjected to Western blot for iNOS (B). Levels of iNOS were normalized α-tubulin levels and expressed as a relative change in comparison with the LPS treatment, which was set to 100% (lane 2). Lastly, culture media of BV-2 microglia cells were collected and subjected to TNFα sandwich ELISA (C). The data were expressed as the mean ± SD (n= 3), and are representative results obtained from three independent experiments. #P < 0.01 versus untreated control; *P < 0.05, **P < 0.01 versus LPS only. iNOS, inducible nitric oxide synthase; IL-1, interleukin 1; TNF, tumour necrosis factor; LPS, lipopolysaccharide; RT-PCR, reverse transcription-polymerase chain reaction.
Obovatol inhibited NF-κB, extracellular signal-regulated kinases (ERK) and c-jun N-terminal kinase (JNK) activation
Nuclear factor κB is complexed with the inhibitory IκB protein in the cytoplasm. Inflammatory stimulation leads to phosphorylation and subsequent degradation of IκB. Free NF-κB translocates into the nucleus and binds to genes with NF-κB binding sites. Expression of a number of proinflammatory cytokines and iNOS is controlled by NF-κB. Thus, the effects of obovatol on the DNA-binding activity of NF-κB were evaluated. LPS-induced DNA binding activity of NF-κB was attenuated by obovatol, whereas the compound alone did not induce any significant change in these activities. The binding specificity of NF-κB was confirmed by supershift assay or by the unlabelled probe containing the NF-κB binding sequence (Figure 4A). Along with NF-κB, MAPKs are known to play an important role in the signalling pathways that induce proinflammatory cytokines in glial cells (Koistinaho and Koistinaho, 2002). Thus, the effect of obovatol on the MAPKs (ERK, JNK and p38) pathways was investigated. Obovatol repressed the phosphorylation of ERK and JNK, whereas p38 phosphorylation was not affected (Figure 4B). However, the results are based on the assessment of obovatol effects on MAPKs at single time point. Further evaluation at different time points is necessary to draw a firm conclusion about the selective regulation. Nevertheless, the results indicated that obovatol inhibited NF-κB, ERK and JNK pathways in this condition. The levels of total JNK, p38 or ERK did not change under these conditions, when normalized to Ponceau staining (data not shown). Although the phosphorylation of ERK seemed to be decreased by the compound alone, these changes were not statistically significant.
Figure 4.
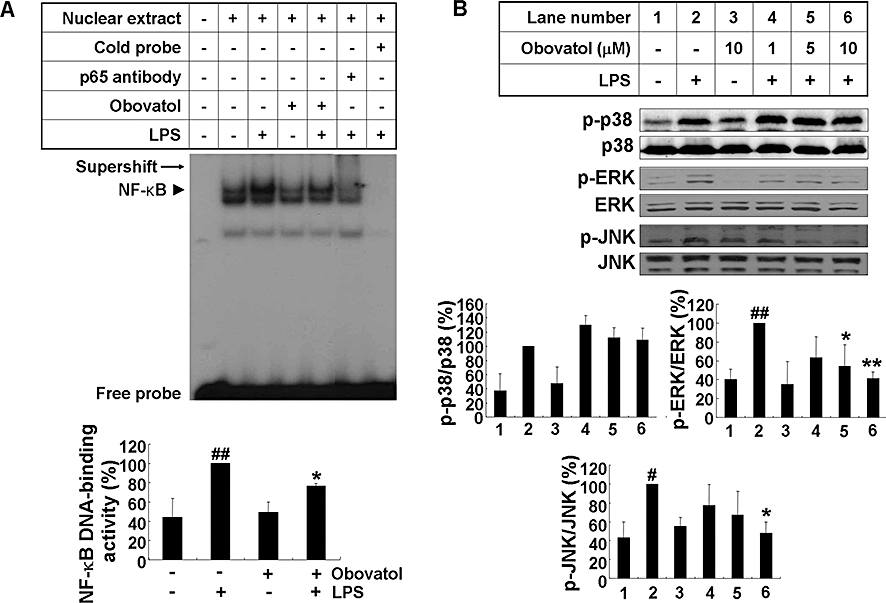
Obovatol suppressed activation of NF-κB, ERK and JNK in LPS-stimulated microglia cells. BV-2 microglia cells were stimulated with 100 ng·mL−1 LPS in the absence or presence of obovatol (10 µM for EMSA, or 1–10 µM for MAPKs Western blot) that had been added 30 min before the activation. After LPS stimulation for 1 h, nuclear extracts were isolated for gel shift assay. LPS-induced NF-κB DNA binding activity was indicated by the arrow head. The supershift of NF-κB-specific band mobility in the gel using antibody against NF-κB confirmed the NF-κB binding specificity as indicated by arrow (A). Total lysates were obtained after 30 min activation with LPS, and phosphorylation of p38, ERK and JNK in the lysates was analysed by Western blotting (B). Levels of phospho-MAPKs were expressed as a relative change in comparison with the LPS treatment, which was set to 100% (lane 2). The data were expressed as the mean ± SD (n= 3). #P < 0.05, ##P < 0.01 versus untreated control; *P < 0.05, **P < 0.01 versus LPS only. NF-κB, nuclear factor κB; ERK, extracellular signal-regulated kinases; JNK, c-jun N-terminal kinase; LPS, lipopolysaccharide; EMSA, electrophoretic mobility shift assay; MAPK, mitogen-activated protein kinase.
Obovatol inhibited the activation of STAT1 and expression of IFN-β
Lipopolysaccharide activates both myeloid differentiation factor 88 (MyD88)-dependent and -independent pathways through the Toll-like receptor 4 (TLR4). The MyD88-independent pathway induces the production of IFN-β and STAT1 activation. Therefore, the effect of obovatol on the STAT1 activation and IFN-β expression was next evaluated. LPS-induced STAT1 phosphorylation was suppressed by obovatol (Figure 5A). The expression of IFN-β gene was also suppressed by obovatol (Figure 5B). Taken together, the results indicate that obovatol inhibits both MyD88-dependent and -independent pathways of microglial LPS/TLR4 signalling.
Figure 5.
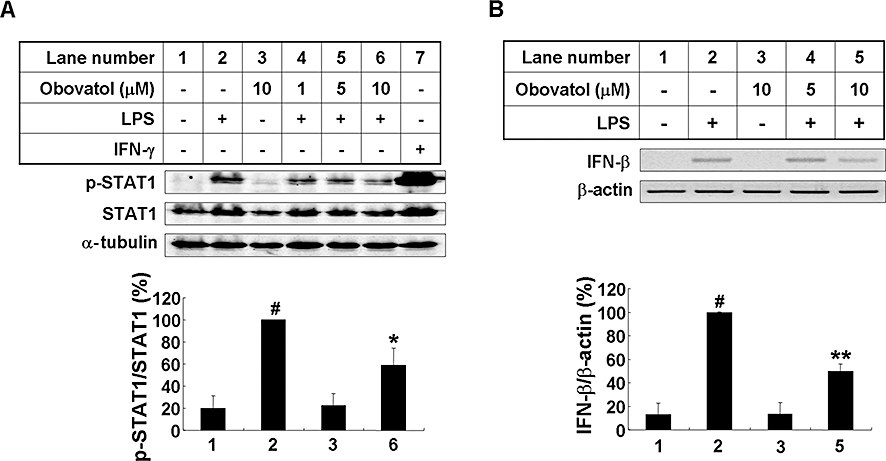
Obovatol suppressed phosphorylation of STAT-1 and mRNA expression of IFN-β in LPS-stimulated microglia cells. BV-2 microglia cells were stimulated with 100 ng·mL−1 LPS in the absence or presence of 1–10 µM obovatol for 4 h. Total lysates were obtained and phosphorylation of STAT-1 in the cellular lysates were analysed by Western blotting. Levels of phospho-STAT1 were normalized to total-STAT1 levels, and expressed as a relative change in comparison with the LPS treatment, which was set to 100% (lane 2) (A). IFN-γ stimulation was used as a control. Alternatively, BV-2 microglia cells were stimulated with 100 ng·mL−1 LPS in the absence or presence of 5–10 µM obovatol for 2 h. Total RNA was isolated and subjected to RT-PCR analysis (B; upper), and then subjected to densitometric quantification (B; lower). Levels of IFN-β was normalized to β-actin levels and expressed as a relative change compared with the LPS treatment, which was set to 100% (lane 2). The data were expressed as the mean ± SD (n= 3). #P < 0.01 versus untreated control; *P < 0.05, **P < 0.01 versus LPS only. STAT1, signal transducers and activators of transcription 1; IFN, interferon; LPS, lipopolysaccharide; RT-PCR, reverse transcription-polymerase chain reaction.
Obovatol inhibited microglial neurotoxicity in the microglia/neuron co-culture
As excessively activated microglia contribute to neuronal damage by releasing various proinflammatory and neurotoxic mediators (Stoll and Jander, 1999), inhibition of microglial activation protects neurons against the cytotoxic effect of activated microglia in microglia/neuron co-cultures (Zheng et al., 2008). Therefore, the potential protective effect of obovatol against microglial neurotoxicity was investigated in either co-cultures of microglia cell line and neuroblastoma cells or those of primary microglia and primary cortical neurons. The co-culture experiments using HAPI microglia cells and EGFP-expressing B35 neuroblastoma cells revealed that LPS-activated microglia reduced the viability of B35-EGFP cells by ∼40% compared with the control co-culture of unstimulated microglia and B35-EGFP cells. Obovatol attenuated the LPS/microglia-induced cell death of B35-EGFP neuroblastoma cells (Figure 6D). The viability of B35-EGFP was not affected by 100 ng·mL−1 LPS alone without microglia co-culture (data not shown). In the co-cultures of primary microglia and neurons, the viability of CMFDA-labelled neurons was reduced by ∼50% compared with the control co-culture of unstimulated microglia and neurons. Obovatol attenuated neuronal cell death induced by the chronically activated microglia (Figure 6D). The representative images of viable neurons (CMFDA-positive) are shown in Figure S1. Moreover, microglia-conditioned media, collected from LPS-treated primary microglia cultures, also reduced the viability of primary neuron cultures by ∼30%. The reduced neuronal viability was restored by treatment of microglia with obovatol prior to microglia-conditioned media preparation under similar conditions (Figure 6D). These results indicate that obovatol may be neuroprotective by suppressing microglial activation and subsequent neurotoxicity.
Figure 6.
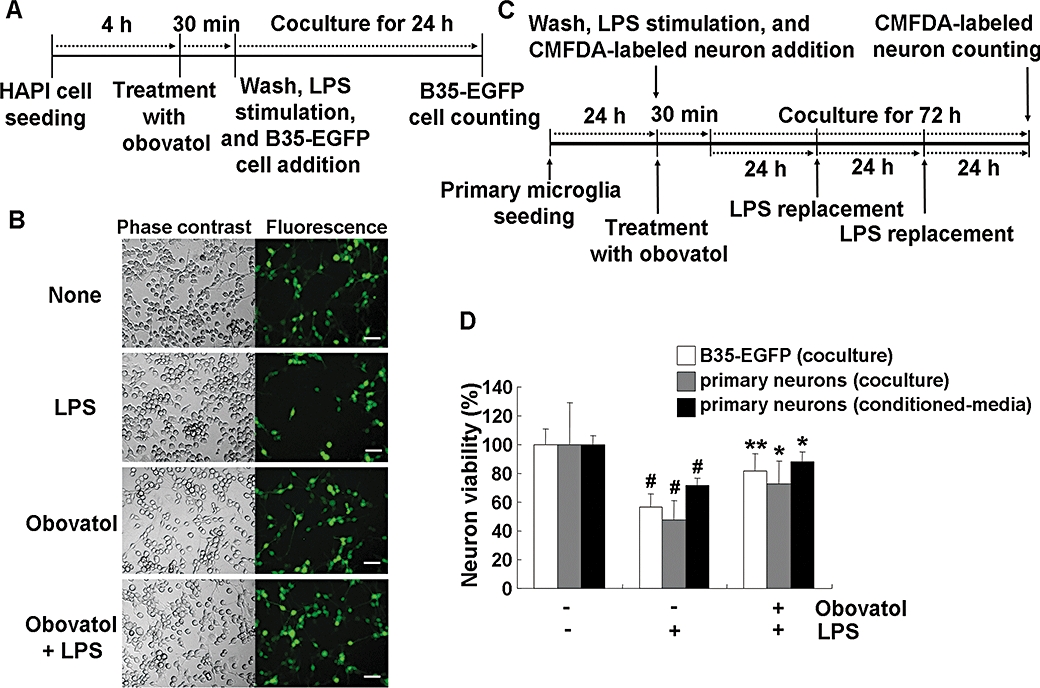
Obovatol suppressed microglial neurotoxicity. HAPI rat microglia cells were pretreated with 10 µM of obovatol for 30 min and washed with PBS. Then, LPS (100 ng·mL−1) and B35-EGFP rat neuroblastoma cells were added to microglia cells for the co-culture for 24 h (A). At the end of co-culture, the number of viable B35-EGFP neuroblastoma cells in the five randomly chosen microscopic fields per well was counted under a fluorescence microscope (D; white bars). Representative fluorescence or phase contrast images of cells are shown (B). Scale bar, 50 µm. Obovatol alone did not exert cytotoxicity against B35-EGFP neuroblastoma cells (data not shown). For the co-culture of primary microglia and primary neurons, microglia were pretreated with 10 µM of obovatol for 30 min. Then, LPS (100 ng·mL−1) and CMFDA-labelled primary neurons were added to microglia cultures. The culture media were replaced with fresh media containing LPS every 24 h, over 72 h (C). After LPS stimulation for 72 h, CMFDA-positive neurons were counted under a fluorescence microscope (D). For the preparation of primary microglia-conditioned media, microglia cultures were treated with LPS (100 ng·mL−1) in the absence or presence of 10 µM obovatol for 6 h. Culture media were removed, and fresh culture media were then added to microglia culture. After additional 24 h incubation, conditioned media were collected and added to primary neurons. After 24 h incubation, the viability of neurons was assessed by MTT assay (D). Results were expressed as a percentage of control (unstimulated microglia cultures+neuroblastoma or neurons) (mean ± SD; n= 3). #P < 0.01 versus control; *P < 0.05, **P < 0.01 versus LPS only. HAPI, highly aggressively proliferating immortalized; PBS, phosphate-buffered saline; LPS, lipopolysaccharide; EGFP, enhanced green fluorescent protein; CMFDA, 5-chloromethylfluorescein diacetate.
Obovatol inhibited microglia-mediated neuroinflammation in vivo
The effect of obovatol on microglia-mediated neuroinflammation was determined by using a mouse neuroinflammation model. Isolectin B4 (IB4) has been used as a specific marker for microglia. Histochemical detection of IB4 in brain tissues indicated that obovatol inhibited LPS-induced microglial activation in vivo as shown by the decrease in the intensity of IB4 staining. Figure 7A shows representative images of IB4 staining of brain tissue sections. The IB4-stained cells whose intensity values are above the arbitrary defined threshold were counted from each of the three regions (Figure 7B). Inhibition of microglia-mediated neuroinflammation by obovatol was further supported by quantitative measurements of the expression of proinflammatory genes such as iNOS, IL-1β, TNFα, monocyte chemotactic protein 1 (MCP-1) and macrophage inflammatory protein (MIP)-1α. RT-PCR analysis of the brain tissues indicated that mRNA levels of these genes were increased by LPS, but were significantly inhibited by obovatol (Figure 7C and D) (Table 1).
Figure 7.
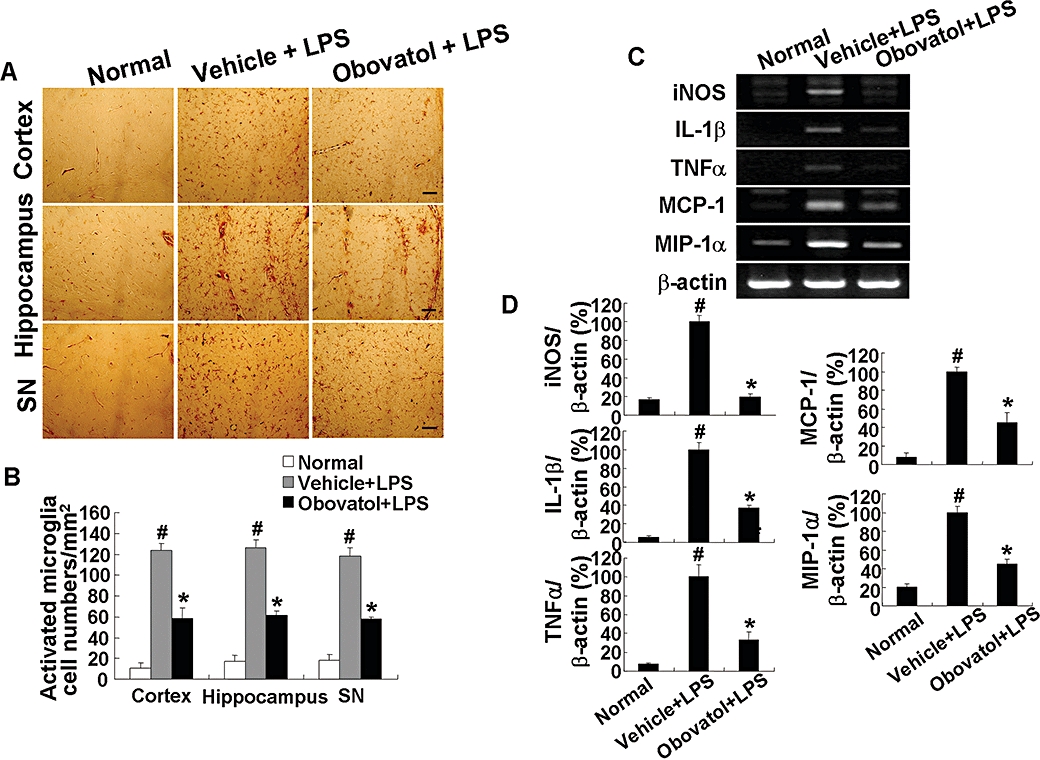
Obovatol suppressed microglial activation in a mouse neuroinflammation model. C57BL/6 mice were injected i.p. with vehicle (saline containing 0.5% DMSO and 5% propylene glycol) or obovatol (diluted in saline containing 5% propylene glycol) once daily at 10 mg·kg−1 for 4 days. At 24 h after the first injection of vehicle or obovatol, mice were injected i.p.with 5 mg·kg−1 LPS. Mice were anesthetized with diethyl ether and transcardially perfused with ice-cold saline 72 h after the LPS injection. Brains were removed and sections were stained with IB4 (a marker for microglia). IB4-positive cells were observed in cortex, hippocampus, and substantia nigra (SN) region of vehicle+LPS- or obovatol+LPS-injected mouse brains (A). Scale bar, 50 µm. The number of IB4-stained cell per mm2 was counted (B). The expression levels of proinflammatory genes were determined by RT-PCR at 6 h after the LPS injection (C). Levels of iNOS, IL-1β, TNFα, MCP-1 and MIP-1α were normalized to β-actin levels and expressed as a relative change in comparison with the LPS treatment, which was set to 100% (D). The data were expressed as the mean ± SD (n= 3 per experimental group). #P < 0.01 versus normal animals; *P < 0.01 versus vehicle+LPS-injected animals. DMSO, dimethyl sulfoxide; LPS, lipopolysaccharide; RT-PCR, reverse transcription-polymerase chain reaction; iNOS, inducible nitric oxide synthase; IL-1, interleukin 1; TNF, tumour necrosis factor; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein.
Table 1.
Primer sequences used for RT-PCR
| Gene | Forward primer | Reverse primer | Product size (bp) |
|---|---|---|---|
| β-actin | 5′-ATC CTG AAA GAC CTC TAT GC-3′ | 5′-AAC GCA GCT CAG TAA CAG TC-3′ | 287 |
| IFN-β | 5′-TAG AAC CTC GCT GGA AAG GAC-3′ | 5′-CAG CTG TTC CAT CTG TTC CTG ACC-3′ | 314 |
| IKKε | 5′-GTA CAG AAT CAC CAC AGA GAA GC-3′ | 5′-GAC CTG CTG AAC AGA GTT AGA GG-3′ | 453 |
| IL-1β | 5′-GCA ACT GTT CCT GAA CTC-3′ | 5′-CTC GGA GCC TGT AGT GCA-3′ | 382 |
| iNOS | 5′-CCC TTC CGA AGT TTC TGG CAG CAG C-3′ | 5′-GGC TGT CAG AGC CTC GTG GCT-3′ | 497 |
| IP10 | 5′-CCT ATC CTG CCC ACG TGT TG-3′ | 5′-CGC ACC TCC ACA TAG CTT ACA-3′ | 431 |
| MCP-1 | 5′-CAG CAG GTG TCC CAA AGA-3′ | 5′-CTT GAG GTG GTT GTG GAA A-3′ | 253 |
| MIP-1α | 5′-TCT GCA ACC AAG TCT TCT CAG-3′ | 5′-GAA GAG TCC CTC GAT GTG GGC TA-3′ | 373 |
| TNFα | 5′-CAT CTT CTC AAA ATT CGA GTG ACA A-3′ | 5′-ACT TGG GCA GAT TGA CCT CAG-3′ | 411 |
RT-PCR, reverse transcription-polymerase chain reaction; IFN, interferon; IKK, inhibitor of kappaB kinase; IL-1, interleukin 1; iNOS, inducible nitric oxide synthase; IP10, interferon-inducible protein 10; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; TNF, tumour necrosis factor.
Obovatol inhibited the transcription of proinflammatory genes in microarray analyses
In order to further assess the effect of obovatol on microglia-mediated neuroinflammation DNA microarray analyses were performed, where a gene expression profile of microglia cells treated with LPS in the absence or presence of obovatol was obtained. Based on the microarray analyses, 275 genes were differentially expressed with more than a 1.5-fold change by the treatment with LPS compared with the unstimulated control, and 108 genes when treated with obovatol showed a recovery pattern to the expression level seen in untreated cells. A complete description of the DNA microarray platform and results is available under Gene Expression Omnibus accession number GSE17069. The gene ontology term analysis of these 108 genes showed that more than 40% of these genes belonged to immunity/defence and signal transduction (Table S1). A hierarchical clustering was performed for the above-mentioned 108 genes, immune/defence-related genes or signal transduction-related genes as shown in Figure S2A and B. From the pathway analysis using GenPlex™ v3.0 software and KEGG database, TLR signalling and cytokine-cytokine receptor interaction pathways have been identified as the KEGG pathways that are modulated by obovatol (Figure S3). As shown in Table 2, stimulation of BV-2 cells with LPS caused a strong up-regulation of chemokines such as MIP-2α, interferon-inducible protein 10 (IP10), MCP-1 and MCP-3. The expression levels of all of these genes were decreased by obovatol treatment. Three down-regulated genes [MCP-1, IP10 and inhibitor of kappaB kinase (IKK)ε] from the list of the differentially expressed genes (Table 2) were subjected to RT-PCR validation (Figure 8). Results of RT-PCR analysis for MCP-1, IP10 and IKKε were consistent with the microarray analysis. However, these results obtained from the BV-2 microglial cell line may not be the same as those from primary microglia cultures, as the BV-2 cell line was established by transfection with a J2 retrovirus carrying a v-raf/v-myc oncogene (Blasi et al., 1990).
Table 2.
Pro-inflammatory genes differentially regulated by obovatol
| Gene name | Fold change (LPS vs. control) | Fold change (obovatol+LPS vs. control) | Gene description |
|---|---|---|---|
| MCP-1 | 3.42 | 0.71 | Monocyte chemotactic protein 1 (CCL 2) |
| MCP-3 | 3.58 | 0.86 | Monocyte chemotactic protein 3 (CCL 7) |
| IP10 | 9.35 | 2.94 | Chemokine (C-X-C motif) ligand 10 (CXCL 10) |
| MIP-2α | 10.19 | 4.24 | Chemokine (C-X-C motif) ligand 2 (CXCL 2) |
| MIP-1β | 3.14 | 1.31 | Chemokine (C-X-C motif) ligand 4 (CXCL 4) |
| IL-1β | 2.26 | 1.02 | Interleukin 1β |
| MCP-5 | 2.12 | 1.10 | Monocyte chemotactic protein 5 (CCL 12) |
| TNFα | 2.28 | 1.26 | Tumour necrosis factor |
| MIP-1 | 2.02 | 1.18 | Chemokine (C-C motif) ligand 3 (CCL 3) |
IL-1, interleukin 1; IP10, interferon-inducible protein 10; MCP, monocyte chemotactic protein; MIP, macrophage inflammatory protein; TNF, tumour necrosis factor.
Figure 8.
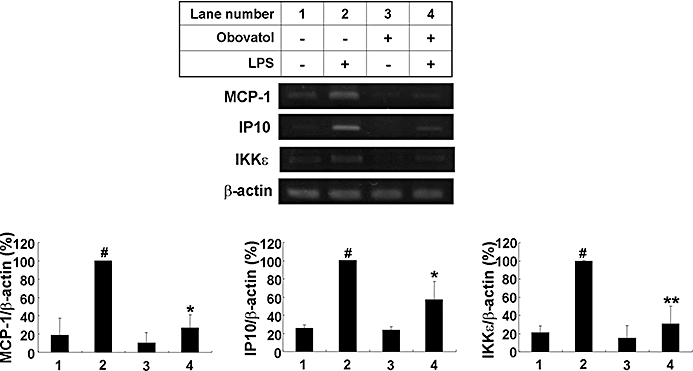
Re-evaluation of microarray-based differential gene expression by RT-PCR. BV-2 microglia cells were stimulated with 100 ng·mL−1 LPS in the absence or presence of 10 µM obovatol for 6 h. After treatment, total RNA was isolated and subjected to DNA microarray analysis. The expression of MCP-1, IP10 and IKKε genes was assessed by RT-PCR (upper). Levels of MCP-1, IP10 and IKKε were normalized to β-actin levels and expressed as a relative change in comparison with the LPS treatment, which was set to 100% (lane 2) (lower). The data were expressed as the mean ± SD (n= 3). #P < 0.01 versus untreated control; *P < 0.05, **P < 0.01 versus LPS only. RT-PCR, reverse transcription-polymerase chain reaction; LPS, lipopolysaccharide; MCP, monocyte chemotactic protein; IP10, interferon-inducible protein 10; IKK, inhibitor of kappaB kinase.
Identification of Prx2 as one of the cellular protein targets of obovatol
The major technical challenge during phenotype-based screening is the identification of the target protein(s) of the bioactive molecule. Affinity purification can be a powerful method for direct target identification. In order to apply this method to the target identification, the chemical structure of obovatol was modified to link the biotin-moiety to obovatol. Before the addition of the biotin-moiety to obovatol, the microglial inhibitory activities of obovatol derivatives (structural analogues) were investigated (data not shown). Based on structure-activity relationship studies, the biotin moiety was linked to position R1 of obovatol (Figure 1). Biotin-conjugation did not affect the microglial inhibitory activity of obovatol based on the measurement of microglial NO production (Figure 9A). For affinity purification of the microglial target of obovatol, whole cell lysates of BV-2 microglia cells were incubated with DMSO (vehicle), biotin-obovatol/obovatol (for competition) or biotin-obovatol, which was then conjugated with avidin-agarose beads in a column. After several extensive washings, bound proteins were eluted and separated by 2-DE and silver staining. Several protein spots were detected on 2-DE gel (Figure 9B). For the quantification of these spots, the 2-DE gel images were subjected to quantitative analysis using Melanie v6.0 software: the intensity of protein spots was normalized according to the total quantity in each gel. These spots were analysed by LC-MS/MS for protein identification. The protein spots that showed differential abundance (Figure 9B) were identified as Prx2 and Prx1 (Table 3). Identity of the eluted proteins was confirmed by Western blot analysis. Prx2 was detected in the eluate of the biotin-obovatol column, which had competition from unlabelled obovatol. On the other hand, Prx2 was detected in the flow-through of all three columns (Figure 9C). As an equal concentration of the proteins was analysed, these results indicate that obovatol specifically binds to Prx2. Although obovatol may also bind to Prx1, this study was focused on Prx2.
Figure 9.
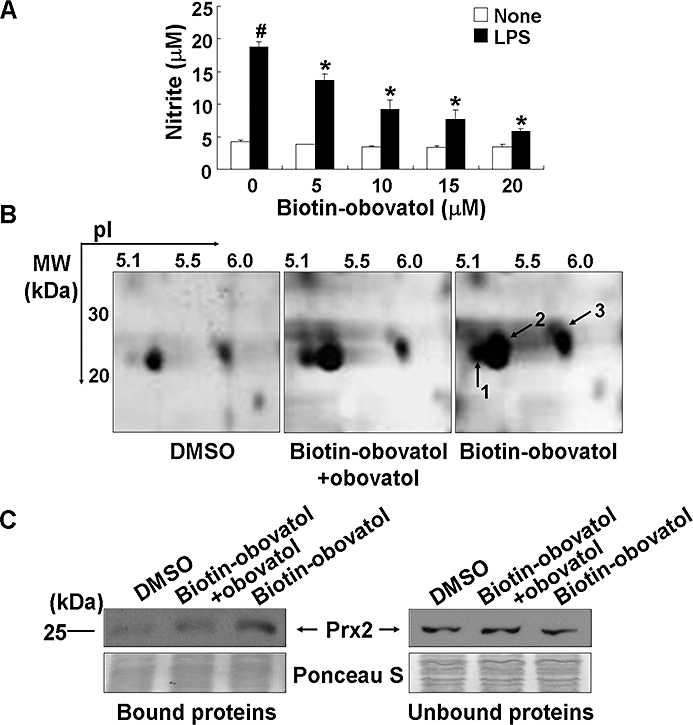
Affinity purification of obovatol-binding protein(s). BV-2 microglia cells were stimulated with 100 ng·mL−1 LPS in the absence or presence of 5–20 µM biotin-obovatol. After 24 h, the amount of nitrite in the culture media was evaluated using Griess reaction (A). The data were expressed as the mean ± SD (n= 3). #P < 0.01 versus untreated control; *P < 0.01 versus LPS only. BV-2 microglia cell lysates were incubated with DMSO, mixture of biotin-obovatol (20 µM) and obovatol (20 µM), or biotin-obovatol (20 µM), which were then applied to a column with avidin beads. The proteins were eluted and subjected to 2-DE analysis followed by silver-staining. The gel image represents the eluted proteins from the column containing DMSO, biotin-obovatol+obovatol or biotin-obovatol (B). Several protein spots, which showed differential intensity in 2-DE gel, were identified as Prx2 (spots 1 and 2) and Prx1 (spot 3). Eluted proteins (bound) and flow-through proteins (unbound) from the each affinity column were analysed by immunoblot for Prx2 (C). DMSO, dimethyl sulfoxide; LPS, lipopolysaccharide; Prx2, peroxiredoxin 2.
Table 3.
Identification of protein spots detected in analysis by two-dimensional gel electrophoresis (2-DE) of the proteins eluted from affinity column
| Spot number | IPI accession number | Protein name | Molecular weight (Da) | Theoretical pI |
Normalized spot density |
||
|---|---|---|---|---|---|---|---|
| DMSO | Biotin-obovatol+obovatol | Biotin-obovatol | |||||
| 1 | IPI00117910 | Peroxiredoxin 2 | 21761.1 | 5.56 | 57 | 92 | 121 |
| 2 | IPI00117910 | Peroxiredoxin 2 | 21761.1 | 5.56 | 0 | 45 | 100 |
| 3 | IPI00121788 | Peroxiredoxin 1 | 22159.2 | 8.26 | 0 | 30 | 101 |
Protein spots detected by were identified by LC-MS/MS, following digestion with trypsin. The peptide masses were matched with the theoretical peptide masses of all proteins from the International Protein Index mouse protein database (version 3.16) using the SEQUEST algorithm (Thermo Electron). Spot density of each gel was analysed by Melanie v6.0 software.
DMSO, dimethyl sulfoxide; IPI, international protein index; pI, isoelectric point.
Obovatol enhanced the Prx-mediated reduction of H2O2
Peroxiredoxin 2 has peroxidase activity against ROS such as H2O2 and peroxynitrite. The identification of Prx2 as the major obovatol-binding protein suggests that obovatol may modulate the peroxidase activity of Prx2. To examine this hypothesis, in vitro Prx activity assay based on the ferrous oxidation reaction was performed to assess the effect of obovatol on the peroxidase activity of Prx2. In this assay, Prx2 reduced H2O2 and this activity of Prx2 was enhanced by the addition of obovatol. BSA or DMSO, which was used as the negative control for Prx2 or obovatol, respectively, was without significant effects (Figure 10A). Furthermore, obovatol alone did not show a direct scavenging effect against H2O2 in a cell-free system (Figure 10B), indicating that the observed effect of obovatol was mediated through Prx2.
Figure 10.
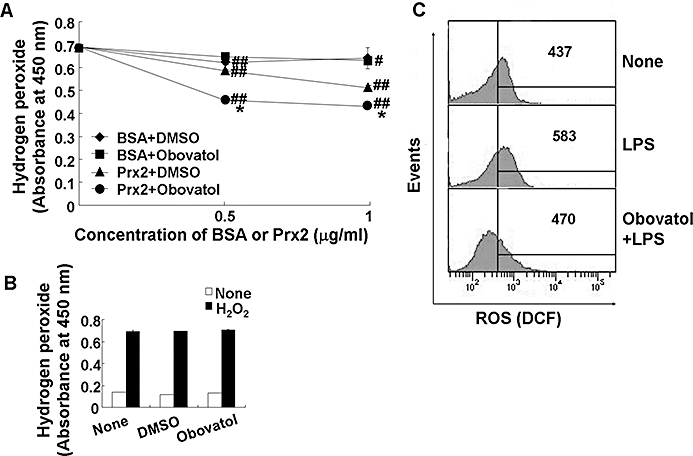
Obovatol enhanced the peroxidase activity of Prx2 and suppressed intracellular ROS production. Ferrous oxidation assay was carried out in 100 µL of reaction mixture containing DTT (2 mM), H2O2 (50 µM), obovatol (10 µM) and recombinant Prx2 protein. After incubation of the mixture for 10 min, the remaining H2O2 was measured based on the color changes after adding Fe(NH4)SO4 and potassium thiocyanate. DMSO and BSA were used as a vehicle control for obovatol and a negative control for Prx2 respectively. The data were expressed as the mean ± SD (n= 3). #P < 0.05, ##P < 0.01 versus H2O2 only; *P < 0.01 versus BSA+DMSO or Prx2+DMSO (A). The direct H2O2-scavenging effect of obovatol was also evaluated by ferrous oxidation assay in the absence of recombinant Prx2 protein (B). BV-2 microglia cells were treated with 100 ng·mL−1 LPS for 30 min in the absence or presence of obovatol (10 µM), followed by 30 min incubation with 10 µM H2DCF-DA. Fluorescence was measured by flow cytometer. The results were expressed as the mean fluorescence intensity (C). DMSO, dimethyl sulfoxide; Prx2, peroxiredoxin 2; ROS, reactive oxygen species; DTT, dithiothreitol; BSA, bovine serum albumin.
Obovatol inhibited the LPS-induced ROS production in microglia
Lipopolysaccharide has been shown to induce the production of ROS such as H2O2 and hydroxyl radicals in microglia through the activation of NADPH oxidase, and these ROS are known to play important roles in the inflammatory signalling pathway (Asehnoune et al., 2004; Block et al., 2007). Thus, the effect of obovatol on LPS-induced ROS generation was examined using the ROS-sensitive indicator H2DCF-DA. The LPS-induced increase in ROS production was diminished by obovatol (Figure 10C). The suppressive effect of obovatol against LPS-induced ROS production was consistently observed up to 18 h (data not shown). These results indicate that obovatol augmented the peroxidase activity of Prx2 via direct interaction, in microglia. The critical role of NADPH oxidase and ROS in the inflammatory activation of microglia and NF-κB activation was confirmed using DPI (an NADPH oxidase inhibitor) and NAC (a ROS scavenger). Both DPI and NAC inhibited NO production in LPS-stimulated BV-2 microglia cells (Figure 11A). The phosphorylation of IκB, which leads to nuclear translocation of NF-κB, was also suppressed by both inhibitors (Figure 11B). These results indicate that LPS activates microglial inflammatory signalling through NADPH oxidase stimulation followed by ROS production. Apart from the fact that ROS play an important role in the microglial signalling, ROS released from activated microglia may also contribute to neuronal degeneration. Therefore, the neurotoxicity of NADPH oxidase-derived microglial ROS was examined by using microglia-conditioned media. The conditioned media prepared from LPS-stimulated microglia induced neuronal death: DPI or NAC attenuated the neuronal death under these conditions (Figure 11C). These results suggest that NADPH oxidase-derived ROS participate in both microglial activation and neurotoxicity. Obovatol may inhibit both aspect by targeting Prx2 and influencing redox regulation.
Figure 11.
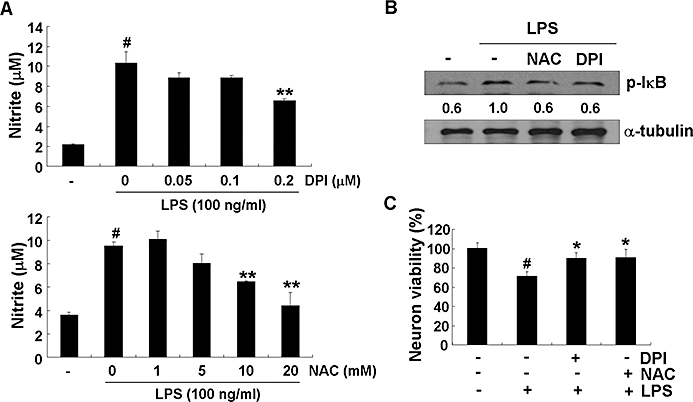
NADPH oxidase mediated LPS-induced microglial activation. BV-2 microglia cells were treated with DPI or NAC for 30 min before the addition of LPS (100 ng·mL−1). After 24 h, the nitrite in the media was measured by Griess reaction (A). After 30 min activation with LPS (100 ng·mL−1), the cells were harvested and analysed by immunoblot for phospho-IκB. Levels of phospho-IκB were normalized to α-tubulin levels and expressed as a fold decrease compared with the LPS treatment (B). Primary microglia were stimulated with LPS (100 ng·mL−1) in the absence or presence of DPI or NAC for 6 h, culture media were replaced with fresh media and further incubated for 24 h. The microglia-conditioned media were collected and transferred to primary neurons followed by additional 24 h incubation. At the end of incubation, cell viability was determined by MTT. Results were expressed as a percentage of control (mean ± SD) (C). The data were expressed as the mean ± SD (n= 3). #P < 0.01 versus untreated control; *P < 0.05, **P < 0.01 versus LPS only. LPS, lipopolysaccharide; DPI, diphenyliodonium; NAC, N-acetyl cysteine.
Discussion
After recognition of LPS by TLR4, both MyD88-dependent and -independent pathways are activated in microglia. In the MyD88-dependent pathway, the expression of inflammatory cytokine genes including TNFα, IL-6 and IL-1β are induced through NF-κB activation. In the MyD88-independent pathway, Toll-IL-1 receptor domain-containing adapter inducing IFN-β (TRIF)-mediated IFN regulatory factor 3 (IRF-3) activation is followed by IFN-β production, which may in turn initiate the STAT1 pathway. This pathway induces the expression of IFN-β-dependent genes such as MCP-5, IP10 and iNOS (Toshchakov et al., 2002; Yamamoto et al., 2003). It has been reported that MCP-1 recruits immune cells to sites of tissue injury and infection (Carr et al., 1994), and IP10 is secreted by several cell types in response to IFN-γ (Luster et al., 1985). IKKε mediates the catalytic activity that leads to phosphorylation of IRF-3 through MyD88-independent pathway in TLR4 signalling (Ikeda et al., 2007). Here, we showed that obovatol, a biphenyl ether lignan isolated from the medicinal plant Magnolia obovata, inhibited microglial activation by suppressing MyD88-dependent and MyD88-independent pathway. In addition, obovatol inhibited inflammatory activation of microglia by regulating intracellular ROS levels. Our results indicate that obovatol targets Prx2 to augment its ability to scavenge ROS, thereby blunting the stimulatory signalling of LPS (Figure 12). ROS such as H2O2, superoxide anions and hydroxyl radicals are commonly produced during inflammatory processes, and they are important components of various intracellular signalling pathways (Deyulia et al., 2005). However, excessive oxidative stress causes cell injury and ultimately cell death (Block et al., 2007). Previously, it has been demonstrated that LPS induces production of ROS via NADPH oxidase activation and leads to activation of NF-κB, MAPKs, STAT and secretion of proinflammatory cytokines (Schreck et al., 1991; Guyton et al., 1996; Ihle, 2001; Asehnoune et al., 2004; Block et al., 2007). Microglia consistently generate ROS when activated by multiple immunological stimuli, which can induce neuronal degeneration (Block et al., 2007). These earlier reports suggested that microglia-derived ROS was implicated in the mechanism of proinflammatory signalling in microglia. However, despite the fact that ROS have been closely related to inflammatory signalling, the molecular mechanisms whereby ROS interact with NF-κB and MAPKs activation pathways and the ensuing inflammatory gene expression remain poorly understood. Previous reports suggest that LPS-stimulated ROS inactivate protein tyrosine phosphatase, which leads to an increase of tyrosine phosphorylation in the key signalling molecules, thereby ultimately enhancing the signal transduction pathways such as IKK, MAPKs and STAT pathway (Rhee et al., 2003; Pawate et al., 2004). The role of TLR4 in LPS-induced ROS generation and their signalling is not consistent. In previous studies, ROS influenced early signalling events of the TLR4 pathway and regulated TLR4-induced gene expression (Asehnoune et al., 2004). Other studies demonstrated that ROS contributed to proinflammatory gene expression of both the TLR4-dependent and TLR4-independent pathways in microglia using TLR4 or NADPH oxidase deficient mice (Qin et al., 2005). Whether ROS directly cross-talk with the TLR4 pathway remains to be further elucidated. However, it is well established that ROS, especially H2O2 as intracellular second messenger, enhance the LPS-induced proinflammatory pathway and play a critical role in microglial activation through NF-κB and MAPK pathway. Therefore, the regulation of ROS levels derived from NADPH oxidase is a key process that dictates the intracellular signalling pathways that are associated with inflammatory activation of microglia. Because LPS stimulation model was mainly used in this study, the effect of obovatol on the LPS/TLR4 pathway has been emphasized. However, obovatol may affect multiple pathways beyond simple MyD88-dependent and -independent TLR4 signalling, as the compound also inhibited IFN-γ- or ATP-induced microglial activation. It is possible that obovatol acts through ROS regulation, which is a common component of multiple inflammatory signalling pathways such as LPS, cytokines and nucleotides (Brown, 2007; Cruz et al., 2007).
Figure 12.
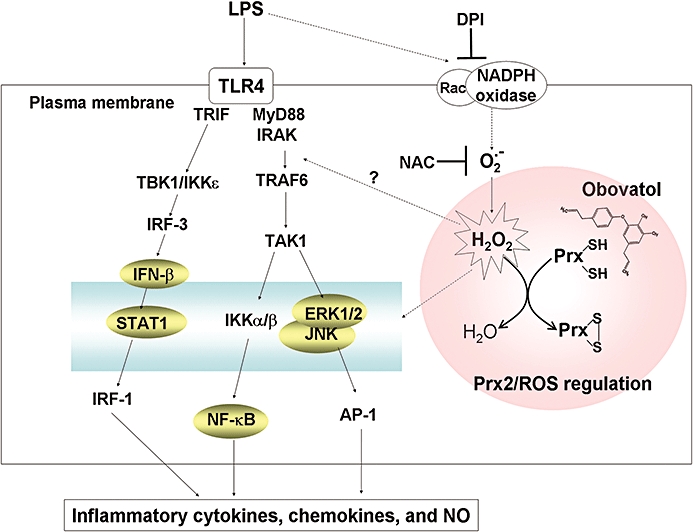
Possible mechanism(s) by which obovatol inhibits microglial inflammatory signalling through targeting Prx2. LPS initiates NADPH oxidase activation and ROS (H2O2) production. H2O2 is believed to modulate phosphorylation of the key signalling proteins in TLR4 signalling. H2O2 may also participate in the activation of NF-κB, MAPK and STAT pathways, which lead to the release of proinflammatory cytokines, chemokines and NO. Prx2 (reduced form) reduces H2O2 through the formation of disulfide bond (oxidized form), thereby suppressing the proinflammatory LPS/TLR4 signalling. Obovatol binds to Prx2 and enhances its peroxidase activity, ultimately down-regulating the LPS/TLR4 signalling. The signalling components on which the effect of obovatol was determined in this study are indicated by shaded circles. Dotted lines represent incompletely defined pathways. Prx2, peroxiredoxin 2; LPS, lipopolysaccharide; ROS, reactive oxygen species; NF-κB, nuclear factor κB; MAPK, mitogen-activated protein kinase; STAT, signal transducers and activators of transcription; NO, nitric oxide; TLR4, Toll-like receptor 4.
Peroxiredoxins are highly abundant in cells and exert their peroxidase activities by catalysing the reduction of H2O2, peroxynitrite and organic peroxides through the oxidation and subsequent reduction of cysteine residues (Rhee et al., 2005). Mammalian Prx proteins consist of six distinct members that can be grouped as 2-Cys or 1-Cys Prx according to their structural characteristics. Of these, Prx1 and Prx2, which were identified as target proteins of obovatol, belong to 2-Cys Prx group and are localized in the cytosol. Although both Prx1 and Prx2 play a role as regulators of H2O2 signalling via their peroxidase activity, Prx1 is involved in tumour suppression (Neumann et al., 2003), while Prx2 is a known regulator of NF-κB and MAPK activation (Kang et al., 1998; Kang et al., 2004). In the current study which focused on Prx2, we demonstrated that Prx2 serves to scavenge LPS-induced ROS, and obovatol enhances this peroxidase activity of Prx2. Thus, the inhibitory effect of obovatol against microglial activation and neuroinflammation is mediated by augmenting the ROS-scavenging activity of Prx2. This contention was supported by Prx2 activity assay in the cell-free system (Figure 10A). Physiologically, the catalytic action of Prx2 requires the reduction of disulfide in their structure by thioredoxin reductase (Kim et al., 2005). These reduced states can also be established by treatment of low molecular weight reductant such as DTT. In this study, H2O2-reducing activity of Prx2 was measured in reaction mixtures containing DTT as an electron donor. Measurement of microglial ROS further supported the proposal that Prx2 is the molecular target of obovatol: LPS-induced ROS production in microglia cells was attenuated by obovatol (Figure 10C). However, one cannot exclude the possibility that obovatol interacts with Prx1 or other proteins to modulate microglial activation. Further studies are needed to elucidate the role of Prx1 as another potential target of obovatol in microglia cells. In addition, the validation of Prx2 as one of the potential targets of obovatol was by indirect experiments. Our results should be verified by using Prx2 knockout cells and mice in further studies. Obovatol may well act on multiple target proteins in microglia cells. The effect of obovatol alone in vivo was not examined in this study. There is not enough evidence to conclude that obovatol alone has no effect in vivo. However, whether the compound alone inhibits the basal level of microglial activation in the control mice would not be likely to change our conclusion that the compound attenuates LPS-induced microglial activation.
In LPS/TLR4 signalling, it is thought that Prx2 regulates phosphatase oxidation and the phosphorylation of key signalling proteins through reduction of LPS-induced H2O2. Previous studies suggested that ROS may regulate any intermediate steps, including TLR4-dependent activation of IL-1 receptor-associated kinase and subsequent TNFα receptor-associated factor 6-induced activation of MAPK kinase kinase family members, which account for the redox sensitivity of these signalling components (Asehnoune et al., 2004; Pawate et al., 2004). Yang et al. (2007) have demonstrated, using Prx knockout cells and mice, that Prx2 is an essential component in the regulation of LPS-induced inflammatory gene expression and for the activation of NF-κB and MAPKs through the modulation of NADPH oxidase activities and ROS signalling. These studies are in agreement with the critical role of Prx2 and ROS in inflammatory signalling of microglia.
In conclusion, we present evidence that the natural product obovatol exerts anti-microglial and anti-neuroinflammatory effects in vitro and in vivo. Subsequent mechanistic studies indicated that obovatol targetted cellular Prx2 protein in order to increase its ROS-scavenging activity. As ROS are the pivotal component of inflammatory signalling of microglia, obovatol-mediated reduction of ROS leads to the inhibition of multiple inflammatory signalling pathways in microglia. In this study, the natural compound obovatol has been successfully used as a chemical probe to interrogate the microglial signalling pathway, and the compound is regarded as a potential drug candidate against neuroinflammatory diseases.
Acknowledgments
This work was supported by the Korea Science and Engineering Foundation (KOSEF) grant funded by the Korea government (MEST) (No. 2009-0078941) and Bio R&D program through the Korea Science and Engineering Foundation funded by the Ministry of Education, Science and Technology (2008-04090). B.-M.K., S.Y.L. and Y.-M.H. were supported by the Plant Diversity Research Center of the 21st Century Frontier Research Program.
Glossary
Abbreviations:
- ERK
extracellular signal-regulated kinases
- HAPI
highly aggressively proliferating immortalized
- IFN
interferon
- IKK
inhibitor of kappaB kinase
- IL-1
interleukin 1
- iNOS
inducible nitric oxide synthase
- IP10
interferon-inducible protein 10
- IRF-3
interferon regulatory factor 3
- JNK
c-jun N-terminal kinase
- LPS
lipopolysaccharide
- MAPK
mitogen-activated protein kinase
- MCP
monocyte chemotactic protein
- MIP
macrophage inflammatory protein
- NF-κB
nuclear factor κB
- Prx2
peroxiredoxin 2
- STAT1
signal transducers and activators of transcription 1
- TLR4
Toll-like receptor 4
- TNF
tumour necrosis factor
Conflict of interest
The authors have no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article:
Appendix S1 Methods.
Figure S1 Obovatol partly prevented neuronal death from chronically activated microglial toxicity. Primary microglia were pretreated with obovatol for 30 min and washed. CMFDA-labelled primary neurons were added to microglia culture containing LPS (100 ng·mL−1). The LPS-containing media were replaced every 24 h over a 72 h period. At the end of the coculture, the CMFDA-positive neurons were examined under a fluorescence microscope. Representative fluorescence or phase contrast images of cells are shown. Scale bar, 50 µm.
Figure S2 Hierarchical clustering of the 108 up- or down-regulated genes by obovatol treatment. Genes that passed appropriate filter criteria were subjected to a hierarchical clustering. The homology tree plot is shown left. The clustering was based on the 108 genes, which showed the recovery pattern for the expression levels when treated with obovatol as compared with LPS-treated condition (A). Hierarchical clustering was also performed for the genes related to immunity/defense (B; left) or signal transduction (B; right) among the 108 genes.
Figure S3 Obovatol influenced the Toll-like receptor signalling pathway and cytokine-cytokine receptor interaction pathways. Differentially expressed genes (LPS alone vs. obovatol+LPS) were analysed using GenPlex™ software and KEGG database. Red-lined boxes represent differentially expressed genes by obovatol treatment compared with LPS treatment. Green filled boxes represent down-regulated genes. The probe ID of differentially expressed gene was shown in blue.
Table S1 Gene ontology (GO) term analysis of the 108 genes, which were differentially expressed by obovatol compared with LPS treatment.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki W, Yuasa K, Takeda S, Shirotani K, Takahashi K, Tabira T. Overexpression of presenilin-2 enhances apoptotic death of cultured cortical neurons. Ann N Y Acad Sci. 2000;920:241–244. doi: 10.1111/j.1749-6632.2000.tb06929.x. [DOI] [PubMed] [Google Scholar]
- Asehnoune K, Strassheim D, Mitra S, Kim JY, Abraham E. Involvement of reactive oxygen species in Toll-like receptor 4-dependent activation of NF-kappa B. J Immunol. 2004;172:2522–2529. doi: 10.4049/jimmunol.172.4.2522. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression in endotoxin-stimulated primary glial cultures. J Neurosci. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasi E, Barluzzi R, Bocchini V, Mazzolla R, Bistoni F. Immortalization of murine microglial cells by a v-raf/v-myc carrying retrovirus. J Neuroimmunol. 1990;27:229–237. doi: 10.1016/0165-5728(90)90073-v. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Brown GC. Mechanisms of inflammatory neurodegeneration: iNOS and NADPH oxidase. Biochem Soc Trans. 2007;35:1119–1121. doi: 10.1042/BST0351119. [DOI] [PubMed] [Google Scholar]
- Carr MW, Roth SJ, Luther E, Rose SS, Springer TA. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc Natl Acad Sci U S A. 1994;91:3652–3656. doi: 10.1073/pnas.91.9.3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheepsunthorn P, Radov L, Menzies S, Reid J, Connor JR. Characterization of a novel brain-derived microglial cell line isolated from neonatal rat brain. Glia. 2001;35:53–62. doi: 10.1002/glia.1070. [DOI] [PubMed] [Google Scholar]
- Choi MS, Lee SH, Cho HS, Kim Y, Yun YP, Jung HY, et al. Inhibitory effect of obovatol on nitric oxide production and activation of NF-kappaB/MAP kinases in lipopolysaccharide-treated RAW 264.7cells. Eur J Pharmacol. 2007;556:181–189. doi: 10.1016/j.ejphar.2006.10.054. [DOI] [PubMed] [Google Scholar]
- Cruz CM, Rinna A, Forman HJ, Ventura AL, Persechini PM, Ojcius DM. ATP activates a reactive oxygen species-dependent oxidative stress response and secretion of proinflammatory cytokines in macrophages. J Biol Chem. 2007;282:2871–2879. doi: 10.1074/jbc.M608083200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeYulia GJ, Jr, Carcamo JM, Borquez-Ojeda O, Shelton CC, Golde DW. Hydrogen peroxide generated extracellularly by receptor-ligand interaction facilitates cell signaling. Proc Natl Acad Sci U S A. 2005;102:5044–5049. doi: 10.1073/pnas.0501154102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton KZ, Gorospe M, Kensler TW, Holbrook NJ. Mitogen-activated protein kinase (MAPK) activation by butylated hydroxytoluene hydroperoxide: implications for cellular survival and tumor promotion. Cancer Res. 1996;56:3480–3485. [PubMed] [Google Scholar]
- Hellendall RP, Ting JP. Differential regulation of cytokine-induced major histocompatibility complex class II expression and nitric oxide release in rat microglia and astrocytes by effectors of tyrosine kinase, protein kinase C, and cAMP. J Neuroimmunol. 1997;74:19–29. doi: 10.1016/s0165-5728(96)00202-0. [DOI] [PubMed] [Google Scholar]
- Ihle JN. The Stat family in cytokine signaling. Curr Opin Cell Biol. 2001;13:211–217. doi: 10.1016/s0955-0674(00)00199-x. [DOI] [PubMed] [Google Scholar]
- Ikeda F, Hecker CM, Rozenknop A, Nordmeier RD, Rogov V, Hofmann K, et al. Involvement of the ubiquitin-like domain of TBK1/IKK-i kinases in regulation of IFN-inducible genes. Embo J. 2007;26:3451–3462. doi: 10.1038/sj.emboj.7601773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Iida T, Ichino K, Tsunezuka M, Hattori M, Namba T. Obovatol and obovatal, novel biphenyl ether lignans from the leaves of Magnolia obovata Thunb. Chem Pharm Bull (Tokyo) 1982;30:3347–3353. doi: 10.1248/cpb.30.3347. [DOI] [PubMed] [Google Scholar]
- Jongeneel CV. Transcriptional regulation of the tumor necrosis factor alpha gene. Immunobiology. 1995;193:210–216. doi: 10.1016/s0171-2985(11)80545-0. [DOI] [PubMed] [Google Scholar]
- Kang SW, Chae HZ, Seo MS, Kim K, Baines IC, Rhee SG. Mammalian peroxiredoxin isoforms can reduce hydrogen peroxide generated in response to growth factors and tumor necrosis factor-alpha. J Biol Chem. 1998;273:6297–6302. doi: 10.1074/jbc.273.11.6297. [DOI] [PubMed] [Google Scholar]
- Kang SW, Chang TS, Lee TH, Kim ES, Yu DY, Rhee SG. Cytosolic peroxiredoxin attenuates the activation of Jnk and p38 but potentiates that of Erk in Hela cells stimulated with tumor necrosis factor-alpha. J Biol Chem. 2004;279:2535–2543. doi: 10.1074/jbc.M307698200. [DOI] [PubMed] [Google Scholar]
- Kim S, Ock J, Kim AK, Lee HW, Cho JY, Kim DR, et al. Neurotoxicity of microglial cathepsin D revealed by secretome analysis. J Neurochem. 2007;103:2640–2650. doi: 10.1111/j.1471-4159.2007.04995.x. [DOI] [PubMed] [Google Scholar]
- Kim JA, Park S, Kim K, Rhee SG, Kang SW. Activity assay of mammalian 2-cys peroxiredoxins using yeast thioredoxin reductase system. Anal Biochem. 2005;338:216–223. doi: 10.1016/j.ab.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Kleinert H, Pautz A, Linker K, Schwarz PM. Regulation of the expression of inducible nitric oxide synthase. Eur J Pharmacol. 2004;500:255–266. doi: 10.1016/j.ejphar.2004.07.030. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Kwon BM, Kim MK, Lee SH, Kim JA, Lee IR, Kim YK, et al. Acyl-CoA: cholesterol acyltransferase inhibitors from Magnolia obovata. Planta Med. 1997;63:550–551. doi: 10.1055/s-2006-957762. [DOI] [PubMed] [Google Scholar]
- Lee SK, Kim HN, Kang YR, Lee CW, Kim HM, Han DC, et al. Obovatol inhibits colorectal cancer growth by inhibiting tumor cell proliferation and inducing apoptosis. Bioorg Med Chem. 2008;16:8397–8402. doi: 10.1016/j.bmc.2008.08.033. [DOI] [PubMed] [Google Scholar]
- Luster AD, Unkeless JC, Ravetch JV. Gamma-interferon transcriptionally regulates an early-response gene containing homology to platelet proteins. Nature. 1985;315:672–676. doi: 10.1038/315672a0. [DOI] [PubMed] [Google Scholar]
- McGeer EG, McGeer PL. Inflammatory processes in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:741–749. doi: 10.1016/S0278-5846(03)00124-6. [DOI] [PubMed] [Google Scholar]
- Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, et al. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424:561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005;308:1314–1318. doi: 10.1126/science.1110647. [DOI] [PubMed] [Google Scholar]
- Ock J, Lee H, Kim S, Lee WH, Choi DK, Park EJ, et al. Induction of microglial apoptosis by corticotropin-releasing hormone. J Neurochem. 2006;98:962–972. doi: 10.1111/j.1471-4159.2006.03933.x. [DOI] [PubMed] [Google Scholar]
- Pawate S, Shen Q, Fan F, Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res. 2004;77:540–551. doi: 10.1002/jnr.20180. [DOI] [PubMed] [Google Scholar]
- Pyo MK, Lee Y, Yun-Choi HS. Anti-platelet effect of the constituents isolated from the barks and fruits of Magnolia obovata. Arch Pharm Res. 2002;25:325–328. doi: 10.1007/BF02976634. [DOI] [PubMed] [Google Scholar]
- Qin L, Li G, Qian X, Liu Y, Wu X, Liu B, et al. Interactive role of the toll-like receptor 4 and reactive oxygen species in LPS-induced microglia activation. Glia. 2005;52:78–84. doi: 10.1002/glia.20225. [DOI] [PubMed] [Google Scholar]
- Qin L, Wu X, Block ML, Liu Y, Breese GR, Hong JS, et al. Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia. 2007;55:453–462. doi: 10.1002/glia.20467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Chang TS, Bae YS, Lee SR, Kang SW. Cellular regulation by hydrogen peroxide. J Am Soc Nephrol. 2003;14:S211–S215. doi: 10.1097/01.asn.0000077404.45564.7e. [DOI] [PubMed] [Google Scholar]
- Saura J, Tusell JM, Serratosa J. High-yield isolation of murine microglia by mild trypsinization. Glia. 2003;44:183–189. doi: 10.1002/glia.10274. [DOI] [PubMed] [Google Scholar]
- Schreck R, Rieber P, Baeuerle PA. Reactive oxygen intermediates as apparently widely used messengers in the activation of the NF-kappa B transcription factor and HIV-1. Embo J. 1991;10:2247–2258. doi: 10.1002/j.1460-2075.1991.tb07761.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Muller MM, Schaffner W. Rapid detection of octamer binding proteins with ‘mini-extracts’, prepared from a small number of cells. Nucleic Acids Res. 1989;17:6419. doi: 10.1093/nar/17.15.6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Heinemann S, Carlisle W, Tarikas H, Kimes B, Patrick J, et al. Clonal cell lines from the rat central nervous system. Nature. 1974;249:224–227. doi: 10.1038/249224a0. [DOI] [PubMed] [Google Scholar]
- Seo JJ, Lee SH, Lee YS, Kwon BM, Ma Y, Hwang BY, et al. Anxiolytic-like effects of obovatol isolated from Magnolia obovata: involvement of GABA/benzodiazepine receptors complex. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1363–1369. doi: 10.1016/j.pnpbp.2007.05.009. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Stoll G, Jander S. The role of microglia and macrophages in the pathophysiology of the CNS. Prog Neurobiol. 1999;58:233–247. doi: 10.1016/s0301-0082(98)00083-5. [DOI] [PubMed] [Google Scholar]
- Thurman RG, Ley HG, Scholz R. Hepatic microsomal ethanol oxidation. Hydrogen peroxide formation and the role of catalase. Eur J Biochem. 1972;25:420–430. doi: 10.1111/j.1432-1033.1972.tb01711.x. [DOI] [PubMed] [Google Scholar]
- Toshchakov V, Jones BW, Perera PY, Thomas K, Cody MJ, Zhang S, et al. TLR4, but not TLR2, mediates IFN-beta-induced STAT1alpha/beta-dependent gene expression in macrophages. Nat Immunol. 2002;3:392–398. doi: 10.1038/ni774. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yang CS, Lee DS, Song CH, An SJ, Li S, Kim JM, et al. Roles of peroxiredoxin II in the regulation of proinflammatory responses to LPS and protection against endotoxin-induced lethal shock. J Exp Med. 2007;204:583–594. doi: 10.1084/jem.20061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng LT, Ock J, Kwon BM, Suk K. Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. Int Immunopharmacol. 2008;8:484–494. doi: 10.1016/j.intimp.2007.12.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.