

Abstract
Mycobacteria use a unique system for covalently modifying proteins based on the conjugation of a small protein, referred to as prokaryotic ubiquitin-like protein (PUP). In this study, we report a proteome-wide analysis of endogenous pupylation targets in the model organism Mycobacterium smegmatis. On affinity capture, a total of 243 candidate pupylation targets were identified by two complementary proteomics approaches. For 41 of these protein targets, direct evidence for a total of 48 lysine-mediated pupylation acceptor sites was obtained by collision-induced dissociation spectra. For the majority of these pupylation targets (38 of 41), orthologous genes are found in the M. tuberculosis genome. Interestingly, approximately half of these proteins are involved in intermediary metabolism and respiration pathways. A considerable fraction of the remaining targets are involved in lipid metabolism, information pathways, and virulence, detoxification and adaptation. Approximately one-third of the genes encoding these targets are located in seven gene clusters, indicating functional linkages of mycobacterial pupylation targets. A comparison of the pupylome under different cell culture conditions indicates that substrate targeting for pupylation is rather dynamic.
Keywords: covalent modification, gene clustering, mycobacteria, proteomics, pupylation
Introduction
The posttranslational modification of proteins by ubiquitin-like modifiers has attracted much interest in recent years. Ubiquitin conjugation, originally linked to protein degradation by the proteasome, is known to control many kinds of biological function and physiological activity (Hershko and Ciechanover, 1986, 1998; Crosas et al, 2006; Grabbe and Dikic, 2009). Recently, a small protein (Rv2111c) from Mycobacterium tuberculosis has been described as a prokaryotic ubiquitin-like protein (PUP) (Pearce et al, 2008; Burns et al, 2009), constituting the first known ubiquitin-like system in bacteria. The PUP-related genes are confined to the actinobacteria phylum and are frequently observed in proximity to genes encoding protein components of the proteasome (Iyer et al, 2008).
To date, two proteins have been identified as part of the PUP conjugation machinery, DOP (Rv2112c) and PafA (Rv2097c), possessing deamidase and putative PUP ligase activities, respectively (Striebel et al, 2009). In addition, three potential mycobacterial PUP substrates have been found (Pearce et al, 2008; Burns et al, 2009): (1) malonyl coenzyme A acyl carrier protein (FabD) that is involved in cell wall biosynthesis (Kremer et al, 2002); (2) myoinositol-1-phosphate synthase (MIPS) that catalyzes the first reaction step of the biosynthesis pathway of L-myo-inositol-1-phosphate (Stieglitz et al, 2005); and (3) superoxide dismutase (SOD) that has a key part in the cellular detoxification of superoxide anions (Kusunose et al, 1976). However, it is not known whether the restricted number of reported targets reflects a current limitation in their detection, or is the result of their relative transience, due to their rapid proteasomal degradation.
To explore the impact of pupylation as a new type of functional regulation in mycobacterial biology, it is essential to explore to what extent components of the mycobacterial proteome are targeted for pupylation. In this study, we used the endogenous PUP conjugation/processing machinery from M. smegmatis to identify potential PUP targets from enriched purified material. Using two complementary proteomics approaches, we have observed a total of 48 pupylation sites in 41 mycobacterial substrates. The majority of these targets are encoded by gene loci clustered in restricted regions of the M. smegmatis genome.
Results and discussion
Identification of PUP substrates from the M. smegmatis proteome
To provide insight into the overall properties of PUP, we first expressed the M. tuberculosis protein (Rv2111c, mtPUP) in Escherichia coli and purified it to homogeneity (Figure 1A). Further biophysical characterization is described in the Supplementary information.
Figure 1.

SDS–PAGE gel analysis of mtPUP, expressed in Escherichia coli and Mycobacterium smegmatis. All lanes contain protein eluates, after the NiNTA purification step. (A) mtPUP expression in E. coli. Lane 1, protein marker; and lane 2, polyhistidine-tagged PUP. (B) mtPUP expression in M. smegmatis. Lane 1, protein marker; lane 2, polyhistidine-tagged mtPUP; and lane 3, control with vector without insertion. Additional control expression in M. smegmatis: lane 4, polyhistidine-tagged mtPUP(Q64A); lane 5, polyhistidine-tagged Rv3874 (mtCFP-10).
To identify potential PUP substrates, we also expressed N-terminally poly-histidine-tagged mtPUP in M. smegmatis, which is a well-established model system (Hatfull et al, 2008). Affinity-purified mtPUP gave rise to multiple bands when assessed by SDS–PAGE (Figure 1B, Supplementary Figure 1A and B) and by western blot analysis (Supplementary Figure 1C). When using a mtPUP(Q64A) variant the multiple band pattern was lacking, indicating that it is caused by specific pupylation involving Gln64 of PUP. The same result was obtained when using a M. tuberculosis control target (Rv3874). Supporting this observation, mtPUP heterologously expressed in E. coli also migrated as a single band after nickel-nitrilotriacetic acid (NiNTA) affinity purification, showing that the effect is specific to M. smegmatis (Figure 1A).
We first used in-solution digestion followed by two-dimensional (2D) liquid chromatography, both by offline strong cation exchange (SCX) followed by online reversed phase (RP), and by electrospray ionization (ESI) tandem mass spectrometry (MS/MS), using a quadrupole-time-of-flight instrument (QqTOF). The aim of this approach was to obtain a high coverage of potential pupylation targets. NiNTA eluate protein fractions from M. smegmatis served as source material for this analysis. To minimize co-enrichment of indirect binders, the purification was carried out under denaturing conditions. To permit the recognition of unspecific binders to the NiNTA matrix, a parallel purification was carried out side-by-side from M. smegmatis cells containing the plasmid without an inserted mtPUP gene. To rule out the possibility that differences in the lists of observed proteins are the result of run-to-run variance during the SCX and RP separations, we incorporated isobaric tagging for relative and absolute quantification (iTRAQ) of the control and specific eluate digests into the workflow. The 2D liquid chromatography served to reduce sample complexity. Only two proteins (GroEL2, MSMEG_1583; CFP29, MSMEG_5830) were found in negative control experiments as well, probably because of the presence of sequence segments with an ability to bind to the NiNTA matrix with high affinity. The corresponding peptide peaks were subtracted from the data set.
Cumulatively, the analysis led to the identification of 1661 peptides that were observed only in the specific sample. They could be assigned to 243 putative pupylation targets, referred to as ‘candidate targets' (Supplementary Table 1). For a protein to be considered as a target, it had to be identified based on the confident assignment of at least two strong collision-induced dissociation (CID) spectra. The analysis allowed a direct assignment of pupylation sites to 32 mycobacterial target proteins, referred to as ‘validated targets' (Table I). For one of these, acyl carrier protein (MSMEG_4326), two distinct pupylation sites were observed. All pupylated peptides that were identified contained an internal lysine residue, which, due to the loss of the positive charge as a result of the bulky PUP conjugation to the side chain ɛ-amine, lost the ability to function as a tryptic cleavage site (Figure 2A and B). This observation independently corroborated the evidence for correct pupylation assignment.
Table 1. Protein targets from M. smegmatis with one or more confirmed PUP modifications.
| Protein targets with pupylated sequences that have been identified by both methods, ESI QqTOF mass spectrometry analysis and 2D gel analysis, are highlighted in gray. Column annotation: Global MS, peptide index from ESI-QqTOF analysis (Supplementary Table 1); 2D gel, peptide index from 2D gel electrophoresis (Supplementary Table 2); App. MW (calculated), apparent MW versus calculated MW (all targets identified by 2D gel electrophoresis show an apparent MW that exceeds the calculated MW of the same target); Gene locus (M. smegmatis); Detected pupylation peptides, PUP GGE sites are highlighted by superscript; Pupylated lysines, sequence residue numbers; Gene locus (M. tuberculosis) for M. smegmatis genes with identified paralogs in M. tuberculosis; Conservation of lysine pupylation site, based on M. smegmatis/M. tuberculosis sequence alignments of targets genes (data not shown); Functional annotation (M. tuberculosis); Functional categorization (M. tuberculosis), category identifiers have been taken from (Camus et al, 2002). 0, virulence, detoxification, adaptation; 1, lipid metabolism; 2, information pathways; 3, cell wall and cell processes; 7, intermediary metabolism and respiration; 8, proteins of unknown function; 9, regulatory proteins; 10, conserved hypothetic proteins. Target gene clusters, pair or multiple pupylation targets that are encoded from genes with neighboring loci, generally found in joint operons, are indicated I–VI. |
|---|
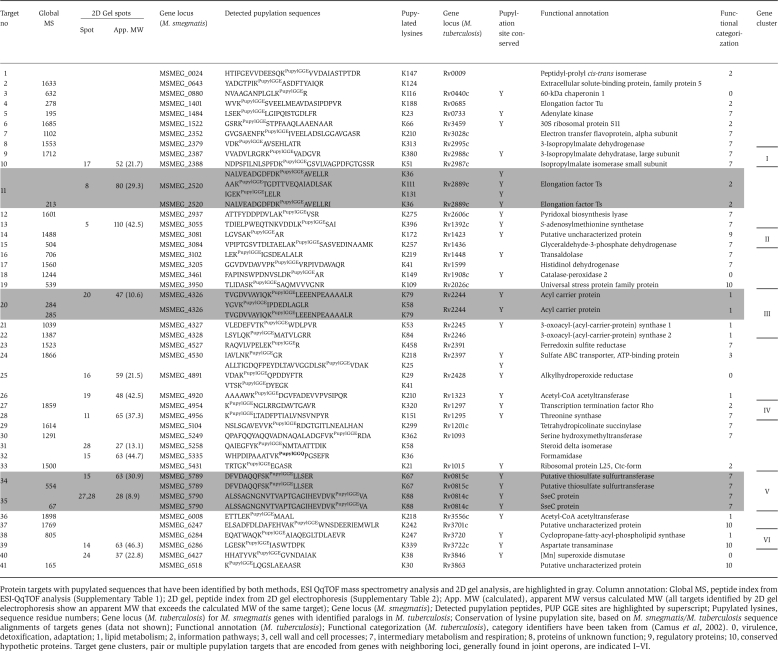 |
Figure 2.

Specific enrichment of pupylated peptide in polyhistidine affinity captured eluate. (A, B) Annotated CID spectra that contributed to the identification of (A) MSMEG_2352 and (B) MSMEG_4326 as a pupylation targets. The CID spectrum of (A) was derived by fragmentation of a quadruple-charged precursor ion observed at m/z [766.91+4H]4+ and was matched to the branched peptide GVGSAENFK(QGG)IVEELADSLGGAVGASR carrying iTRAQ labels at the N-termini of both its main and branched chain. Please note the absence of a detectable iTRAQ114 signature mass peak (inset), indicating that this peptide was exclusively contributed by the pupylated iTRAQ 115-labeled sample. Also note the double charged nature of branched peptide fragments due to the retention of an additional charge by the primary amine present within the N-terminal glycine of the GGQ pupylation stub. The CID spectrum of (B) was derived by fragmentation of a triple charged precursor ion observed at m/z [693.37 +3H]3+ and was matched to the branched peptide YGVK(QGG)IPDEDLAGLR, carrying iTRAQ labels at the N-termini of both its main and branched chain. For information on additional peptides see Table I. (C) Complementary 2D gel analysis. Pupylated substrates were separated on a 2D gel and spots are numbered, which are subjected to tandem MS/MS analyses. The numbering of the spots corresponds to the numbering used in the tables. The pH range of the first dimension and apparent molecular weight of second dimension are denoted.
In parallel, the same samples were subjected to a complementary 2D gel electrophoresis analysis (Figure 2C). Single spots from the 2D gel were trypsinized and the resulting peptides were analyzed by ESI MS/MS (Supplementary Table 2). This approach led to the identification of 133 peptides that could be assigned to 13 validated pupylation substrates, covering a total of 17 confirmed pupylation sites (Table I). Three substrates were observed to be pupylated on more than a single lysine site: elongation factor Ts (MSMEG_2520, three sites, total number of lysines: 24), acyl carrier protein (MSMEG_4326, two sites, total number of lysines: 5) and alkylhydroperoxide reductase (MSMEG_4391, three sites, total number of lysines: 11).
For further analysis, we pooled the identified pupylated protein sequences and sorted them with respect to the gene loci organization in M. smegmatis (see Table I for pooled validated targets and Supplementary Table 3A for pooled candidate targets). The resulting list comprises 41 validated mycobacterial pupylation targets with a total of 48 non-redundant lysine-mediated pupylation sites. Owing to the direct demonstration of this type of lysine conjugation, these sequences can be considered to be unambiguously pupylated under the chosen experimental conditions. All candidate pupylation substrates revealed by 2D gel analysis were also detected in the ESI QqTOF analysis, except for MSMEG_4298, confirming the validity of the data (Supplementary Table 1). Remarkably, for four new validated targets we independently found direct evidence for pupylation at identical sites by both methods: elongation factor Ts (MSMEG_2520), acyl carrier protein (MSMEG_4326), a putative thiosulfate sulfurtransferase (MSMEG_5789) and SseC (MSMEG_5790). All three previously identified pupylation targets (FabD, MIPS and SOD) were also observed in the complete list of potential candidate pupylation targets (Supplementary Table 1).
We also observed auto-pupylation at two sites, K31 and K61. However, as we were not able to distinguish between auto-pupylation of endogenous PUP from M. smegmatis and overexpressed mtPUP, due to the high sequence similarity of the two proteins (92% identity), we cannot rule out the possibility that some of the observed auto-pupylation may not reflect a physiological situation.
To analyze to what extent the M. smegmatis pupylome may vary during cell growth and under stress conditions, we monitored M. smegmatis culture growth in the presence of established stress inducing agents and protocols (Darwin et al, 2003), including hydrogen peroxide (to mimic oxidative stress), sodium nitrite (to mimic nitrosative stress) and epoxomicin (to inhibit proteasomal degradation; Supplementary Figure 2). Under the latter two conditions, the M. smegmatis growth was completely inhibited, whereas applied oxidative stress did not lead to any significant growth rate changes. In addition, it is worth mentioning that PUP overexpression on its own reduced M. smegmatis growth about three-fold, when compared with wild-type M. smegmatis cell cultures lacking a heterologous expression vector. In a subsequent 2D DIGE experiment (Supplementary Figure 3, Supplementary Table 4), we have identified and validated a total of six spots observed in all three gels. These spots show substantial intensity variations under the three different tested conditions, indicating that the mycobacterial pupylome is dynamic.
Genetic and functional clustering of pupylation targets
Although the genomes of several M. smegmatis strains have been sequenced (http://www.ncbi.nlm.nih.gov/nuccore/CP000480), no systematic functional annotation and categorization has yet been published. However, the vast majority (36/41) of our validated M. smegmatis targets with pupylation sites is mirrored by homologous proteins in M. tuberculosis H37Rv (Table I). Therefore, we have made use of available analyses of the related genome from the M. tuberculosis H37Rv strain, in which the proteome was divided into ten different functional categories (Camus et al, 2002). A total of 19 of these targets (47%) with homologs in M. tuberculosis H37Rv are involved in intermediary metabolism and respiration pathways. Substantial numbers of targets are also found for categories ‘lipid metabolism' (six targets), ‘virulence, detoxification, adaptation' (four targets) and ‘information pathways' (five targets). These categories are thought to be rich in potential targets that could become useful for future drug discoveries (Zhang, 2005).
When expanding this analysis to the complete list of M. smegmatis candidate targets we have identified as candidate pupylation substrates (Supplementary Table 3B), we notice that many of our observations for either overrepresentation or underrepresentation of specific functional target categories in the M. tuberculosis proteome serving as potential pupylation substrates are preserved (Figure 3). In our analysis, there is a distinct accumulation of potential pupylation substrate targets involved virulence, detoxification and adaptation (category 0), lipid metabolism (category 1), information pathways (category 2) and intermediary metabolism and regulation (category 7), when compared with the overall presentation of targets from these categories within the entire proteome. In contrast, the number of potential pupylation substrate targets involved in cell wall and cell processes (category 3) is small, compared with its overall presentation within the complete proteome. Finally, we have observed no evidence for any pupylation substrates that are identified as targets in functional categories 4, 5 and 6.
Figure 3.

Functional analysis of pupylation targets. Relative distribution of validated PUP targets (gray bars, for details see Table I) and candidate PUP targets (black bars, for details see Supplementary Tables 3A and B) with M. tuberculosis homologs for different functional categories (Camus et al, 2002). The functional distribution of the M. tuberculosis proteome is shown for comparison (white bars).
We have also carried out an analysis of the pupylated targets in terms of gene locus organization. Strikingly, a substantial fraction of the identified targets with identified conjugation sites (13/41) is located in six gene clusters, I–VI (Table I), containing at least two genes encoded within one operon or in close proximity. This observation suggests that protein target pupylation could be correlated with functional linkages (Dandekar et al, 1998; Overbeek et al, 1999; Pellegrini et al, 1999). Noticeably, gene locus clustering of potential pupylation targets becomes even more obvious when the complete list of candidate pupylation targets is taken into account (Supplementary Table 3A).
Outlook
The availability of the complete genome and the corresponding proteome of M. tuberculosis has revolutionized fundamental research and generated new approaches to study disease mechanisms (Cole et al, 1998; Mattow et al, 2001). However, little is known about posttranslational modifications of targets from the M. tuberculosis proteome and related mycobacteria, except for phosphorylation (Greenstein et al, 2005; Wehenkel et al, 2008). In this contribution, we have shown for M. smegmatis that a substantial number of proteins may serve as targets for covalent modification by PUP conjugation, ultimately leading to alteration of their functional status and perhaps their fate for controlled proteasome-mediated degradation.
An important task for the future is to experimentally map the available evidence for pupylation of M. smegmatis targets onto the proteome of pathogenic M. tuberculosis strains and other pathogenic mycobacteria. Furthermore, it will be of specific interest to determine to what extent pupylation of targets may vary during different stages of the mycobacterial life cycle and within the host environment; this will require studies in appropriate in vivo models. Future studies exploring the significance of pupylation for fundamental biological processes in mycobacteria will be needed to elucidate aspects of pupylation biology that may find applications in ongoing efforts to overcome tuberculosis and other mycobacterial diseases.
Materials and methods
M. smegmatis growth under different experimental conditions
Bacterial growth was monitored by measuring the optical densities at 600 nm (OD600) as a function of time at a wavelength of 600 nm, in triplicates. Cultures were prepared in identical triplicates for each time point, thereby ensuring that the growth of bacteria was not disturbed until the measurement time. Each cell density with an OD600 value exceeding 1.5 was diluted to allow density measurements within the linear range of the detector. Specific stress conditions were generated, by adding 2 mM H2O2, 3 mM NaNO2 (pH 5.5) or 20 μM epoxomicin (Enzo Life Sciences) to the growth medium.
Expression and purification of mtPUP
The ORF of PrcB, PrcA and PUP (Rv2109c, Rv2110c and Rv2111c) was amplified from the H37Rv genomic DNA by PCR, using the forward primer 5′-GAGCCATGGCGCAAGAGCAGACCAA-3′ and the reverse primer 5′-GAGAAGCTTCAGCCCGACGATTCGCC-3′. For cloning of the mtPUP(Q64A) variant, the forward primer 5′-GAGCCATGGCGCAAGAGCAGACCAA-3′ and reverse primer 5′-GCGGGATCCTTATCACGCTCCGCCCTTTTGGAC-3′ were used. The PCR-introduced restriction sites, NcoI and HindIII were used to clone the PCR product into a modified pSD26 vector in M. smegmatis (Daugelat et al, 2003). For heterologous expression in E. coli, the PCR product was cloned into a pETM-11 vector (Dummler et al, 2005). We used DNA sequencing to verify that both constructs contained the correct insert. For mtPUP expression in M. smegmatis, we used a previously established protocol (Ma et al, 2006). The pMyNT construct and empty pMyNT vector were transformed into M. smegmatis MC2 155 by electroporation and grown on 7H10 agar plates supplemented with 0.5% glycerol, 0.05% Tween 80, ADS (5 g/l BSA, 2 g/l D-dextrose and 0.8 g/l NaCl). In both plates and medium, 50 μg/ml hygromycin was added as a selection marker. Overnight pre-cultures in 7H9 broth supplemented with 0.5% glycerol, 0.05% Tween 80 and ADS were added to 2 l of expression medium (7H9 broth supplemented with 0.2% glycerol, 0.05% Tween 80 and 2 g/l glucose). Induction was started, by adding 30 mM acetamide. Depending on the experimental conditions, induction was carried out at a different cell density as specified in the result section. For pupylome analysis the cells were collected by centrifugation and resuspended in lysis buffer containing 50 mM Tris–HCl (pH 8.0), 8 M urea, protease inhibitors cocktail (Serva #39106) containing AEBSF-HCl, aprotinin, E64, and leupeptin, and 40 mM imidazole. Lysis was carried out by sonication; after centrifugation, the soluble fraction was filtered through a 0.22-μm filter and passed over a 200-μl NiNTA resin column. Affinity-bound proteins were eluted with elution buffer containing 300 mM imidazole and 6 M urea.
For mtPUP expression in E. coli, the pETM-11 construct was transformed into BL21 DE3 RIL and plated on agar plates containing 50 μg/ml kanamycin and 37 μg/ml chloramphenicol. Pre-cultures were added to LB medium and 1 mM IPTG was used to induce the expression at an OD600 value of 0.8. The induced cells were left overnight at 20°C before collecting. The cells were resuspended in lysis/washing buffer: 50 mM Tris–HCl (pH 8.0), 300 mM NaCl, 1 M urea and protease inhibitors. The protein was purified as described, but in the absence of denaturing agents. Eluted protein was exchanged to 50 mM Tris–HCl (pH 8.0) and 100 mM NaCl for TEV protease cleavage. Uncleaved His-tagged protein, TEV and other contaminants were removed by passing the sample through NiNTA resin.
For immunoblot analysis, purified proteins were separated on SDS–PAGE. The bands were electrotransferred on to a polyvinylidene difluoride membrane, which was treated with blocking solution (Sigma) in PBST buffer (25 mM phosphate buffer (pH 7.4), 125 mM NaCl and 0.05% Tween-20). Polyhistidine-tagged proteins were detected with a horseradish peroxidase (HRP)-linked anti-penta-his antibody (Qiagen), using the Enhanced Chemiluminescence (ECL) detection kit (GE Healthcare).
ESI QqTOF mass spectrometry analysis for identification of PUP-conjugated targets
Protein-containing fractions were denatured in the presence of 6 M urea, 20 mM NH4HCO3 (pH 8.0), followed by reduction with 1 mM Tris-(2-carboxyethyl)-phosphine for 30 min at 60°C and alkylation with 2.5 mM 4-vinylpyridine for 1 h at room temperature in the dark. Samples were diluted four-fold to ensure that the concentration of urea did not exceed 1.5 M. Tryptic digestion was initiated by the addition of 1% (wt/wt) of side chain-modified, TPCK-treated porcine trypsin and allowed to proceed at 37°C for 6 h.
Individual iTRAQ labeling reagents (Applied Biosystems, Foster City, CA, USA) were reconstituted in ethanol, added to peptide mixtures derived from the tryptic digestion of NiNTA eluate fractions (control, iTRAQ 114; sample, TRAQ 115) and incubated at room temperature in the dark for 3 h.
The column effluent was coupled directly via a fused silica capillary transfer line to a QSTAR XL hybrid quadrupole/time-of-flight tandem mass spectrometer (Applied Biosystems; MDS Sciex, Concord, ON, Canada) equipped with a MicroIonSpray source. The progress of each LC/MS run was monitored by recording the total ion current (TIC) as a function of time for ions in the m/z range 300–1800. At 5-s intervals through the gradient, a mass spectrum was acquired for 1 s, followed by one CID acquisition of 4 s each on ions selected by preset parameters of the information-dependent acquisition (IDA) method, using nitrogen as the collision gas. Singly charged ions were excluded from CID selection. The collision energy was adjusted automatically for each CID spectrum using an empirically optimized formula that considers the charge state and m/z value of the precursor ion.
Strong cation exchange (SCX) chromatography was used to achieve peptide fractionation of the complex digest mixture. Samples digested with trypsin were adjusted to 25% acetonitrile and acidified (pH 3.0) by 20-fold dilution in 25% acetonitrile and 20 mM KH2PO4 (pH 3.0). High performance liquid chromatography was carried out using the Ultimate System (Dionex, Sunnyvale, CA, USA), equipped with a microflow calibration cartridge, a Valco injection port and a 180 nl UV cell. Separation was achieved on a self-packed 0.5 mm × 110 mm Luna SCX column (Phenomenex, Torrance, CA, USA), at a flow rate of 18 μl/min and with a salt gradient of 0–400 mM NH4Cl, in 25% acetonitrile and 20 mM KH2PO4 (pH 3.0). Fractions eluted from the SCX column were desalted with C18 Empore (3 M, Minneapolis, MN, USA) stop and go extraction tips (Rappsilber et al, 2003), and subsequently subjected to a nano-flow RP-HPLC using the Ultimate LC system (Dionex) equipped with a nano-flow calibration cartridge at a flow rate of 250 nl/min. Peptides were separated on a 75-μm ID self-packed column containing a Proteo C12 reverse-phase matrix (Phenomenex), using a 100-min gradient of 2–34% acetonitrile in water, with 0.1% (w/v) formic acid as ion-pairing agent.
The analysis of the iTRAQ data was carried out with the software program ProteinPilot (Applied Biosystems; MDS Sciex). Raw iTRAQ ratios were corrected for impurity levels of individual reagent lots, determined by the manufacturer. Alternatively, peak lists for database searching were created using Mascot Distiller (Version 1; MatrixScience, London, UK), and searches were performed using designated MS/MS data interpretation algorithms within Mascot (Version 2.2; MatrixScience). Modifications considered were the attachment of GGQ or GGE motifs to lysine residues, assuming a condensation reaction that proceeds with the concomitant loss of a water molecule and possible deamidation. In a negative control data analysis, a similar modification was assumed. For ProteinPilot searches, the algorithm assumes by default all possible unexpected cleavages and many of the more common modifications. Searches further considered up to two missed cleavages and charge states ranging from +2 to +4. In the few instances in which confidence values for assigned pupylation sites were not meeting or exceeding 95% confidence levels, the lower confidence, and thus higher assumed error rate, was partially offset by the presence of corroborating missed cleavages at the pupylated lysine residues and manual inspection of CID spectra. In particular, the latter approach led in a subset of spectra to the detection of candidate pupylation fragments that increased the confidence of assignments but were not considered by the algorithms for the calculation of scores. The experimental data have been submitted to the PRIDE data base (accession number: 11999).
For the analysis of 2D gel spots, these were manually isolated from the gel, washed three times with 10 mM ammonium bicarbonate buffer (pH 7.8) and in-gel digested with trypsin (Promega, Mannheim, Germany) at 37°C overnight. For nano-HPLC/ESI-MS/MS analyses, tryptic peptides were extracted twice from gel pieces with 50% ACN in 0.1% trifluoroacetic acid. The MS/MS analyses were performed on a high-capacity ion trap system (HCT ultra, Bruker Daltonics), in conjunction with an online RP nano-HPLC system (Dionex U3000, Dionex LC Packings, Idstein, Germany).The mass spectrometer was operated in the sensitive mode with the following parameters: capillary voltage, 1400 V; end plate offset, 500 V; dry gas, 8.0 l/min; dry temperature, 160°C; aimed ion charge control, 150 000; maximal fill-time, 500 ms. The nano-ESI source (Bruker Daltonics) was equipped with distal coated silica tips (FS360–20–10-D; New Objective). The MS spectra were recorded from the sum of seven individual scans, ranging from m/z 300 to m/z 1500, with a scanning speed of 8100 (m/z)/s. The three most intense, multiple-charged peptide ions detected within the MS spectra were selected in a data-dependent manner (HCT ultra, Esquire Control) to subsequently perform MS/MS. To generate fragment ions, low-energy CID was performed on previously isolated peptide ions by applying a fragmentation amplitude of 0.6 V. Generally, MS/MS spectra were the sum of four scans ranging from m/z 100 to m/z 2200, at a scan rate of 26 000 (m/z)/s. Exclusion limits were automatically placed on previously selected mass-to-charge ratios for 1.2 min. The ion trap instrument was externally calibrated with commercially available standard compounds.
For protein identification, raw MS/MS data were searched using the Mascot algorithm (Matrix Science, v.2.2.0) against the NCBInr database (v.23.01.2009, 7 699 833 protein entries), restricted to bacteria, with the following search parameters: fixed modification on cysteines, due to propionamidation; variable modifications due to mass shift of 243.085521 Da on pupylated lysines, resulting from a tryptic fragment of mtPUP (sequence GGE) attached via an isopeptide bond (Pearce et al, 2008), and methionine oxidation. In addition, two maximal missed cleavage sites, in case of incomplete trypsin hydrolysis and a mass tolerances of ±1.2 Da for parent ions and ±0.4 Da fragment ions, were considered for database searches. The reliability of protein identification was verified manually; a mascot score >60 was required to consider protein identification as significant. The experimental data have been submitted to the PRIDE data base (accession number: 12078).
2D gel electrophoresis
For 2D gel electrophoresis, protein eluate from the NiNTA preparation was treated three times with a buffer (30 mM Tris, 7 M urea, 2 M thiourea and 4% CHAPS (pH 8.5)) compatible with isoelectric focusing using Microcons (Millipore, Schwalbach, Germany) with a 3-kDa cut-off. For IEF, a 20-μg sample was prepared by adding 0.7 μl DTT (1.08 g/ml; Bio-Rad) and 0.7 μl Servalyt 2–4 (Serva). Carrier ampholyte-based IEF was performed in a self-made IEF chamber, using tube gels (20 cm × 1.5 mm) as described elsewhere (Sitek et al, 2006). Briefly, after running a 21-h voltage gradient, the ejected tube gels were incubated in equilibration buffer (125 mM Tris, 40% (w/v) glycerol, 3% (w/v) SDS and 65 mM DTT (pH 6.8)) for 10 min. The second dimension was performed in a Desaphor VA 300 system (Sarstedt, Nümbrecht, Germany) using polyacrylamide gels (15.2% total acrylamide and 1.3% bisacrylamide) as described elsewhere (Sitek et al, 2006). Therefore, the IEF tube gels were placed onto the polyacrylamide gels (20 cm × 30 cm × 1.5 mm) and fixed using 1.0% (wt/vol) agarose containing 0.01% (wt/vol) bromophenol blue dye (Riedel deHaen, Seelze, Germany). Silver staining was performed using an MS-compatible protocol (Blum et al, 1986).
To analyze the differential expression of proteins under different growth conditions, we have used the 2D DIGE method (Unlu et al, 1997). Before electrophoresis, the protein samples were labeled with three different fluorescence CyDyes (GE Healthcare). The fluorescence dye stock solutions (1 nmol/μl) were diluted with anhydrous DMF (400 pmol/μl) and 8 pmol dye was added per microgram protein. The samples were mixed thoroughly, cleared by centrifugation and the labeling reaction was carried out in the dark by incubating on ice for 0.5 h. The reaction was stopped by adding 1 μl of 10 mM L-lysine per 8 pmol dye.
Multiple PUP sequence alignment
All PUP homologs of mtPUP (Rv2111) were obtained in a global BLAST search. All unique sequences below an E-value of 1.0 were selected for the alignment. The alignment was carried out with ClustalX (2.0.9; Larkin et al, 2007). The secondary structure prediction was carried out using the computational server of Pred and PROF (Ouali and King, 2000).
Functional annotation and gene clustering
The identifiers of M. smegmatis and M. tuberculosis genes have been taken from the Comprehensive Microbial Resources (http://cmr.jcvi.org). The identification of genes with paralogs in M. smegmatis and M. tuberculosis were carried out with the Artemis Comparison Tool (Carver et al, 2005). Functional annotation and categorization (M. tuberculosis) has been retrieved from TubercuList (http://tuberculist.epfl.ch). Category identifiers were defined by (Camus et al, 2002). Pupylation targets were grouped in one gene cluster according to the following criteria: (a) genetic vicinity, for example, consecutive location and/or encoded in the same operon; and (b) conservation of gene order in M. smegmatis and M. tuberculosis.
Supplementary Material
Supplementary figures S1–5
Supplementary text, Supplementary tables S1–4
Acknowledgments
YA thanks DAAD (German Academic Exchange Service) for a long-term doctoral fellowship. We thank Morlin Milewski for technical assistance. This study was supported by grants from the Canadian Institutes of Health Research (CIHR; MOP-74734 (GSU)), from BMBF (Pathogenomics-Plus, PTJ-BIO 0313801L) and from the European Commission Framework VII (NATT, 222965) to MW.
Footnotes
The authors declare that they have no conflict of interest.
References
- Blum H, Beier H, Gross HJ (1986) Improved silver staining of plant proteins, RNA and DNA in polyacrylamide gels. Electrophoresis 8: 93–99 [Google Scholar]
- Burns KE, Liu WT, Boshoff HI, Dorrestein PC, Barry CE III (2009) Proteasomal protein degradation in Mycobacteria is dependent upon a prokaryotic ubiquitin-like protein. J Biol Chem 284: 3069–3075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus JC, Pryor MJ, Medigue C, Cole ST (2002) Re-annotation of the genome sequence of Mycobacterium tuberculosis H37Rv. Microbiology 148: 2967–2973 [DOI] [PubMed] [Google Scholar]
- Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J (2005) ACT: the artemis comparison tool. Bioinformatics 21: 3422–3423 [DOI] [PubMed] [Google Scholar]
- Cole ST, Brosch R, Parkhill J, Garnier T, Churcher C, Harris D, Gordon SV, Eiglmeier K, Gas S, Barry CE III, Tekaia F, Badcock K, Basham D, Brown D, Chillingworth T, Connor R, Davies R, Devlin K, Feltwell T, Gentles S et al. (1998) Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537–544 [DOI] [PubMed] [Google Scholar]
- Crosas B, Hanna J, Kirkpatrick DS, Zhang DP, Tone Y, Hathaway NA, Buecker C, Leggett DS, Schmidt M, King RW, Gygi SP, Finley D (2006) Ubiquitin chains are remodeled at the proteasome by opposing ubiquitin ligase and deubiquitinating activities. Cell 127: 1401–1413 [DOI] [PubMed] [Google Scholar]
- Dandekar T, Snel B, Huynen M, Bork P (1998) Conservation of gene order: a fingerprint of proteins that physically interact. Trends Biochem Sci 23: 324–328 [DOI] [PubMed] [Google Scholar]
- Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF (2003) The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science 302: 1963–1966 [DOI] [PubMed] [Google Scholar]
- Daugelat S, Kowall J, Mattow J, Bumann D, Winter R, Hurwitz R, Kaufmann SH (2003) The RD1 proteins of Mycobacterium tuberculosis: expression in Mycobacterium smegmatis and biochemical characterization. Microbes Infect 5: 1082–1095 [DOI] [PubMed] [Google Scholar]
- Dummler A, Lawrence AM, de Marco A (2005) Simplified screening for the detection of soluble fusion constructs expressed in E. coli using a modular set of vectors. Microb Cell Fact 4: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabbe C, Dikic I (2009) Functional roles of ubiquitin-like domain (ULD) and ubiquitin-binding domain (UBD) containing proteins. Chem Rev 109: 1481–1494 [DOI] [PubMed] [Google Scholar]
- Greenstein AE, Grundner C, Echols N, Gay LM, Lombana TN, Miecskowski CA, Pullen KE, Sung PY, Alber T (2005) Structure/function studies of Ser/Thr and Tyr protein phosphorylation in Mycobacterium tuberculosis. J Mol Microbiol Biotechnol 9: 167–181 [DOI] [PubMed] [Google Scholar]
- Hatfull GF, Cresawn SG, Hendrix RW (2008) Comparative genomics of the mycobacteriophages: insights into bacteriophage evolution. Res Microbiol 159: 332–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A (1986) The ubiquitin pathway for the degradation of intracellular proteins. Prog Nucleic Acid Res Mol Biol 33: 19–56, 301 [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A (1998) The ubiquitin system. Annu Rev Biochem 67: 425–479 [DOI] [PubMed] [Google Scholar]
- Iyer LM, Burroughs AM, Aravind L (2008) Unraveling the biochemistry and provenance of pupylation: a prokaryotic analog of ubiquitination. Biol Direct 3: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer L, Dover LG, Carrere S, Nampoothiri KM, Lesjean S, Brown AK, Brennan PJ, Minnikin DE, Locht C, Besra GS (2002) Mycolic acid biosynthesis and enzymic characterization of the beta-ketoacyl-ACP synthase A-condensing enzyme from Mycobacterium tuberculosis. Biochem J 364: 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusunose E, Ichihara K, Noda Y, Kusunose M (1976) Superoxide dismutase from Mycobacterium tuberculosis. J Biochem 80: 1343–1352 [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- Ma Q, Zhao X, Eddine AN, Geerlof A, Li X, Cronan JE, Kaufmann SH, Wilmanns M (2006) The Mycobacterium tuberculosis LipB enzyme functions as a cysteine/lysine dyad acyltransferase. Proc Natl Acad Sci USA 103: 8662–8667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattow J, Jungblut PR, Schaible UE, Mollenkopf HJ, Lamer S, Zimny-Arndt U, Hagens K, Muller EC, Kaufmann SH (2001) Identification of proteins from Mycobacterium tuberculosis missing in attenuated Mycobacterium bovis BCG strains. Electrophoresis 22: 2936–2946 [DOI] [PubMed] [Google Scholar]
- Ouali M, King RD (2000) Cascaded multiple classifiers for secondary structure prediction. Protein Sci 9: 1162–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N (1999) The use of gene clusters to infer functional coupling. Proc Natl Acad Sci USA 96: 2896–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH (2008) Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science 322: 1104–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellegrini M, Marcotte EM, Yeates TO (1999) A fast algorithm for genome-wide analysis of proteins with repeated sequences. Proteins 35: 440–446 [PubMed] [Google Scholar]
- Rappsilber J, Ishihama Y, Mann M (2003) Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal Chem 75: 663–670 [DOI] [PubMed] [Google Scholar]
- Sitek B, Potthoff S, Schulenborg T, Stegbauer J, Vinke T, Rump LC, Meyer HE, Vonend O, Stuhler K (2006) Novel approaches to analyse glomerular proteins from smallest scale murine and human samples using DIGE saturation labelling. Proteomics 6: 4337–4345 [DOI] [PubMed] [Google Scholar]
- Stieglitz KA, Yang H, Roberts MF, Stec B (2005) Reaching for mechanistic consensus across life kingdoms: structure and insights into catalysis of the myo-inositol-1-phosphate synthase (mIPS) from Archaeoglobus fulgidus. Biochemistry 44: 213–224 [DOI] [PubMed] [Google Scholar]
- Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, Weber-Ban E (2009) Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol 16: 647–651 [DOI] [PubMed] [Google Scholar]
- Unlu M, Morgan ME, Minden JS (1997) Difference gel electrophoresis: a single gel method for detecting changes in protein extracts. Electrophoresis 18: 2071–2077 [DOI] [PubMed] [Google Scholar]
- Wehenkel A, Bellinzoni M, Grana M, Duran R, Villarino A, Fernandez P, Andre-Leroux G, England P, Takiff H, Cervenansky C, Cole ST, Alzari PM (2008) Mycobacterial Ser/Thr protein kinases and phosphatases: physiological roles and therapeutic potential. Biochim Biophys Acta 1784: 193–202 [DOI] [PubMed] [Google Scholar]
- Zhang Y (2005) The magic bullets and tuberculosis drug targets. Annu Rev Pharmacol Toxicol 45: 529–564 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figures S1–5
Supplementary text, Supplementary tables S1–4