

Abstract
IL-17–producing T cells infiltrate kidneys of patients with lupus nephritis, and IL-23–treated lymph node cells from lupus-prone mice may transfer disease to Rag1-deficient mice. In this study, we show that IL-23R–deficient lupus-prone C57BL/6–lpr/lpr mice display decreased numbers of CD3+CD4−CD8− cells and IL-17A–producing cells in the lymph nodes and produce less anti-DNA Abs. In addition, clinical and pathology measures of lupus nephritis are abrogated. The presented experiments document the importance of IL-23R–mediated signaling in the development of lupus nephritis and urge the consideration of proper biologics for the treatment of the disease.
T cells are central to systemic lupus erythematosus pathogenesis, as they provide help to autoreactive B cells, infiltrate target tissues, and fail to perform appropriate regulatory functions (1). The precise effector mechanisms used by T cells to promote disease pathology are still unclear. Recently IL-17, the signature cytokine for the proinflammatory T cell subset Th17, was found overexpressed in systemic lupus erythematosus, whereas IL-17–producing cells were found in the kidneys of patients with lupus nephritis (2). Lupus-prone mice have also increased numbers of Th17 cells, which can instigate disease when transferred into Rag1−/− mice after being conditioned with IL-23 in vitro (3, 4). IL-23 is important for the stabilization and differentiation of Th17 cells (5), and IL-23R–deficient mice are resistant to experimental autoimmune encephalomyelitis (6, 7).
Based on the above observations, we hypothesized that IL-23R–mediated signaling is central to the expression of lupus pathology. In this study, we demonstrate that IL-23R−/−– deficient C57BL/6–lpr/lpr (B6/lpr) mice do not mount an autoimmune response and are protected from lupus nephritis.
Materials and Methods
Mice
C57BL/6J, B6.MRL-Faslpr/J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The IL-23R–deficient C57BL/6 mice (IL-23R−/− C57BL/6) were generated as previously described (6). All mice were housed at the Beth Israel Deaconess Medical Center pathogen-free animal facility (Boston, MA). Our protocol was approved by the Beth Israel Deaconess Medical Center institutional animal care and use committee.
We generated IL-23R–deficient B6.MRL-Faslpr/J mice using a backcross-intercross scheme. After six generations of breeding, the mice were PCR screened for the Faslpr and mutated IL-23R gene. Primers for Faslpr genetic screen: 5′-GTAAATAATTGTGCTTCGTCAG-3′, 5′-TAGAAAGGTGCACGGGTGTG- 3′, and 5′-CAAATCTAGGCATTAACAGTG-3′; IL-23R genetic screen (6): 5′-ACCCCTAGGAATGCTCGTCAAG-3′ and 5′-TGGTTGCCTGCACCAATTTAAAAG-3′; and for homozygous versus heterozygous mutated: 5′-GATCATCTTATGGCTGGTCCTC-3′ and 5′-GAGTGAGACAGTGTAGCCACAGAT-3′.
Histopathology
The kidneys were fixed in 10% formalin overnight at 4°C, embedded in paraffin, and sectioned at 6 μM before being stained with H&E and periodic acid-Schiff reagent. We evaluated glomerular pathology by assessing 100 glomeruli per kidney and scored each glomerulus using a semiquantitative scale (8).
Intracellular staining and confocal microscopy was performed as previously described (3). IgG and anti-dsDNA Abs were measured using ELISA (Immunology Consultants Lab, Newberg, OR). The Mann-Whitney U two-tailed test was used for statistical significance.
Results and Discussion
IL-23R deficiency prevents hyperplasia of secondary lymphoid organs in lupus-prone mice
We have shown (3) that when B6/lpr-derived lymphocytes are cultured in the presence of IL-23, they acquire the capacity to become pathogenic. Accordingly, we hypothesized that IL-23–mediated signaling is central in the pathogenesis of lupus. To address this question, we generated lupus-prone B6/lpr mice lacking IL-23R. These IL-23R−/−B6/lpr mice were healthy and fertile. We observed the mice for a period of 9 mo for the development of signs of lupus and lupus nephritis. We found that the IL-23R−/−B6/lpr mice developed milder proteinuria than age- and sex-matched B6/lpr wild-type mice; IL-23R−/−B6/lpr mice were similar to control B6 mice in terms of proteinuria (IL-23R−/−B6/lpr versus B6/lpr versus B6, protein in urine: 100 mg/dl versus 2000 mg/dl versus 100 mg/dl; n = 5). More importantly, there was no evidence of pyuria in the IL-23R−/−B6/lpr mice as opposed to the B6lpr wild-type mice.
We then sacrificed 9-mo-old B6/lpr, IL-23R−/−B6/lpr, and B6 mice. Although at this age B6/lpr mice exhibit severe disease manifestations including hyperplasia of the secondary lymphoid organs and glomerulonephritis, mice that lacked IL-23R had significantly smaller spleens and lymph nodes than wildtype lupus-prone mice (Fig. 1A). The size of the lymphoid organs approximated those of a normal B6 mouse (not shown in this paper). The reduced size of the lymphoid organs in the IL-23R−/−B6/lpr mice was associated with a profound reduction in the cell population in these organs by ~50% in the spleen and 90% in the lymph nodes [B6/lpr (n = 5) versus IL-23R−/−B6/lpr (n = 5) versus B6 (n = 4) total splenocytes × 106 ± SD: 186.4 ± 8.5 versus 112.26 ± 2.58 versus 124.15 ± 5.08; p = 0.01; B6/lpr (n = 5) versus IL-23R−/−B6/lpr (n = 5) versus B6 (n = 4) total lymph node cells × 106 ± SD: 672.3 ± 35.5 versus 66 ± 9.17 versus 40 ± 8.27; p = 0.016; Fig. 1B).
FIGURE 1.

IL-23R−/−B6/lpr mice have smaller secondary lymphoid organs and significantly reduced number of CD3+ and CD3+CD4−CD8− cells as compared with B6/lpr mice. A, The secondary lymphoid organs were harvested from 9-mo-old B6/lpr and IL-23R−/−B6/lpr mice. A representative spleen and an inguinal lymph node from these two strains are shown. B, Single-cell suspensions were made from the spleens and lymph nodes of five B6/lpr, five IL-23R−/−B6/lpr, and four B6 mice. We present the total number of cells in the spleens and lymph nodes ± SD for each group. C, Single-cell suspensions were made from B6/lpr and IL-23R−/−B6/lpr spleens and lymph nodes. The cells were stained with fluorochrome-labeled Abs against CD3, CD4, and CD8. A representative FACS analysis plot of cells that are CD3+ is presented. D, The total number of CD3+CD4+, CD3+CD8+, and CD3+CD4−CD8− (DNT) was calculated using the FACS analyses of individual cell populations shown in C and the total number of cells shown in B. We show the average number of CD3+ cells and the CD3 subsets from the spleens and lymph nodes of five B6/lpr, five IL-23R−/−B6/lpr, and four B6 mice (see text for exact means ± SD for each cell population). *Statistically significant difference.
In evaluating the subsets of lymphocytes in the IL-23R−/−B6/lpr mice, we found that the total number ofCD3+ T cells in both spleens and lymph nodes are reduced when compared with wildtype B6/lpr animals [B6/lpr (n = 5) versus IL-23R−/−B6/lpr (n = 5) versus B6 (n =4)CD3+ splenocytes × 106 ± SD: 36.42 ± 3.5 versus 27.41 ± 3.8 versus 28.44 ± 1.4; p = 0.04 for B6/lpr versus IL-23R−/−B6/lpr. B6/lpr (n=5) versus IL-23R−/−B6/lpr (n=5) versus B6 (n = 4)CD3+ lymph node cells × 106 ± SD: 338.49 ± 21.4 versus 26.07 ± 1.12 versus 12.6 ± 0.45; p = 0.04 for B6/lpr versus IL-23R−/−B6/lpr; Fig. 1D]. In analyzing T cell subsets, we found that the IL-23R−/−B6/lpr mice had a significant decrease in the number of CD3+CD4−CD8− (double-negative T cells [DNTs]) that accumulate abnormally in the lymphoid organs of lupus-prone mice that bear the lpr mutation [B6/lpr (n = 5) versus IL-23R−/−B6/lpr (n = 5) versus B6 (n = 4) DNT percent ofCD3+ splenocytes: 24.5 ± 2.4 versus 17 ± 1.78 versus 6.73 ± 1.04; p = 0.025 for B6/lpr versus IL-23R−/−B6/lpr; DNT percent among CD3+ lymph node cells: 32.89 ± 2.78 versus 9.87 ± 1.19 versus 6.8 ± 0.84; p = 0.0002 for B6/lpr versus IL-23R−/−B6/lpr). A representative experiment is shown in Fig. 1C and cumulative data in Fig. 1D. Conversely, we observed a significant rise in the percentage of CD8 cells in both spleen (p = 0.0079) and lymph nodes (p = 0.0317) of IL-23R−/−B6/lpr versus B6/lpr mice (Fig. 1C). We did not observe significant changes in the percentage of CD4+ cells between the two groups (Fig. 1C). It has to be stated, although, that because the overall number of lymphocytes in the lymph nodes of the IL-23R– deficient mice was profoundly reduced, the overall number of DNT, CD4, and CD8 T cells in the lymph nodes was significantly decreased (Fig. 1D). The percentage of regulatory T (CD4+CD25hiFoxp3+) (Supplemental Fig. 1), γδ T, B, dendritic, and monocyte lineage cells did not differ significantly between the IL-23R−/−B6/lpr and B6/lpr mice. Additionally, there was no difference in T cell subtypes, such as DNT and regulatory T cells, between B6 and IL-23R−/−B6 (Supplemental Fig. 2).
This set of experiments shows that in the absence of IL-23R, the accumulation of lymphocytes and the generation of the abnormal double-negative cells are profoundly reduced.
IL-23R deficiency resulted in reduced production of inflammatory T cell-derived cytokines and autoantibodies
It has been shown that the development of lupus nephritis in mice depends on the production of proinflammatory cytokines, such as IFN-γ (9) and, more recently, IL-17. Because IL-23R deficiency resulted in profound changes in the T cell phenotype in lupus-prone mice, we hypothesized that the production of proinflammatory cytokines and, in particular, IL-17A was impaired in the IL-23R−/−B6/lpr mice.
Indeed, as shown in Fig. 2A, the overall number of cells producing IL-17A is profoundly decreased in the IL-23R−/−B6/lpr mice (B6/lpr versus IL-23R−/−B6/lpr CD3+IL-17+ lymph node cells ± SD: 813,800 ± 204,800 versus 8,020 ± 1,880; n = 5; Fig. 2A, lower panel). Both CD4+IL-17+ and CD3+CD4−CD8−IL-17+ cells were decreased in the IL-23R−/−B6/lpr mice versus B6/lpr mice. Additionally, the percentage of DNTs expressing IL-17+ was significantly decreased in IL-23R−/−B6/lpr mice (Fig. 2A, right panel). In the CD4+ population, there were no significant differences between the two mice in percentage of IL-17+ cells.
FIGURE 2.

IL-23R−/−B6/lpr mice have decreased number of IL-17+ cells, serum IgG, and anti-dsDNA Ab levels as compared with B6/lpr mice. A, Lymph node-derived cells from both IL-23R−/−B6/lpr and B6/lpr mice were stained for intracellular IL-17A as previously described (3). We show a representative FACS plot of IL-17A production (top panels). In the lower panel, we show the total number of IL-17+ cells in lymph node extracts from IL-23R−/−B6/lpr and B6/lpr mice (n = 5). B, Lymph node-derived cells from both IL-23R−/−B6/lpr and B6/lpr mice (n = 5) were stained for intracellular IFN-γ. We show a representative FACS plot of IFN-γ production. C, Serum was collected from 9-mo-old B6/lpr (n = 8), IL-23R−/−B6/lpr (n = 7), and B6 (n = 3). We show mean levels ± SD of total IgG. D, Serum was collected from 9-mo-old B6/lpr (n = 6), IL-23R−/−B6/lpr (n = 7), and B6 (n = 4). We show mean levels ± SD of anti-dsDNA. *Statistically significant difference.
Because development of lupus depends on IFN-γ, we evaluated expression of this cytokine by both CD4+ and DNTs. As shown in Fig. 2B, both CD4+ and DNTs from IL-23R−/−B6/lpr mice had reduced expression of IFN-γ when compared with control B6/lpr mice. These findings suggest not only that DNTs (and to a lesser extent CD4+ cells) are reduced in number in IL-23R−/−B6/lpr mice, but also that the production of proinflammatory cytokines is significantly decreased. When comparing B6 to IL-23R−/−B6 mice, there was a nonstatistically significant decrease of the expression of IFN-γ and IL-17A by DNTs in the IL-23R–deficient mice (Supplemental Fig. 3). It has to be stated, although, that both cytokines were expressed at very low levels by DNTs in both strains.
We have shown in the past that IL-23 treatment of B6/lpr-derived lymphocytes that were later transferred to Rag-1−/− mice results in IgG deposition in the kidneys of recipient mice. We asked the question whether abrogation of the IL-23–mediated signaling would affect the production of Ig and, more importantly, of pathogenic autoantibodies. Indeed, as shown in Fig. 2C, the levels of IgG in the serum of IL-23R−/−B6/lpr mice is significantly reduced when compared with control B6/lpr mice [B6/lpr (n = 8) versus IL-23R−/−B6/lpr (n = 7) versus B6 (n = 3) total serum IgG (ng/ml): 394418.75 ± 28155.85 versus 182571.42 ± 11330.09 versus 135250 ± 19440.99; p = 0.0002 for B6/lpr versus IL-23R−/−B6/lpr]. More importantly, the IL-23R–deficient mice produced less anti-dsDNA Abs than B6/lpr mice and at similar levels to normal B6 mice [B6/lpr (n = 9) versus IL-23R−/−B6/lpr (n = 10) versus B6 (n = 4) serum anti-dsDNA Ab levels (IU/ml): 3424.44 ± 481.31 versus 1365.29 ± 143.52 versus 836.23 ± 75.54; p = 0.0003 for B6/lpr versus IL-23R−/−B6/lpr; Fig. 2D].
The above findings point to the fact that in the absence of IL-23R, lupus-prone mice do not exhibit some of the readily recognizable features of immune dysregulation characteristic of lupus.
IL-23R deficiency mitigates glomerulonephritis in lupus-prone mice
As shown above, IL-23R−/−B6/lpr mice are characterized by significantly decreased spontaneous expression of inflammatory cytokines and production of autoantibodies. Therefore, we evaluated whether glomerular and interstitial inflammation that characterizes B6/lpr lupus-prone mice is decreased in the IL-23R−/−B6/lpr mice. As shown in Fig. 3A, glomeruli from IL-23R−/−B6/lpr mice displayed reduced mesangial cell proliferation and membrane thickening when compared with B6/lpr [glomerular damage grade: B6/lpr (n = 8) versus IL-23R−/−B6/lpr (n = 7) versus B6 (n = 4): 2.2 ± 0.14 versus 0.8 ± 0.14 versus 0.7 ± 0.13; p = 0.0001 for B6/lpr versus IL-23R−/−B6/lpr]. In parallel, the IL-23R−/−B6/lpr mice had decreased perivascular infiltration with lymphocytes when compared with B6/lpr [B6/lpr (n = 8) versus IL-23R−/−B6/lpr (n=7) versusB6(n = 4): 1.86 ± 0.31 versus 0.57 ± 0.28 versus 0.75 ± 0.5; p = 0.02 for B6/lpr versus IL-23R−/−B6/lpr; Fig. 3B]. Although all B6/lpr kidneys had perivascular infiltration with inflammatory cells, only one out of seven IL-23R−/−B6/lpr mice and one out of four B6 mice had evidence of perivascular inflammation in the kidneys.
FIGURE 3.
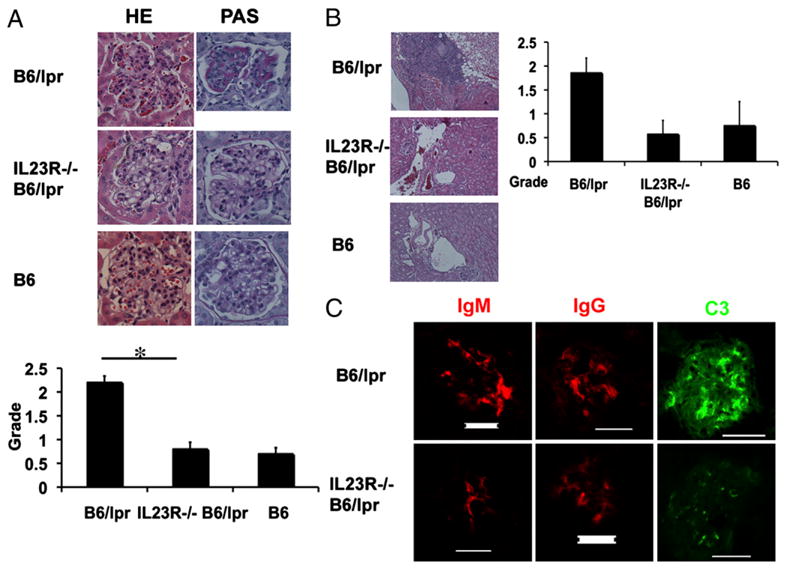
IL-23R−/−B6/lpr mice do not develop significant nephritis. Nine-month-old IL-23R−/−B6/lpr, B6/lpr, and B6 mice were sacrificed and their kidneys harvested and stained. A, H&E and periodic acid-Schiff staining of a representative glomerulus from all three groups is depicted. In the bar graph, we show the glomerulonephritis pathologic scores (mean ± SD) from eight B6/lpr, seven IL-23R−/−B6/lpr, and four B6 mice. Original magnification ×40. B, H&E staining of a representative perivascular area is shown. Original magnification ×40. In the bar graph, we show the perivascular cell infiltration pathologic scores (mean ± SD) from eight B6/lpr, seven IL-23R−/−B6/lpr, and four B6 mice. C, Immunofluorescent staining for IgM, IgG, and C3 deposition in a representative kidney section from IL-23R−/−B6/lpr and B6/lpr mice. The staining was done as previously described (3). Scale bar, 50 μM. *Statistically significant difference.
Because we found that the IL-23R−/−B6/lpr mice had also decreased Ig and anti-dsDNA Ab levels in the serum when compared with B6/lpr, we stained the glomeruli for IgG, IgM, and C3 deposition. As shown in Fig. 3C, the intensity of the staining for both was significantly reduced in the IL-23R– deficient mice-derived kidneys when compared with B6/lpr. We also stained the kidneys for IL-17A and found rare IL-17+ cells infiltrating the kidneys of B6/lpr mice but not the kidneys of IL-23R−/−B6/lpr mice (data not shown).
Given these findings in IL23R−/−B6/lpr mice, we used an Ab against IL-23 to treat 12-wk-old MRL/lpr mice for 4 wk. We found that at the end of the treatment, the anti–IL-23 Ab-injected mice had decreased proteinuria compared with IgG control-treated mice (100 mg/dl versus 2000 mg/dl; n = 3 for each group). The anti–IL-23 Ab-treated mice did not have any hematuria or pyuria, whereas the control IgG-treated mice had both (Z. Zhang, V.C. Kyttaris, and G.C. Tsokos, unpublished observation). Moreover, IL-17A production was significantly decreased in the anti–IL-23–treated mice as compared with controls (Supplemental Fig. 4).
Our previous studies showed that treatment with IL-23– pretreated B6/lpr lymph node-derived cells were capable of causing glomerulonephritis when transferred to Rag-1−/− mice. This study expands our previous understanding of the contribution of IL-23 to the pathogenesis of lupus by showing for the first time that IL-23R is necessary for lymphoid hyperplasia, production of pathogenic autoantibodies, and infiltration of kidneys by IL-17+ cells in lupus-prone mice. Furthermore, the spontaneous secretion of IFN-γ is decreased in the IL-23R−/− lupus-prone animals as a consequence of global decrease in spontaneous immune system activation. These findings coupled with the fact that proteinuria was decreased and pyuria was not present in the IL-23R–deficient mice point to the fact that IL-23R represents a key molecule in the chain reaction that leads to lupus nephritis. This conclusion is further reinforced by our preliminary data on treatment of lupus-prone mice with an anti–IL-23R Ab showing reversal of the proinflammatory phenotype and clinical improvement of these mice.
The most striking immunologic feature of the IL-23R−/−B6/lpr mouse was the significant decrease of DNTs, the major source of IL-17A in lupus-prone mice. Our data strongly suggest that these cells may act as effector cells (10), orchestrating the inflammatory response in the kidneys and/or providing help to B cells to produce pathogenic autoantibodies (11). The extent to which each of these pathways contributes to the development of lupus nephritis remains to be determined. Additional studies will also be needed to further elucidate the exact cytokines secreted by Th17 cells that contribute to lupus pathology.
In summary, this study demonstrates that IL-23R deficiency limits the lymphoid hyperplasia, abrogates the accumulation of DNTs, and prevents the development of nephritis in lupus-prone mice. Inhibition of IL-23 and/or IL-17A in vivo at various stages of lupus will enable us to determine the optimal time to block this pathway to prevent lupus nephritis.
Supplementary Material
Acknowledgments
We thank Dr. Li Li (BD Biosciences) for the generous gift of the anti–IL-23 Ab.
This work was supported by National Institutes of Health Grants RO1 AI42269, RO1 AI 49954 (to G.C.T.), RO1 AI49954 (to M.O.), and K23 AR055672 (to V.C.K.).
Abbreviations used in this paper
- B6/lpr
C57BL/6–lpr/lpr
- DNT
double-negative T cell
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.Kyttaris VC, Juang YT, Tsokos GC. Immune cells and cytokines in systemic lupus erythematosus: an update. Curr Opin Rheumatol. 2005;17:518–522. doi: 10.1097/01.bor.0000170479.01451.ab. [DOI] [PubMed] [Google Scholar]
- 2.Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. 2008;181:8761–8766. doi: 10.4049/jimmunol.181.12.8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Z, V, Kyttaris C, Tsokos GC. The role of IL-23/IL-17 axis in lupus nephritis. J Immunol. 2009;183:3160–3169. doi: 10.4049/jimmunol.0900385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kang HK, Liu M, Datta SK. Low-dose peptide tolerance therapy of lupus generates plasmacytoid dendritic cells that cause expansion of autoantigen-specific regulatory T cells and contraction of inflammatory Th17 cells. J Immunol. 2007;178:7849–7858. doi: 10.4049/jimmunol.178.12.7849. [DOI] [PubMed] [Google Scholar]
- 5.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 6.Awasthi A, Riol-Blanco L, Jäger A, Korn T, Pot C, Galileos G, Bettelli E, Kuchroo VK, Oukka M. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 2009;182:5904–5908. doi: 10.4049/jimmunol.0900732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 8.Kikawada E, Lenda DM, Kelley VR. IL-12 deficiency in MRL-Fas (lpr) mice delays nephritis and intrarenal IFN-gamma expression, and diminishes systemic pathology. J Immunol. 2003;170:3915–3925. doi: 10.4049/jimmunol.170.7.3915. [DOI] [PubMed] [Google Scholar]
- 9.Lawson BR, Prud’homme GJ, Chang Y, Gardner HA, Kuan J, Kono DH, Theofilopoulos AN. Treatment of murine lupus with cDNA encoding IFN-gammaR/Fc. J Clin Invest. 2000;106:207–215. doi: 10.1172/JCI10167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crispín JC, Tsokos GC. Human TCR-alpha beta+ CD4− CD8− T cells can derive from CD8+ T cells and display an inflammatory effector phenotype. J Immunol. 2009;183:4675–4681. doi: 10.4049/jimmunol.0901533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, Craft J. ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med. 2008;205:2873–2886. doi: 10.1084/jem.20080840. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.