

Abstract
It is well established that insulin-like growth factor (IGF)-I is critical for the regulation of peak bone mineral density (BMD) and bone width. However, the role of systemic vs. local IGF-I is not well understood. To determine the role local IGF-I plays in regulating BMD and bone width, we crossed IGF-I flox/flox mice with procollagen, typeIIαI-Cre mice to generate conditional mutants in which chondrocyte-derived IGF-I was disrupted. Bone parameters were measured by dual X-ray absorptiometry at 2, 4, 8, and 12 wk of age and peripheral quantitative computed tomography at 12 wk of age. Body length, areal BMD, and bone mineral content (BMC) were reduced (P < 0.05) between 4 and 12 wk in the conditional mutant mice. Bone width was reduced 7% in the vertebrae and femur (P < 0.05) of conditional mutant mice at 12 wk. Gains in body length and total body BMC and BMD were reduced by 27, 22, and 18%, respectively (P < 0.05) in conditional mutant mice between 2 and 4 wk of age. Expression of parathyroid hormone related protein, parathyroid hormone receptor, distal-less homeobox (Dlx)-5, SRY-box containing gene-9, and IGF binding protein (IGFBP)-5 were reduced 27, 36, 45, 33, and 45%, respectively, in the conditional mutant cartilage (P < 0.05); however, no changes in Indian hedgehog, Dlx-3, growth hormone receptor, IGF-I receptor, and IGFBP-3 expression were observed (P ≥ 0.20). In conclusion, IGF-I from cells expressing procollagen type IIαI regulates bone accretion that occurs during postnatal growth period.
Keywords: cre-loxP, postnatal growth, transgenic mice
THE INSULIN-LIKE GROWTH FACTORS (IGF) are the most abundant growth factors stored in bone and produced by osteoblasts (38). It has been well established that IGF-I plays an important role in the regulation of peak bone mineral density (BMD) and bone width (31, 32), as demonstrated by the severe deficit in femur BMD and periosteal circumference observed in IGF-I knockout mice (32). Regarding the mechanism by which IGF-I regulates skeletal growth and maintenance, following the discovery of IGF-I as a sulfation factor/somatomedin by Daughaday (7), it has been proposed that IGF-I functions in an endocrine manner whereby it is released from the liver, after stimulation by growth hormone (GH), and travels to target tissues, such as bone (17, 23). To evaluate the role of endocrine IGF-I actions, recent studies have used the Cre/loxP approach to conditionally delete IGF-I in the liver. Surprisingly, a 75% reduction in circulating IGF-I in the liver-derived IGF-I knockout (KO) mice had very little effect on growth and skeletal parameters (45, 46, 52). However, in a recent study, disruption of both the acid-labile subunit (ALS) and IGF-I in the liver led to a dramatic 90% reduction in circulating IGF-I and a 35% reduction in periosteal circumference (53), thus suggesting that a threshold concentration of circulating IGF-I is necessary for normal bone growth in mice. In addition to the endocrine actions of IGF-I, studies in our laboratory and others' have suggested that locally produced IGF-I acts in an autocrine/paracrine manner to regulate proliferation, differentiation, and apoptosis of osteoblast cells (46, 52, 54). Accordingly, it was shown in a recent study that disruption of the IGF-I receptor in mature osteoblasts led to a significant reduction in bone volume (54), suggesting that local signaling of IGF-I is important in the regulation of bone accretion as well.
Past studies using transgenic mouse models lacking IGF-I or overexpressing IGF-I have shown that IGF-I influences bone accretion by regulating longitudinal growth, bone width, and material bone density (3, 18, 31, 32, 42, 46, 49, 53, 54). However, the question of whether endocrine or local sources of IGF-I play a predominant role in regulating one or more of the above processes remains to be determined. Furthermore, it is also not known whether IGF-I produced by different cell types within the bone (i.e., osteoblast, chondroblast, osteoclast, or marrow cells) exerts differential effects on longitudinal growth, bone width, and mineral deposition. In this regard, IGF-I produced by chondrocytes has been proposed to play a significant role in the regulation of longitudinal bone growth (33, 47). This prediction is based on the findings that 1) chondrocytes both produce and respond to IGF-I (34, 40), and 2) the rate of longitudinal growth is dependent on the rate of chondrocyte proliferation, as well as hypertrophic differentiation, which is accompanied by cell enlargement, which is disturbed in the IGF-I KO mice (49, 50, 53). To evaluate whether IGF-I produced locally by cells that express procollagen, type IIαI [Col2α1 (36)], which are primarily chondrocyte cells, exerts a significant role in promoting longitudinal growth, we used the Cre/loxP approach to disrupt IGF-I production in cells that express Col2α1. Accordingly, we determined that production of IGF-I from Col2α1-expressing cells not only regulates longitudinal bone growth, but also bone width.
MATERIALS AND METHODS
Generation of Cre/loxP mice
Breeding pairs of transgenic mice in which Cre recombinase is driven by the procollagen type IIαI gene (Col2α1-Cre) were generated as previously described in a C57BL/6 × SJL background (36). Breeding pairs of transgenic mice in which exon 4 of the IGF-I gene is flanked by the loxP gene (IGF-I flox/flox) in a C57BL/6 background were kindly provided by Dr. Derek LeRoith. Crosses to generate IGF-I flox/flox, Col2α1-Cre mice (hereafter called Col2α1 conditional mutants or simply conditional mutants), were performed according to the following breeding scheme. IGF-I flox/flox mice were bred to Col2α1-Cre-positive (+) mice to generate IGF-I flox/−; Col2α1-Cre+ mice. These mice were then bred to IGF-I flox/flox mice to generate conditional mutants (IGF-I flox/flox; Col2α1-Cre+/−). The conditional mutants were bred to IGF-I flox/flox mice to generate littermate conditional mutant and control offspring that were used for our experiments. All the Cre+ mice were heterozygous for Cre; therefore, any variation observed within the conditional mutant group was not due to a difference in gene dosage. Since the mice used for experiments were of a mixed genetic background, we used littermate controls to account for variation due to the mixed genetic background.
The experimental procedures performed in this study were in compliance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the Animal Studies Subcommittee at the Jerry L. Pettis Memorial Veterans Affairs Medical Center.
Genotyping of Cre/loxP mice
At 3 wk of age, DNA was extracted from ear or tail tissue using a PUREGENE DNA Purification Kit (Gentra Systems, Minneapolis, MN) according to the manufacturer's protocol. PCR was performed to identify mice with Cre recombinase and/or loxP sites. Primers specific for the Cre recombinase gene (forward: 5′-GTGTAGAGAAGGCACTTAGC-3′ and reverse: 5′-CTGACCAGAGTCATCCTTAG-3′) were used under the following conditions: 94°C for 4 min; 41 cycles at 94°C for 1 min, 60°C for 1 min, 72°C for 2 min; and 72°C for 10 min. loxP sites were amplified using primers previously described (26) under the following conditions: 93°C for 2 min; 30 cycles at 93°C for 20 s, 57°C for 1 min, 70°C for 1 min; and 70°C for 10 min. The PCR products were run on a 2% agarose gel, and the image was taken with a ChemiImager 4400 (Alpha Innotech, San Leandro, CA).
Bone densitometry by dual X-ray absorptiometry
Bone mineral content (BMC) and areal (a) BMD were measured by dual X-ray absorptiometry (DXA), using the PIXImus instrument (Lunar, Madison, WI). The precision for the BMC and BMD was ± 1% for repeat measurements of the same bones several times (29). Animals were anesthetized by a ketamine-xylazine (50/5 mg/kg body wt) injection prior to measurement.
Volumetric BMD and geometric parameters
Volumetric BMD (vBMD) and geometric parameters at the middiaphysis of the femur and distance between the fourth and fifth lumbar vertebrae of bones isolated at 12 wk of age were determined by peripheral quantitative computed tomography (pQCT; Norland Stratec XCT, Stratec Medzizintechnik, Madison, WI). Analysis of the scans was performed using the manufacturer-supplied software program (Stratec Medzizintechnik Bone Density Software, version 5.40 C). Total BMD and geometric parameters were estimated with Loop analysis. The threshold was set at 230–630 mg/cm3. For femur analysis, nine scans per bone were measured, and the data presented are the average of the fourth, fifth, and six scans (middiaphysis region). For the vertebrae analysis, six to nine scans at a set distance of 0.7 mm between each scan were measured, and the data presented are the average of the four to six scans that ran through the fourth and fifth vertebrae (scans between the two vertebrae or at either end of the vertebrae were eliminated). The coefficient of variation for total BMD, periosteal circumference, and endosteal circumference for repeat measurements of four mouse femurs (2–5 measurements) was <3%, <1, and <2%, respectively (29). The longitudinal lengths of the femurs and the entire distance between and including the fourth and fifth lumbar vertebrae were measured with a caliper.
Histological measurements
Growth rate of femurs between 3 and 4 wk of age was determined as previously described (18, 43) using calcein (20 mg/kg body wt) as the double label. The length of hypertrophic and proliferating zones in the tibias was also determined at 4 wk of age. Specifically, four measurements were manually taken at each region, while avoiding the perichondrium, and averaged together for each mouse.
Serum IGF-I radioimmunoassay
IGF-I was measured by radioimmunoassay (RIA) using rabbit polyclonal antiserum and recombinant IGF-I as standard and tracer, respectively. IGF binding proteins (IGFBP) were removed from serum prior to RIA by acid gel filtration protocol (30).
RNA extraction
RNA was extracted from the tissues using a Lipid Tissue Mini Kit (Qiagen, Valencia, CA) according to the manufacturer's protocol. For bone sample collection (metaphysis and diaphysis regions), muscle and tissue were removed, and bone marrow was flushed from the femur and tibia bones. With mice at both 2 and 12 wk of age, we collected the region between the femur and tibia (epiphysis), which was made up of primarily cartilage tissue. Although the diaphysis/metaphysis of femur/tibia did express low levels of Col2α1, there was about a 14-fold greater expression of Col2α1 in the cartilage samples. Therefore, the cartilage samples from the epiphysis at 2 wk were used for further gene expression analysis. Liver and kidney tissues were also collected. Following RNA extraction, residual DNA was removed from up to 10 μg of RNA with a DNA-free kit (Ambion, Austin, TX). RNA quality was determined using a 2100 Bioanalyzer (Agilent, Palo Alto, CA) and RNA was quantified using a NanoDrop Spectrophotometer (Wilmington, DE).
Gene expression analysis
Quantitative real-time RT-PCR analysis was used to determine the expression levels of IGF-I, Cre, Col2α1, GH receptor (GHR), IGF-I receptor (IGF-IR), parathyroid hormone (PTH)-related protein (PTHrP), Indian hedgehog (Ihh), PTH/PTHrP receptor (PTH1R), distal-less homeobox (Dlx)3, Dlx5, SRY-box containing gene (Sox)9, IGFBP-3 and -5, and PPIA (peptidylprolyl isomerase A; endogenous control) as previously described (13). PPIA was chosen as an internal control based on the determination that its expression is not different between different tissues or control and conditional mutant mice (data not shown). Briefly, Stratagene Brilliant SYBRGreen Master Mix (Stratagene, La Jolla, CA) and a 7000 ABI Prism sequence detection system (Applied Biosystems, Foster City, CA) were used to determine gene expression. Primers were validated as previously described (13), and primer sequences are located in Table 1. Delta cycle threshold (Ct) values were determined (Ct value for gene of interest minus Ct value for control gene), and comparisons of the Ct values were used for relative quantification of gene expression (8). This provides a quantitative value for the expression of the gene of interest, adjusted for any variation in total RNA concentrations. Data are presented as fold change or relative to control using the well accepted 2−ΔΔCt method (27).
Table 1.
Primer sequences used for real-time RT-PCR
| Gene | Forward (5′ to 3′) | Reverse (5′ to 3′) |
|---|---|---|
| IGF-I | GCTCTTCAGTTCGTGTGTGGAC | CATCTCCAGTCTCCTCAGATC |
| Cre recombinase | GTGTAGAGAAGGCACTTAGC | CTGACCAGAGTCATCCTTAG |
| Col2α1 | TGGCTTCCACTTCAGCTATG | AGGTAGGCGATGCTGTTCTT |
| GHR | TCTTAACCTTGGCACTGGCA | GGGCATTCTTTCCATTCCTG |
| IGF-IR | TGCCTTGGTCTCCTTGTCCT | TATGTCCCCTTTGCTCTGGC |
| PTHrP | TCCACACAGCCGAAATCAGA | TGCCTTTCTTCTTCTTCCCG |
| Indian hedgehog | CCCCAACTACAATCCCGACATC | CGCCAGCAGTCCATACTTATTTCG |
| PTH1R | ACTCCTTCCAGGGATTTTTTGTT | GAAGTCCAATGCCAGTGTCCA |
| Dlx3 | CACCTACCACCACCAGTTCAA | GCTCCTCTTTCACCGACACTG |
| Dlx5 | AGAAGAGTCCCAAGCATCCGA | GCCATAAGAAGCAGAGGTAGG |
| Sox9 | CCGCATCTGCACAACGC | TCCTCCACGAAGGGTCTCTTC |
| IGFBP-3 | CCAGAACTTCTCCTCCGAGTCTAAG | CTCAGCACATTGAGGAACTTCAGAT |
| IGFBP-5 | ATACAACCCAGAACGCCAGCT | ACCTGGGCTATGCACTTGATG |
| PPIA | TCCTGGACCCAAAACGCTCC | CCATGGCAAATGCTGGACCA |
Dlx, distal-less homeobox; GHR, growth hormone receptor; IGF-I, insulin-like growth factor-I; IGF-IR, IGF-I receptor; Col2α1, procollagen, typeIIα1; PTHrP, parathyroid hormone related peptide; IGFBP, insulin-like growth factor binding protein; PPIA, peptidylprolyl isomerase A; endogenous control; Sox, SRY-box containing gene.
Primary chondroblast culture
Primary mouse chondrocytes were isolated from conditional mutant and control mice as previously described (24, 35) with the following modifications. In brief, rib cartilage was isolated from 2- to 4-day-old pups and rinsed with 0.25% trypsin at 37°C for 20 min, following by addition of DMEM + 10% FBS. Tissues were rinsed with PBS and incubated with collagenase D (3 mg/ml; Roche, Indianapolis, IN) for 60–90 min on a shaker at 37°C until all soft tissue was detached, and the majority of the perichondrial cells were removed. Following incubation, ribs were washed with PBS and incubated for an additional 4 h in collagenase D. Following incubation, cells were passed through a 70-μm filter using DMEM + 10% FBS + 50 μg/ml AA2P (ascorbic acid-2-P). Cells were plated at a density of 1,000,000 cells per well in six-well plates and grown for 2 days. For experiments, cells were passaged by incubating in collagenase D for 90 min, counted, and plated at a density of 400,000 cells per well in six-well plates. Cells were allowed to attach for 24 h and left in serum-free conditions for 48 h, and then RNA was extracted as presently described.
Primary osteoblast culture
Calvaria were removed from 2- to 4-day-old pups, and tissue was removed. Calvaria were cut into small pieces and incubated in collagenase A (2 mg/ml, Sigma) for 90 min at 37°C and shaken every 15 min. Supernatant was collected and plated in α-MEM + 10% FBS + 1% fungizone. After 2 days, cells were passaged with 0.25% trypsin and plated at a density of up to 400,000 cells per well in six-well plates. Cells were allowed to attach for 24 h and left in serum-free conditions for 48 h, and then RNA was extracted as presently described.
Statistical analysis
Bone parameters analyzed by DXA analysis at 4, 8, and 12 wk of age were analyzed by repeated-measures ANOVA. Bone parameters and gene expression data at 2 and 12 wk of age, histological analysis, and in vitro data were analyzed by ANOVA, and post hoc analysis was performed by Newman-Keuls analysis. When appropriate, the interaction between transgenic line (control and conditional mutant) versus sex (male and female) was included in the analysis. However, for presentation of data, if a significant interaction was not observed, the male and female data were averaged together. Statistical analysis presented is based on the analysis performed with main effects and the interaction. Data were analyzed using Statistica 6 software (StatSoft, Tulsa, OK). Data are presented as means ± SE, and a significant difference was determined at P ≤ 0.05.
RESULTS
Circulating and mRNA expression of IGF-I
To confirm that Cre recombinase expression was specific to tissues that express Col2α1, we determined Cre expression and Col2α1 expression in different tissues at 2 wk of age. Accordingly, expression of Cre was greater in tissues (cartilage) that had the greatest expression of Col2α1 and the least in tissues (diaphysis/metaphysis of femur/tibia, calvarium, and liver) with the lowest expression of Col2α1 (Table 2). Accordingly, expression of Cre was positively correlated with Col2α1 expression (R = 0.5905, P < 0.001). To determine if Cre expression driven by the Col2α1 promoter leads to disruption of the IGF-I gene specifically in tissues that express high levels of Col2α1, we measured IGF-I mRNA expression by real-time RT-PCR in bone and liver tissues at 2 wk of age. IGF-I PCR primers for real-time RT-PCR were targeted against exon 4, which would be deleted by Cre recombinase action in the conditional mutant mice. Accordingly, we determined that IGF-I expression was reduced 40% (P < 0.05) in cartilage and ~30% (P < 0.05) in the diaphysis/metaphysis of femur/tibia, which are tissues that express Col2α1 in the conditional mutants at 2 wk of age (Fig. 1). This reduction in mRNA expression was confirmed in the whole femur and tibia bones at 12 wk of age (P < 0.05, data not shown). In contrast, there was no change in IGF-I expression in the liver and kidney tissues of the mice at 2 wk of age (P = 0.75). To determine if chondrocyte disruption of IGF-I in Col2α1-expressing cells leads to a reduction in circulating IGF-I, we measured IGF-I in the serum at 12 wk of age. Accordingly, there was no difference in serum concentrations of IGF-I (P = 0.78) between conditional mutant mice and control mice (248 ± 8 vs. 252 ± 13 ng/ml, respectively).
Table 2.
Cre is highly expressed in tissues that express high levels of Col2α1
| Tissue | Cre | Col2α1 |
|---|---|---|
| Cartilage | 1.0±0.16 | 1.0±0.06 |
| Diaphysis/metaphysis of femur/tibia | 0.10±0.03* | 0.16±0.035* |
| Calvarium | 0.03±0.01* | 0.0032±0.0006* |
| Liver | 0.04±0.01* | 0.395 × 10−7±0.63 × 10−7* |
Data are expressed as relative expression compared with cartilage for Cre or Col2α1 based on the 2−ΔΔCt method (27) and presented as means ± SE.
Significant difference from cartilage at P < 0.05. Based on the ΔCt values, expression of Cre was positively correlated with Col2α1 expression (R = 0.5905; P < 0.001).
Fig. 1.
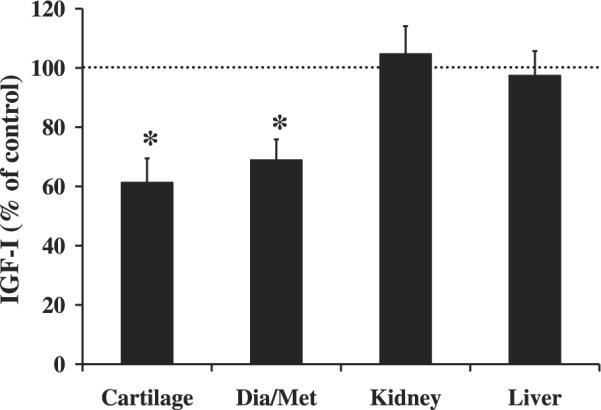
Reduced insulin-like growth factor (IGF)-I expression in the bones of conditional mutant mice at 2 wk of age. Gene expression was determined by real-time RT-PCR and expressed as a percent of controls. A 50% change is equal to a 2-fold change in expression. Data are presented as means ± SE. *Significant difference from controls at P < 0.05. Conditional mutant mice (n = 7) and control mice (n = 8). Dia/Met, diaphysis/metaphysis of femur/tibia.
To confirm the specificity of the Cre expression in chondrocytes, we isolated primarily chondrocytes and osteoblasts from conditional mutant mice. To verify the primary cell types, we evaluated expression of Col2α1 and determined its expression was 32-fold greater in the primary chondrocytes than in the primary osteoblasts (P < 0.05). Consistent with these data, Cre expression was observed in the primary chondrocytes but not observed in the primary osteoblasts (Fig. 2), thus confirming the specificity of Cre expression in our model.
Fig. 2.
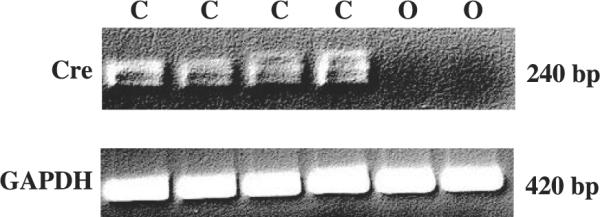
Cre expression is specific to chondrocyte cells. Image presented is the PCR product of Cre and GAPDH for primary chondrocytes (C) and osteoblasts (O). We amplified 30 and 15 ng of RNA for 40 cycles for Cre and GAPDH, respectively. Cre expression was not detectable in primary osteoblast cells by PCR analysis or real-time RT-PCR.
Growth and skeletal parameters
Changes in growth and skeletal parameters are shown in Fig. 3 and Table 3. All pups born were at the expected ratio of 50% conditional mutant and 50% control. In addition, body weights were not significantly different between conditional mutant and control mice at 3–4 days of age (data not shown). Interestingly, there were no significant differences for any of the growth and skeletal parameters at 2 wk of age (Fig. 3 and Table 3). However, at 8 wk of age, body weights of the conditional mutants were reduced 10% (P < 0.01) compared with their littermate controls, and this remained significant through 12 wk of age (P < 0.05, Fig. 3A). In addition, body lengths of the conditional mutant mice were reduced by 7% beginning at 4 wk of age and remained significant through 12 wk of age (P ≤ 0.001, Fig. 3B). Total body BMC was reduced 14, 13, and 11% at 4, 8, and 12 wk, respectively (P < 0.01), in the conditional mutant mice, while total body aBMD was reduced 5% at all three time points (P < 0.05). During prepubertal growth, a period of rapid bone accretion, there was a 27, 22, and 18% reduction in the rate of gain for body length, total body BMC, and total body aBMD, respectively, (P < 0.05, Fig. 4) in the conditional mutants compared with controls. Spine BMC was reduced 19 and 12% at 8 and 12 wk of age (P < 0.05, Table 3), respectively, in the conditional mutant mice. Spine aBMD was reduced 11% (P < 0.05) and 8% (P = 0.06) at 8 and 12 wk of age, respectively (Table 3).
Fig. 3.

Body weight and length are reduced in conditional mutant mice. Body weights (A) and lengths (B) were determined between 2 and 12 wk of age. Diamond with solid line, conditional mutant females; diamond with dashed line, conditional mutant males; circle with solid line, control females; circle with dashed line, control males. *Significant difference between conditional mutant and control mice at P < 0.05.
Table 3.
Body and skeletal changes in conditional mutant mice and age-matched control mice
| Age, wk |
|||||||||
|---|---|---|---|---|---|---|---|---|---|
| 2‡ |
4§ |
8§ |
12§ |
||||||
| Parameter | Sex | Control | Conditional Mutant | Control | Conditional Mutant | Control | Conditional Mutant | Confol | Conditional Mutant |
| Total body BMC, (mg) | female | 35.40±2.84 | 50.58±6.52 | 196.8±12.2† | 164.5±8.0* † | 330.5±15.5† | 280.9±12.8* † | 404.1±16.8† | 360.3±15.2* † |
| male | 42.83±5.41 | 47.88±5.93 | 225.0±9.1 | 197.0±11.5* | 405.4±17.3 | 360.2±13.7* | 465.8±13.7 | 415.1±12.8* | |
| Total body aBMD, mg/cm2 | female | 21.23±0.41 | 22.85±0.55 | 35.25±0.65 | 33.48±0.57* | 44.03±0.88† | 42.01±0.55* † | 49.45±1.30 | 47.45±0.75* |
| male | 22.08±0.87 | 22.75±0.60 | 35.78±0.67 | 33.86±0.51* | 46.81±0.97 | 45.38±0.64* | 51.81±1.27 | 48.79±0.58* | |
| Femur BMC, mg | female | 1.33±0.19 | 2.10±0.38 | 6.64±0.44† | 5.60±0.26† | 11.91±0.82† | 10.58±0.28† | 15.76±0.82† | 14.49±0.78† |
| male | 1.48±0.19 | 1.65±0.13 | 7.51±0.58 | 7.28±0.33 | 15.80±0.93 | 14.70±0.66 | 18.74±1.06 | 17.60±0.89 | |
| Femur aBMD, mg/cm2 | female | 20.63±0.86 | 24.05±1.38 | 44.93±0.88† | 45.14±1.52† | 59.01±2.59† | 59.41±2.09† | 69.35±1.52† | 68.48±1.84† |
| male | 21.30±0.51 | 21.85±0.77 | 50.96±3.15 | 49.08±1.17 | 73.75±2.36 | 72.15±2.60 | 80.44±2.60 | 75.90±2.20 | |
| Spine BMC, mg | female | 1.55±0.88 | 1.92±0.22 | 8.18±0.34 | 7.94±0.85 | 13.79±0.83† | 11.99±0.83* † | 15.63±0.55 | 14.61±1.20* |
| male | 1.85±0.36 | 1.80±0.25 | 9.30±0.66 | 7.34±0.42 | 16.98±1.20 | 12.88±0.77* | 16.70±0.89 | 13.89±0.64* | |
| Spine aBMD, mg/cm2 | female | 25.58±0.85 | 24.40±1.29 | 57.23±2.26 | 56.63±5.18 | 71.18±2.52 | 67.41±3.78* | 80.09±2.40 | 77.19±4.42 |
| male | 25.35±1.89 | 26.08±2.04 | 60.35±4.24 | 48.31±2.19 | 75.68±3.68 | 63.21±3.03* | 80.34±4.36 | 69.84±1.79 | |
Growth and bone parameters were determined by dual X-ray absorptiometry analysis between 2 and 12 wk of age. a, Areal; BMC, bone mineral content; BMD, bone mineral density
Significant main effect for treatment (control vs. conditional mutants) at P ≤ 0.05.
Significant main effect for sex at P ≤ 0.05.
Conditional mutant female mice (n = 6), conditional mutant male mice (n = 4), control female mice (n = 4), control male mice (n = 4)
n = 8 mice/sex/treatment group (N = 32).
Fig. 4.
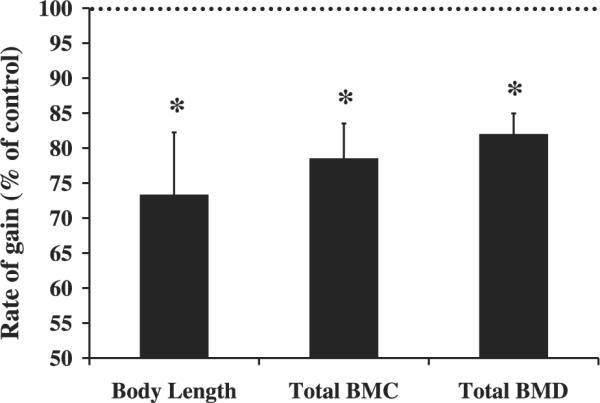
Reduced rate of gain in body length, total body bone mineral density (BMD), and total body bone mineral content (BMC) in conditional mutant mice during prepubertal growth. Growth and bone parameters were determined by dual X-ray absorptiometry analysis and rate of gain was calculated between 2 and 4 wk of age. Data are expressed as conditional mutants as a percent of controls and presented as means ± SE. *Significant difference from controls at P < 0.05. At 2 wk of age [conditional mutant female mice (n = 6), conditional mutant male mice (n = 4), control female mice (n = 4), control male mice (n = 4)]. At 4 wk of age, n = 8 mice/sex/treatment group.
vBMD and geometric parameters
Since the conditional mutants were slightly smaller than the control mice, which can influence BMD measurements by DXA analysis, we also measured vBMD by pQCT. Femur length was reduced 3.5% in the conditional mutants (P = 0.01); however, no difference in total vBMD at the middiaphysis was observed at 12 wk of age (Table 4). Interestingly, a 4 and 9% reduction in periosteal and endosteal circumference, respectively (P < 0.05, Table 4), was observed in the conditional mutant mice compared with their littermate controls.
Table 4.
Reduced femur length and width in conditional mutant mice
| Females |
Males |
|||||
|---|---|---|---|---|---|---|
| Parameter | Control | Conditional Mutant | Control | Conditional Mutant | Treatment P Value | Sex P Value |
| Length, mm | 13.73±0.17 | 13.24±0.13 | 14.51±0.25 | 14.01±0.16 | 0.0113 | 0.0002 |
| Total vBMD, mg/cm3 | 669.68±14.81 | 712.79±25.32 | 700.16±12.68 | 711.09±7.58 | 0.0967 | 0.3675 |
| Peri. Circ., mm | 4.50±0.07 | 4.27±0.02 | 4.86±0.09 | 4.74±0.09 | 0.0264 | 0.0000 |
| Endo. Circ., mm | 2.68±0.05 | 2.35±0.05 | 2.90±0.08 | 2.75±0.06 | 0.0006 | 0.0000 |
Bone parameters were determined by peripheral quantitative computed tomography at 12 wk of age. vBMD, volumetric bone mineral density; Peri. Circ., periosteal circumference; Endo. Circ., endosteal circumference; n = 8 mice per treatment group.
The lengths of the fourth and fifth vertebrae in the conditional mutants was reduced 6% compared with controls (Table 5). We observed a 14, 10, and 18% reduction in total, trabecular, and cortical content, respectively (P ≤ 0.01), in the conditional mutants (data not shown). Conditional mutant mice had a 4% reduction in cortical density compared with controls (P = 0.05, Table 5). Total, trabecular, and cortical area were reduced 13, 12, and 14%, respectively (P < 0.01), in the conditional mutants compared with controls (data not shown). Similar to the femurs, a 6.5% reduction (P < 0.001) in periosteal and endosteal circumference was observed in the spines of the conditional mutants (Table 5).
Table 5.
Reduced length and width of vertebrae in conditional mutant mice
| Females |
Males |
|||||
|---|---|---|---|---|---|---|
| Parameter | Control | Conditional Mutant | Control | Conditional Mutant | Treatment P Value | Sex P Value |
| Length, mm | 5.45±0.19 | 5.35±0.16 | 6.26±0.05 | 5.61±0.12 | 0.0031 | 0.0001 |
| Total vBMD, mg/cm3 | 428.00±9.68 | 421.19±5.35 | 422.73±7.03 | 415.40±6.47 | 0.3325 | 0.4471 |
| Trabecular vBMD, mg/cm3 | 313.61±11.70 | 326.59±9.54 | 329.60±8.57 | 335.35±9.36 | 0.3507. | 0.2201 |
| Cortical vBMD, mg/cm3 | 533.47±12.37 | 511.98±5.84 | 519.99±13.07 | 499.42±9.48 | 0.0512 | 0.2169 |
| Peri. Circ., mm | 8.21±0.09 | 7.64±0.08 | 8.62±0.11 | 8.08±0.10 | 0.0000 | 0.0001 |
| Endo. Circ., mm | 6.12±0.06 | 5.70±0.07 | 6.54±0.08 | 6.16±0.11 | 0.0001 | 0.0000 |
Bone parameters were determined by peripheral quantitative computed tomography at 12 wk of age. Control female mice (n = 7), control male mice (n = 7), conditional mutant female mice (n = 8), conditional mutant male mice (n = 8).
For all growth and skeletal parameters, the expected sex differences were observed with males being greater than females for several of the parameters measured, beginning at 4 or 8 wk of age. However, an interaction between the main effects (sex and treatment) was not observed for any of the parameters, except spine length, which was due to a greater length in the control males.
Histological analysis
Histomorphometric measurements were made between 3 and 4 wk of age by use of a calcein double label. We did not observe a significant difference between conditional mutants and controls for bone formation rate (data not shown). To determine if the reduced longitudinal growth in the conditional mutants was due to reduced number or size of hypertrophic chondrocytes, as previously observed in the total IGF-I knockout mice (49, 50), we measured the proliferation and hypertrophic region of the growth plate in the tibias at 4 wk of age. Although we observed a 15% reduction in the size of the hypertrophic region, in the conditional mutants compared with control mice, this difference was not statistically significant.
mRNA expression of cartilage growth factors and IGFBPs
To determine the molecular basis for the skeletal deficit in the conditional mutant mice, we proposed in the current study, that IGF-I regulates longitudinal growth and bone width, in part, via regulating the expression of one or more messenger molecules (PTHrP, Ihh, and transcription factors) that are involved in chondrocyte proliferation and/or differentiation. To test this hypothesis we determined the expression of PTHrP, PTH1R, Ihh, Sox9, Dlx3, and Dlx5 in conditional mutant and control cartilage tissue at 2 wk of age. We chose 2 wk of age because the rate of gain in various skeletal parameters were significantly reduced in the conditional mutants between 2 and 4 wk of age, and we anticipated that the expression of key regulatory genes, known to be actively involved in regulating bone development, to proceed before skeletal changes. Interestingly expression of PTHrP, PTH1R, Dlx5, and Sox9 were reduced 27, 36, 45, and 33%, respectively in the conditional mutants (P < 0.05, Fig. 5); however, no change in Ihh and Dlx3 expression was observed (P = 0.44, Fig. 5).
Fig. 5.
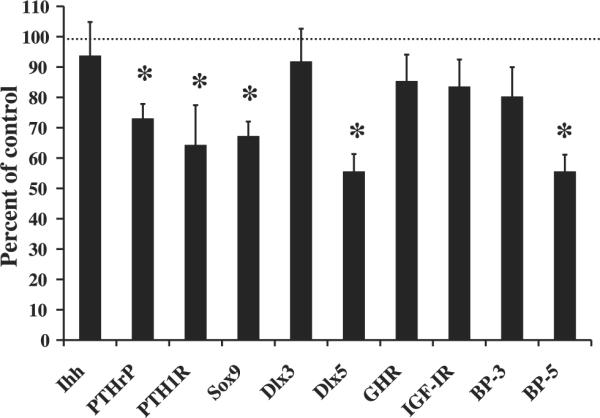
Reduced PTHrP, PTH1R, Sox9, Dlx5, and IGFBP-5 expression in the cartilage of conditional mutant mice at 2 wk of age. Gene expression was determined by real-time RT-PCR and expressed as a percent of control. A 50% change is equal to a 2-fold change in expression. Data are presented as means ± SE. *Significant difference from controls at P < 0.05. Conditional mutant mice (n = 7) and control mice (n = 8). Ihh, Indian hedgehog; PTHrP, parathyroid hormone-related protein; PTH1R, PTHrP/PTH receptor; Sox, SRY-box containing gene; Dlx, distal-less homeobox; GHR, growth hormone receptor; IGF-IR, IGF-I receptor; BP, IGF binding protein.
The GH/IGF axis is regulated by several feedback loops. Therefore, to determine if disruption of local IGF-I alters other local members of the axis, we determined expression of GHR and IGF-IR. The expression of neither IGF-IR nor GHR was significantly altered in the cartilage tissue of the conditional mutant mice (P = 0.20, Fig. 5).
Since the IGFBPs are key regulators of IGF-I action in bone as well as other tissues, we hypothesized that IGF-I from Col2α1-expressing cells regulates IGFBP expression in cartilage tissue. Accordingly, expression of IGFBP-5 was reduced 45% in the conditional mutant mice (P < 0.01, Fig. 5). Although the expression of IGFBP-3 was reduced by 20%, this reduction was not statistically significant (P = 0.20, Fig. 5).
DISCUSSION
In the current study, we have provided the first in vivo evidence that local chondrocyte-produced IGF-I is an important regulator of longitudinal growth and bone width in mice. Previous experiments have demonstrated the importance of IGF-I in regulating growth and BMD using the total IGF-I KO mouse model (1, 3, 32, 37, 50). Interestingly, when liver-derived IGF-I was disrupted, the skeletal phenotype was only slightly compromised despite a 75–80% reduction in circulating IGF-I (52, 53). However, when liver-derived IGF-I KO mice were crossed with ALS KO mice, a significant reduction in bone density and width was observed. These findings suggest that circulating IGF-I levels may need to be reduced below a certain threshold to produce a change in the skeletal phenotype (53). However, the liver-derived IGF-I KO/ALS KO crossed mice still did not account for the even greater reduction in growth and BMD observed in the total IGF-I KO mouse, thus suggesting that local IGF-I may be critical for growth and bone development in mice. As demonstrated in the current study, IGF-I derived from Col2α1-expressing cells, which were previously determined to be primarily chondrocytes (36), and this was confirmed in the present study by demonstrating Cre expression in primary chondroblasts, but not osteoblasts, is required for optimal longitudinal growth and bone width in mice. However, since bone contains other cells besides chondrocytes, such as osteoblasts, osteoclasts, and bone marrow cells, the relative contribution of each of these other cell types alone and in combination needs to be evaluated to completely determine the relative contribution of local vs. systemic IGF-I in the regulation of BMD. Studies are currently in progress to determine the role of osteoblast-derived IGF-I in attaining peak BMD.
The Cre/loxP method is a well-established method that is often used to determine the role of genes, and when this model is used to conditionally disrupt a gene of interest, the efficiency of the Cre recombinase, as well as the disruption of the gene of interest, is critical. We demonstrated that Cre recombinase expression was positively correlated with Col2α1 expression and confirmed that expression of IGF-I was significantly reduced in cartilage tissue, which expresses high levels of Col2α1, but did not alter circulating concentrations of IGF-I. Therefore, our model specifically evaluates the role of local IGF-I on skeletal tissue. Surprisingly, IGF-I expression was reduced in diaphysis/metaphysis of femur/tibia. We speculate that this may be caused by the following: 1) albeit at lower levels compared with cartilage, Col2α1 is expressed in these tissues; 2) the samples collected were whole tissue and, therefore, may contain other cell types, including chondrocytes; and 3) the loss of IGF-I in chondrocytes may alter the expression of IGF-I in other cell types, such as osteoblasts, through paracrine signaling. Importantly, we confirmed that Cre was expressed in primary chondrocytes, but not osteoblasts, in our conditional mutants and that IGF-I expression was decreased in bone and cartilage, but not in liver and kidney, thus providing evidence for the specificity of the promoter used for Cre expression to disrupt IGF-I in tissues that express Col2α1.
In a previous study, we found that disruption of total IGF-I resulted in reduced bone accretion during prepubertal, pubertal, and postpubertal growth in mice (25, 32), thus demonstrating that IGF-I is critical for bone accretion during all three periods of growth. Accordingly, we found that disruption of IGF-I in chondrocytes led to a 27% reduction in the rate of gain in body length between 2 and 4 wk of age. Furthermore, rate of gain in total body BMC and BMD were reduced by 18 to 22%. Surprisingly, we did not observe an effect of reduced IGF-I expression in chondrocytes during early states of development when chondrocytes are more present. This may be due to the predominant role of IGF-II during early postnatal development. Importantly, these data demonstrate that IGF-I from Col2α1-expressing cells is an important determinant of bone accretion that occurs during postnatal growth in mice.
Surprisingly, we observed a reduction in periosteal and endosteal circumference in the conditional mutant mice. Although these parameters are thought to be primarily regulated by osteoblasts and osteocytes, we hypothesize that the decreased bone width observed with disruption of IGF-I in Col2α1 expressing cells may be due to 1) decreased proliferation and/or differentiation of perichondrial cells caused by disruption of IGF-I produced by the cells. Since Col2α1 is known to be expressed in the perichondrium during embryonic development (14, 44), Col2α1 promoter-driven Cre expression could lead to disruption of IGF-I gene and/or 2) the reduction in local IGF-I from chondrocytes possibly affecting growth of cells in the periochondrium in a paracrine manner. In addition, there is recent evidence that factors produced by chondrocytes can regulate perichondrial cells (bone collar) and in turn influence bone width (5). Further studies are needed to determine the specific mechanism(s) by which local IGF-I regulates periosteal and endosteal circumference in growing mice.
The mechanism by which IGF-I regulates longitudinal bone growth was previously demonstrated by Wang et al. (49, 50), in which a lack of IGF-I reduced chondrocyte hypertrophy but did not affect proliferation in the growth plate. Consistent with these findings, Long et al. (28) demonstrated that chondrocyte proliferation was not altered in IGF-IR null mice. However, more recent evidence suggests that lack of local and circulating IGF-I reduces proliferation of chondrocytes (51). Consistent with these data, overexpression, by retroviral vector, of IGFBP-2 in developing long bones of chicks reduced chondrocyte proliferation by blocking IGF-I action (11). In addition, administration of IGF-I to hypophysectomized rats increased both chondrocyte proliferation and differentiation (16). That we did not find a significant difference by histomorphometric analysis may be due to either the moderate reduction in bone length and width observed in the conditional mutants or the variation associated with histomorphometric analysis, and/or the number of mice used may be too small to detect a significant difference. Similarly, a significant reduction in the growth plate size was not observed in the liver-derived IGF-I KO mouse in which the phenotypic changes were small (46). Therefore, further analysis is needed to determine whether chondrocyte-derived IGF-I or another source of local IGF-I is responsible for the reduction in chondrocyte hypertrophy observed in the total IGF-I knockout mouse.
Previous findings demonstrate that IGFBPs are important regulators of IGF actions (10, 17). In this regard, it is known that IGFBP-5 potentiated IGF-I actions on chondrocyte differentiation (21). Furthermore, it has also been shown that IGF-I stimulates IGFBP-5 expression in chondrocytes (22). Our finding that expression of IGFBP-5 was reduced in the cartilage derived from conditional mutant mice is consistent with these previous findings and further demonstrates an important role for IGFBP-5 in regulating cartilage growth.
It is well established that long bones are formed by endochondral ossification in which cartilage is replaced by bone. Based on the reduction in bone length and width in response to disruption of IGF-I from Col2α1-expressing cells, we hypothesized that IGF-I regulates longitudinal growth and bone width, in part, via regulating the expression of one or more messenger molecules (PTHrP, Ihh, and transcription factors) that are involved in chondrocyte proliferation and/or differentiation (39, 48). The reduction in PTHrP and PTH1R, but no change in Ihh, which is upstream of PTHrP, suggests that IGF-I regulates chondrocyte proliferation and/or differentiation through factors downstream of Ihh. Our data are consistent with previous findings that IGF-I affects PTHrP production and/or action (4, 41). The reduced expression of PTH1R is opposite from previous reports in osteoblast cells that demonstrate that IGF-I suppresses PTH1R (19, 20), thus suggesting that IGF-I may differentially regulate PTH1R in chondroblasts vs. osteoblasts. In addition, our data suggest that Sox9 may mediate IGF-I action in cartilage development; however, this may be an indirect effect of IGF-I through the PTHrP pathway since Sox9 is downstream of PTHrP (15). The reduced expression of Dlx5, an important regulator of chondrocyte differentiation (2), suggests a potentially important role for Dlx5 in mediating IGF-I actions in cartilage development. Dlx3, which is upregulated in prehypertrophic chondrocytes, is reduced in hypertrophic chondrocytes and has similar expression patterns to Ihh (12). However, it does not appear to play a critical role in IGF-I regulation of chondrocyte proliferation and differentiation. It should be noted that these were whole tissue samples and may also contain bone, perichondrium, and periosteum cells in addition to cartilage cells. Therefore, the differences observed between control and conditional mutant expression of these genes may be due to expression in one or more of these cell types. Overall, our data suggest that these messenger molecules may play a role in mediating local IGF-I action in bone; however, further analysis is needed to determine their specific roles in mediating IGF-I in chondrocytes.
In conclusion, using the Cre/loxP method, we demonstrated that local IGF-I from Col2α1-expressing cells is important for long bone formation, as well as bone width. In addition, we have provided preliminary evidence that IGF-I from Col2α1-expressing cells appears to be involved in the regulation of chondrocyte development, longitudinal growth, and bone width by interacting with or regulating the expression of PTHrP, PTH1R, Sox9, Dlx5, and IGFBP-5. Further studies are in progress to determine how IGF-I interacts with these factors to regulate cartilage growth, as well as to elucidate the relative contributions of chondroblast- vs. osteoblast-derived IGF-I in regulating peak BMD and bone width.
ACKNOWLEDGMENTS
The authors acknowledge the contribution of Professor Derek LeRoith (Mt. Sinai School of Medicine, New York, NY) for providing us with IGF-1 loxP mice for these studies. The authors also thank Erica Winter, Catrina Raynor, Shamyne Hover, Sheila Pourteymoor, and Joe Rung-Aroon for technical assistance and Terry Dent for secretarial assistance.
All work was performed in facilities provided by the Department of Veterans Affairs.
GRANTS This work was supported by National Institute of Arthritis and Musculo-skeletal and Skin Diseases Grants AR-048139 (S. Mohan) and AR-42919 (R. R. Behringer).
REFERENCES
- 1.Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73–82. [PubMed] [Google Scholar]
- 2.Bendall AJ, Hu G, Levi G, Abate-Shen C. Dlx5 regulates chondrocyte differentiation at multiple stages. Int J Dev Biol. 2003;47:335–344. [PubMed] [Google Scholar]
- 3.Bikle DD, Sakata T, Leary C, Elalieh H, Ginzinger D, Rosen CJ, Beamer W, Majumdar S, Halloran BP. Insulin-like growth factor I is required for the anabolic actions of parathyroid hormone on mouse bone. J Bone Miner Res. 2002;17:1570–1578. doi: 10.1359/jbmr.2002.17.9.1570. [DOI] [PubMed] [Google Scholar]
- 4.Canalis E, McCarthy TL, Centrella M. Differential effects of continuous and transient treatment with parathyroid hormone related peptide (PTHrp) on bone collagen synthesis. Endocrinology. 1990;126:1806–1812. doi: 10.1210/endo-126-4-1806. [DOI] [PubMed] [Google Scholar]
- 5.Chung UI, Schipani E, McMahon AP, Kronenberg HM. Indian hedgehog couples chondrogenesis to osteogenesis in endochondral bone development. J Clin Invest. 2001;107:295–304. doi: 10.1172/JCI11706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collett-Solberg PF, Cohen P. Genetics, chemistry, and function of the IGF/IGFBP system. Endocrine. 2000;12:121–136. doi: 10.1385/ENDO:12:2:121. [DOI] [PubMed] [Google Scholar]
- 7.Daughaday WH. Growth hormone axis overview–somatomedin hypothesis. Pediatr Nephrol. 2000;14:537–540. doi: 10.1007/s004670000334. [DOI] [PubMed] [Google Scholar]
- 8.Dorak MT. Real-time PCR. http://dorakmt.tripod.com/genetics/real-time.html ( http://dorakmttripodcom/genetics/realtimehtml, 2005)
- 9.Duan C, Xu Q. Roles of insulin-like growth factor (IGF) binding proteins in regulating IGF actions. Gen Comp Endocrinol. 2005;142:44–52. doi: 10.1016/j.ygcen.2004.12.022. [DOI] [PubMed] [Google Scholar]
- 10.Firth SM, Baxter RC. Cellular actions of the insulin-like growth factor binding proteins. Endocr Rev. 2002;23:824–854. doi: 10.1210/er.2001-0033. [DOI] [PubMed] [Google Scholar]
- 11.Fisher MC, Meyer C, Garber G, Dealy CN. Role of IGFBP2, IGF-I and IGF-II in regulating long bone growth. Bone. 2005;37:741–750. doi: 10.1016/j.bone.2005.07.024. [DOI] [PubMed] [Google Scholar]
- 12.Ghoul-Mazgar S, Hotton D, Lezot F, Blin-Wakkach C, Asselin A, Sautier JM, Berdal A. Expression pattern of Dlx3 during cell differentiation in mineralized tissues. Bone. 2005;37:799–809. doi: 10.1016/j.bone.2005.03.020. [DOI] [PubMed] [Google Scholar]
- 13.Govoni KE, Amaar YG, Kramer A, Winter E, Baylink DJ, Mohan S. Regulation of insulin-like growth factor binding protein-5, four and a half lim-2, and a disintegrin and metalloprotease-9 expression in osteoblasts. Growth Horm IGF Res. 2006;16:49–56. doi: 10.1016/j.ghir.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haaijman A, D'Souza RN, Bronckers AL, Goei SW, Burger EH. OP-1 (BMP-7) affects mRNA expression of type I, II, X collagen, and matrix Gla protein in ossifying long bones in vitro. J Bone Miner Res. 1997;12:1815–1823. doi: 10.1359/jbmr.1997.12.11.1815. [DOI] [PubMed] [Google Scholar]
- 15.Huang W, Chung UI, Kronenberg HM, de Crombrugghe B. The chondrogenic transcription factor Sox9 is a target of signaling by the parathyroid hormone-related peptide in the growth plate of endochondral bones. Proc Natl Acad Sci USA. 2001;98:160–165. doi: 10.1073/pnas.011393998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hunziker EB, Wagner J, Zapf J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J Clin Invest. 1994;93:1078–1086. doi: 10.1172/JCI117058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones JI, Clemmons DR. Insulin-like growth factors and their binding proteins: biological actions. Endocr Rev. 1995;16:3–34. doi: 10.1210/edrv-16-1-3. [DOI] [PubMed] [Google Scholar]
- 18.Kasukawa Y, Baylink DJ, Guo R, Mohan S. Evidence that sensitivity to growth hormone (GH) is growth period and tissue type dependent: studies in GH-deficient lit/lit mice. Endocrinology. 2003;144:3950–3957. doi: 10.1210/en.2002-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kawane T, Horiuchi N. Insulin-like growth factor I suppresses parathyroid hormone (PTH)/PTH-related protein receptor expression via a mitogen-activated protein kinase pathway in UMR-106 osteoblast-like cells. Endocrinology. 1999;140:871–879. doi: 10.1210/endo.140.2.6517. [DOI] [PubMed] [Google Scholar]
- 20.Kawane T, Mimura J, Fujii-Kuriyama Y, Horiuchi N. Identification of the promoter region of the parathyroid hormone receptor gene responsible for transcriptional suppression by insulin-like growth factor-I. Arch Biochem Biophys. 2005;439:61–69. doi: 10.1016/j.abb.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 21.Kiepe D, Ciarmatori S, Haarmann A, Tonshoff B. Differential expression of IGF system components in proliferating vs. differentiating growth plate chondrocytes: the functional role of IGFBP-5. Am J Physiol Endocrinol Metab. 2006;290:E363–E371. doi: 10.1152/ajpendo.00363.2005. [DOI] [PubMed] [Google Scholar]
- 22.Kiepe D, Ciarmatori S, Hoeflich A, Wolf E, Tonshoff B. Insulin-like growth factor (IGF)-I stimulates cell proliferation and induces IGF binding protein (IGFBP)-3 and IGFBP-5 gene expression in cultured growth plate chondrocytes via distinct signaling pathways. Endocrinology. 2005;146:3096–3104. doi: 10.1210/en.2005-0324. [DOI] [PubMed] [Google Scholar]
- 23.Le Roith D, Bondy C, Yakar S, Liu JL, Butler A. The somatomedin hypothesis: 2001. Endocr Rev. 2001;22:53–74. doi: 10.1210/edrv.22.1.0419. [DOI] [PubMed] [Google Scholar]
- 24.Lefebvre V, Garofalo S, Zhou G, Metsaranta M, Vuorio E, De Crombrugghe B. Characterization of primary cultures of chondrocytes from type II collagen/beta-galactosidase transgenic mice. Matrix Biol. 1994;14:329–335. doi: 10.1016/0945-053x(94)90199-6. [DOI] [PubMed] [Google Scholar]
- 25.Libanati C, Baylink DJ, Lois-Wenzel E, Srinvasan N, Mohan S. Studies on the potential mediators of skeletal changes occurring during puberty in girls. J Clin Endocrinol Metab. 1999;84:2807–2814. doi: 10.1210/jcem.84.8.5905. [DOI] [PubMed] [Google Scholar]
- 26.Liu JL, Grinberg A, Westphal H, Sauer B, Accili D, Karas M, LeRoith D. Insulin-like growth factor-I affects perinatal lethality and postnatal development in a gene dosage-dependent manner: manipulation using the Cre/loxP system in transgenic mice. Mol Endocrinol. 1998;12:1452–1462. doi: 10.1210/mend.12.9.0162. [DOI] [PubMed] [Google Scholar]
- 27.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-(delta delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 28.Long F, Joeng KS, Xuan S, Efstratiadis A, McMahon AP. Independent regulation of skeletal growth by Ihh and IGF signaling. Dev Biol. 2006;298:327–333. doi: 10.1016/j.ydbio.2006.06.042. [DOI] [PubMed] [Google Scholar]
- 29.Miyakoshi N, Kasukawa Y, Linkhart TA, Baylink DJ, Mohan S. Evidence that anabolic effects of PTH on bone require IGF-I in growing mice. Endocrinology. 2001;142:4349–4356. doi: 10.1210/endo.142.10.8436. [DOI] [PubMed] [Google Scholar]
- 30.Mohan S, Baylink DJ. Development of a simple valid method for the complete removal of insulin-like growth factor (IGF)-binding proteins from IGFs in human serum and other biological fluids: comparison with acid-ethanol treatment and C18 Sep-Pak separation. J Clin Endocrinol Metab. 1995;80:637–647. doi: 10.1210/jcem.80.2.7531716. [DOI] [PubMed] [Google Scholar]
- 31.Mohan S, Baylink DJ. Impaired skeletal growth in mice with haploin-sufficiency of IGF-I: genetic evidence that differences in IGF-I expression could contribute to peak bone mineral density differences. J Endocrinol. 2005;185:415–420. doi: 10.1677/joe.1.06141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mohan S, Richman C, Guo R, Amaar Y, Donahue LR, Wergedal J, Baylink DJ. Insulin-like growth factor regulates peak bone mineral density in mice by both growth hormone-dependent and -independent mechanisms. Endocrinology. 2003;144:929–936. doi: 10.1210/en.2002-220948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ohlsson C, Bengtsson BA, Isaksson OG, Andreassen TT, Slootweg MC. Growth hormone and bone. Endocr Rev. 1998;19:55–79. doi: 10.1210/edrv.19.1.0324. [DOI] [PubMed] [Google Scholar]
- 34.Ohlsson C, Nilsson A, Isaksson OG, Lindahl A. Effect of growth hormone and insulin-like growth factor-I on DNA synthesis and matrix production in rat epiphyseal chondrocytes in monolayer culture. J Endocrinol. 1992;133:291–300. doi: 10.1677/joe.0.1330291. [DOI] [PubMed] [Google Scholar]
- 35.Okubo Y, Reddi AH. Thyroxine downregulates Sox9 and promotes chondrocyte hypertrophy. Biochem Biophys Res Commun. 2003;306:186–190. doi: 10.1016/s0006-291x(03)00912-4. [DOI] [PubMed] [Google Scholar]
- 36.Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- 37.Powell-Braxton L, Hollingshead P, Warburton C, Dowd M, Pitts-Meek S, Dalton D, Gillett N, Stewart TA. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- 38.Rajaram S, Baylink DJ, Mohan S. Insulin-like growth factor-binding proteins in serum and other biological fluids: regulation and functions. Endocr Rev. 1997;18:801–831. doi: 10.1210/edrv.18.6.0321. [DOI] [PubMed] [Google Scholar]
- 39.Razzzaque MS, Soegiarto DW, Chang D, Long F, Lanske B. Conditional deletion of Indian hedgehog from collagen type 2alpha1-expressing cells results in abnormal endochondral bone formation. J Pathol. 2005;207:453–461. doi: 10.1002/path.1870. [DOI] [PubMed] [Google Scholar]
- 40.Reinecke M, Schmid AC, Heyberger-Meyer B, Hunziker EB, Zapf J. Effect of growth hormone and insulin-like growth factor I (IGF-I) on the expression of IGF-I messenger ribonucleic acid and peptide in rat tibial growth plate and articular chondrocytes in vivo. Endocrinology. 2000;141:2847–2853. doi: 10.1210/endo.141.8.7624. [DOI] [PubMed] [Google Scholar]
- 41.Rizzoli R, Feyen JH, Grau G, Wohlwend A, Sappino AP, Bonjour JP. Regulation of parathyroid hormone-related protein production in a human lung squamous cell carcinoma line. J Endocrinol. 1994;143:333–341. doi: 10.1677/joe.0.1430333. [DOI] [PubMed] [Google Scholar]
- 42.Rutter MM, Markoff E, Clayton L, Akeno N, Zhao G, Clemens TL, Chernausek SD. Osteoblast-specific expression of insulin-like growth factor-1 in bone of transgenic mice induces insulin-like growth factor binding protein-5. Bone. 2005;36:224–231. doi: 10.1016/j.bone.2004.10.005. [DOI] [PubMed] [Google Scholar]
- 43.Salih DA, Mohan S, Kasukawa Y, Tripathi G, Lovett FA, Anderson NF, Carter EJ, Wergedal JE, Baylink DJ, Pell JM. Insulin-like growth factor-binding protein-5 induces a gender-related decrease in bone mineral density in transgenic mice. Endocrinology. 2005;146:931–940. doi: 10.1210/en.2004-0816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Savontaus M, Ihanamaki T, Perala M, Metsaranta M, Sandberg-Lall M, Vuorio E. Expression of type II and IX collagen isoforms during normal and pathological cartilage and eye development. Histochem Cell Biol. 1998;110:149–159. doi: 10.1007/s004180050276. [DOI] [PubMed] [Google Scholar]
- 45.Sjogren K, Liu JL, Blad K, Skrtic S, Vidal O, Wallenius V, LeRoith D, Tornell J, Isaksson OG, Jansson JO, Ohlsson C. Liver-derived insulin-like growth factor I (IGF-I) is the principal source of IGF-I in blood but is not required for postnatal body growth in mice. Proc Natl Acad Sci USA. 1999;96:7088–7092. doi: 10.1073/pnas.96.12.7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sjogren K, Sheng M, Moverare S, Liu JL, Wallenius K, Tornell J, Isaksson O, Jansson JO, Mohan S, Ohlsson C. Effects of liver-derived insulin-like growth factor I on bone metabolism in mice. J Bone Miner Res. 2002;17:1977–1987. doi: 10.1359/jbmr.2002.17.11.1977. [DOI] [PubMed] [Google Scholar]
- 47.Smink JJ, Koster JG, Gresnigt MG, Rooman R, Koedam JA, Van Buul-Offers SC. IGF and IGF-binding protein expression in the growth plate of normal, dexamethasone-treated and human IGF-II transgenic mice. J Endocrinol. 2002;175:143–153. doi: 10.1677/joe.0.1750143. [DOI] [PubMed] [Google Scholar]
- 48.Van der Eerden BC, Karperien M, Wit JM. Systemic and local regulation of the growth plate. Endocr Rev. 2003;24:782–801. doi: 10.1210/er.2002-0033. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Zhou J, Bondy CA. Igf1 promotes longitudinal bone growth by insulin-like actions augmenting chondrocyte hypertrophy. FASEB J. 1999;13:1985–1990. doi: 10.1096/fasebj.13.14.1985. [DOI] [PubMed] [Google Scholar]
- 50.Wang J, Zhou J, Cheng CM, Kopchick JJ, Bondy CA. Evidence supporting dual, IGF-I-independent and IGF-I-dependent, roles for GH in promoting longitudinal bone growth. J Endocrinol. 2004;180:247–255. doi: 10.1677/joe.0.1800247. [DOI] [PubMed] [Google Scholar]
- 51.Wang Y, Nishida S, Sakata T, Elalieh HZ, Chang W, Halloran BP, Doty SB, Bikle DD. Insulin-like growth factor-I is essential for embryonic bone development. Endocrinology. 2006;147:4753–4761. doi: 10.1210/en.2006-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B, LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci USA. 1999;96:7324–7329. doi: 10.1073/pnas.96.13.7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yakar S, Rosen CJ, Beamer WG, Ackert-Bicknell CL, Wu Y, Liu JL, Ooi GT, Setser J, Frystyk J, Boisclair YR, LeRoith D. Circulating levels of IGF-1 directly regulate bone growth and density. J Clin Invest. 2002;110:771–781. doi: 10.1172/JCI15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang M, Xuan S, Bouxsein ML, von Stechow D, Akeno N, Faugere MC, Malluche H, Zhao G, Rosen CJ, Efstratiadis A, Clemens TL. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]