

Abstract
The genomics revolution has spurred the undertaking of HapMap studies of numerous species, allowing for population genomics to increase the understanding of how selection has created genetic differences between subspecies populations. The objectives of this study were to (1) develop an approach to detect signatures of selection in subsets of phenotypically similar breeds of livestock by comparing single nucleotide polymorphism (SNP) diversity between the subset and a larger population, (2) verify this method in breeds selected for simply inherited traits, and (3) apply this method to the dairy breeds in the International Bovine HapMap (IBHM) study. The data consisted of genotypes for 32,689 SNPs of 497 animals from 19 breeds. For a given subset of breeds, the test statistic was the parametric composite log likelihood (CLL) of the differences in allelic frequencies between the subset and the IBHM for a sliding window of SNPs. The null distribution was obtained by calculating CLL for 50,000 random subsets (per chromosome) of individuals. The validity of this approach was confirmed by obtaining extremely large CLLs at the sites of causative variation for polled (BTA1) and black-coat-color (BTA18) phenotypes. Across the 30 bovine chromosomes, 699 putative selection signatures were detected. The largest CLL was on BTA6 and corresponded to KIT, which is responsible for the piebald phenotype present in four of the five dairy breeds. Potassium channel-related genes were at the site of the largest CLL on three chromosomes (BTA14, -16, and -25) whereas integrins (BTA18 and -19) and serine/arginine rich splicing factors (BTA20 and -23) each had the largest CLL on two chromosomes. On the basis of the results of this study, the application of population genomics to farm animals seems quite promising. Comparisons between breed groups have the potential to identify genomic regions influencing complex traits with no need for complex equipment and the collection of extensive phenotypic records and can contribute to the identification of candidate genes and to the understanding of the biological mechanisms controlling complex traits.
RECENT advances in genomics have greatly expanded our ability to study the genetics of organisms. Numerous “HapMap” studies have been undertaken, whereby subpopulations of a given species are genotyped and compared for genomic differences. In livestock, HapMap studies can provide insight into the differentiation of breeds and long-term selection for complex traits. When a favorable mutation occurs within a population under directional selection, the frequency of the favorable allele is likely to increase over time. Because DNA is composed of linear molecules and the probability of recombination is inversely proportional to the distance separating them, nucleotides adjacent to the favorable mutation also tend to increase in frequency, in a sort of “hitch-hiking” process (Maynard Smith and Haigh 1974; Fay and Wu 2000). This process leads to “signatures of selection” that are characterized by distributions of nucleotides around favorable mutations that differ statistically from that expected purely by chance (Kim and Stephan 2002). Detection of selection signatures can increase the understanding of the evolution and biology underlying a given phenotype and may provide tools to increase efficiency of selection.
Various methods have been developed for detection of selection signatures through genomic analysis. In general, most of these methods are based on comparison of the distribution of allelic frequencies, either directly, or indirectly, by calculating population genetics statistics that are a function of allelic or genotypic frequencies. As examples of the latter, FST (e.g., Weir et al. 2006; Bovine HapMap Consortium et al. 2009) and linkage disequilibrium (e.g., Parsch et al. 2001; Przeworski 2002; Kim and Nielsen 2004; Ennis 2007) have been used. In addition, specific tests for detecting signatures have been developed (e.g., Tajima 1989; Fu and Li 1993; Fay and Wu 2000; Kim and Stephan 2002; Voight et al. 2006).
With many of these methods, constructing a significance test is not straightforward, especially when searching for selection signatures within a single population. Determining the null distribution of the test statistic often requires making assumptions about the null distribution and applying a parametric test based on statistical theory. An alternative approach is to use simulation to derive a distribution of the test statistic under the assumption of no selection. For example, Kim and Stephan (2002) proposed the use of a coalescent simulation. The use of simulation, however, requires that the simulation model accurately mimics the dynamics of the population of interest and that the model is robust in its underlying assumptions. Another factor that complicates significance testing is that methods to identify selection signatures often involve many tests, on nonindependent loci, across multiple chromosomes or even entire genomes.
When data are available from a large number of populations, and one desires to search for a signature of selection within a subset of similar populations, construction of a permutation test may be possible. Livestock breeds selected for various phenotypic traits may offer one such opportunity. For example, the International Bovine HapMap (IBHM) project (Bovine HapMap Consortium et al. 2009) evaluated a range of breeds that have been historically selected, both naturally and artificially, for different phenotypic traits.
The primary objective of this study was to develop a test for selection signatures in a subset of breeds sharing a similar phenotype. Randomly drawn sets of individuals from the whole population of breeds were used to establish a null distribution of marker alleles of animals that were not undergoing artificial selection for a specific quantitative phenotype, such as dairy production. In addition, the test was designed to account for the multiple testing across a complete genome consisting of multiple chromosomes.
This method was first tested by using it to identify selection signatures for discrete phenotypes determined by a single well-characterized locus. The method was then applied to identify putative signatures within breeds of dairy cattle. In a final step, a brief and subjective evaluation was undertaken of the potential biological significance of several of the genes located closest to the center of regions carrying putative selection signatures.
MATERIALS AND METHODS
The data used in this study were from the IBHM (Bovine HapMap Consortium et al. 2009) and are available to the public at www.bovinehapmap.org. The IBHM evaluated genotypes of animals from 19 breeds of cattle (see Table 1) plus single animals of two outgroup species (Anoa and Water Buffalo), which were not included in this study. Sampling included Bos taurus, B. indicus and synthetic breeds from different geographic locations and historically different breeding goals (Table 1). The study included 497 animals. The IBHM sampled 24 animals per breed, with the exception of Red Angus (12), Holstein (53), and Limousin (42). Animals were generally unrelated, with the exception of a few breeds for which parent–offspring trios were included to help validate genotyping. The offspring of these trios were not considered in this study.
TABLE 1.
Breeds included in the study and their respective locations of origin and sampling
| Breed | Breeding goal | Land of origin | Country of sampling |
|---|---|---|---|
| Angus | Beef | Scotland | USA and New Zealand |
| Brown Swiss | Dairy | Switzerland | USA |
| Charolais | Beef | France | United Kingdom |
| Guernsey | Dairy | Channel Islands | USA and United Kingdom |
| Hereford | Beef | United Kingdom | USA and New Zealand |
| Holstein | Dairy | Netherlands | USA and New Zealand |
| Jersey | Dairy | Channel Islands | USA and New Zealand |
| Limousin | Beef | France | USA and France |
| N'dama | Multiple | West Africa | Guinea |
| Norwegian Red | Dairy | Norway | Norway |
| Piedmontese | Beef | Italy | Italy |
| Red Angus | Beef | Scotland | USA and Canada |
| Romagnola | Beef | Italy | Italy |
| Sheko | Multiple | East Africa | Ethiopia |
| Brahman | Beef | USA | USA and Australia |
| Gir | Dairy | India | Brazil |
| Nelore | Beef | India | Brazil |
| Beefmaster | Beef | USA | USA |
| Santa Gertrudis | Beef | USA | USA |
For the IBHM, genotypes were obtained for 37,470 single nucleotide polymorphisms (SNPs). Only those SNPs that had been assigned to a chromosome (29 autosomes and X) in the Btau_4.0 build of the bovine genome were considered in this analysis, however, leaving 32,689 SNPs. The distribution of SNPs across chromosomes is in Table 2. Chromosomes 6, 14, and 25 had more SNPs in the IBHM, because these chromosomes were specially targeted, as they have genes affecting economically important phenotypic traits in cattle (Khatkar et al. 2004).
TABLE 2.
Information regarding number and density of SNP and significance tests for each Chromosome (BTA)
| bp/SNP |
CCLL for P < 0.01a |
|||||
|---|---|---|---|---|---|---|
| BTA | Length (Mbp) | SNP (N) | Mean | Maximum | 1 breed | 5 breeds |
| 1 | 161 | 1730 | 93,064 | 978,843 | 129.23 | 72.24 |
| 2 | 141 | 1562 | 90,269 | 783,395 | 137.98 | 84.34 |
| 3 | 128 | 1409 | 90,845 | 863,367 | 135.75 | 81.79 |
| 4 | 124 | 1341 | 92,468 | 830,579 | 111.72 | 88.82 |
| 5 | 126 | 1338 | 94,170 | 954,295 | 110.82 | 81.94 |
| 6 | 123 | 2517 | 48,868 | 1,107,425 | 178.66 | 115.85 |
| 7 | 112 | 1165 | 96,137 | 986,266 | 106.61 | 76.48 |
| 8 | 117 | 1286 | 90,980 | 831,673 | 100.92 | 75.76 |
| 9 | 108 | 1074 | 100,559 | 870,580 | 108.89 | 70.09 |
| 10 | 106 | 1203 | 88,113 | 1,124,898 | 110.74 | 83.84 |
| 11 | 110 | 1305 | 84,291 | 655,880 | 104.62 | 78.63 |
| 12 | 85 | 932 | 91,202 | 999,234 | 109.34 | 96.61 |
| 13 | 84 | 1030 | 81,553 | 903,334 | 122.46 | 81.06 |
| 14 | 81 | 2806 | 28,867 | 664,832 | 178.29 | 123.76 |
| 15 | 85 | 892 | 95,291 | 988,771 | 105.13 | 82.24 |
| 16 | 78 | 906 | 86,093 | 888,460 | 118.22 | 83.86 |
| 17 | 77 | 891 | 86,420 | 783,294 | 97.15 | 71.80 |
| 18 | 66 | 717 | 92,050 | 1,014,891 | 130.42 | 78.83 |
| 19 | 65 | 748 | 86,898 | 805,252 | 110.21 | 79.19 |
| 20 | 76 | 895 | 84,916 | 1,411,900 | 104.42 | 71.93 |
| 21 | 69 | 716 | 96,369 | 909,403 | 88.44 | 65.56 |
| 22 | 62 | 736 | 84,239 | 1,126,708 | 133.81 | 83.94 |
| 23 | 53 | 651 | 81,413 | 629,834 | 94.33 | 68.35 |
| 24 | 65 | 772 | 84,197 | 657,655 | 93.83 | 78.44 |
| 25 | 44 | 1280 | 34,375 | 887,633 | 161.46 | 102.13 |
| 26 | 52 | 619 | 84,006 | 1,069,536 | 89.44 | 66.36 |
| 27 | 49 | 531 | 92,279 | 953,380 | 79.00 | 63.18 |
| 28 | 46 | 552 | 83,333 | 593,124 | 87.14 | 71.15 |
| 29 | 52 | 544 | 95,588 | 1,533,744 | 106.07 | 81.58 |
| X | 89 | 541 | 164,510 | 2,170,289 | 138.19 | 118.71 |
For each chromosome (BTA), the length in base pairs (Mbp), the number of evaluated genotypes (SNP), SNP density statistics, and the critical values of the negative composite log likelihood (CCLL) above which significance was declared as P < 0.01 on a genome-wide level for single- and 5-breed subpopulations.
P < 0.01 on a genome-wide basis.
Test statistic:
The test statistic used in this study was derived from work by Kim and Stephan (2002) and modified by Nielsen et al. (2005). The approach is based on the calculation of a composite likelihood of the allelic frequencies of SNP observed across “sliding windows,” of adjacent loci. The approaches of Kim and Stephan (2002) and Nielsen et al. (2005) relied on a composite likelihood ratio to test for significance, whereas our method employed permutation testing. The three methods differ in the proposed theoretical distribution of allelic frequencies. Kim and Stephan (2002) used a genetic model, whereas Nielsen et al. (2005) compared two approaches: (a) the observed discrete distribution of allelic frequencies across all loci and (b) a parametric distribution assumed to describe allelic frequencies of loci in the absence of selection. In the present study, SNP allelic frequencies were modeled to follow a simple binomial distribution. The permutation test approach was presumed to be more robust, by basing it upon the specific distribution of allelic frequencies observed in the data, rather than on a theoretical distribution.
To construct the test, the frequency of the major allele was calculated for each locus on each chromosome across all breeds to obtain the expected frequencies in cattle selected for no particular phenotypic trait. Because some breeds differed in the number of animals included, frequencies were first calculated within breed and then averaged across breeds. These allelic frequencies (when expressed as a proportion) can be denoted p′ij for the jth SNP (j = 1 to ni) on the ith chromosome (i = 1 to 30), where ni is the number of SNPs on chromosome i.
Then, the process was repeated for the subset of breeds with the common phenotype for which selection signatures were being searched. These frequencies were denoted pij.
Starting at locus j = 1 of BTA1, (negative) parametric composite log likelihoods (CLL) were calculated for sliding windows of w SNP, according to the following formula:
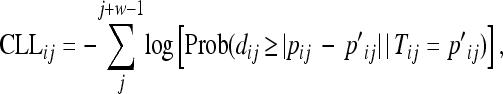 |
(1) |
where dij is a random draw from a distribution of allelic frequencies with true mean = Tij. For all loci where p′ij or pij ≥ 0.95, exact probabilities were calculated according to the binomial distribution. For loci where p′ij and pij < 0.95, the normal approximation to the binomial distribution was used.
The CLL was calculated for three sliding window sizes: w = 5, 9, and 19 SNPs.
Permutation test:
The permutation testing procedure was inspired by the method developed by Churchill and Doerge (1994) for significance testing in multilocus linkage mapping. Thresholds of critical values for type I error were established for each chromosome. For a given chromosome i, the procedure started by randomly selecting without replacement n × 24 individuals from the full dataset of 497 individuals in 19 breeds, where n is the number of breeds with a common phenotype (or selection goal) for which signatures of selection are being searched. To choose these individuals, first the breed was chosen randomly, and then an individual from that breed was chosen. This two-step process was necessary to avoid over- (under-) representation of the breeds with > (<) 24 animals in the full dataset. Then, CLLs were calculated for sliding windows of SNP, according to Equation 1. The maximum CLL was then recorded for each of 50,000 permutations. This process was repeated for each chromosome and for subsets of different numbers of n breeds. Establishing the distribution of CLL for each chromosome was necessary to account for differences among chromosomes in physical length and number of SNP, as well as any differences in linkage disequilibrium. Critical values (critical composite log likelihood, CCLL) for significance testing at the α = 0.25, 0.10, 0.05, and 0.01 levels were established at a genome-wide level by sorting the 50,000 maximum CLLs for each chromosome and storing the 416th, 166th, 83rd, and 16th greatest values, respectively. These CCLLiα (for chromosome BTA i and respective levels of α) were then compared to the CLLij to identify genomic regions with significantly different allelic frequencies than those expected in a random sample of individuals. Such regions were considered to harbor signatures of selection.
This permutation testing approach provides some advantages over other methods based on construction of likelihood ratios. First, it precludes the need for making specific assumptions about the genetic model underlying the real data or simulated data to be used for constructing the likelihood ratio. Second, this permutation testing approach can be applied to other test statistics, such as FST or measures of linkage disequilibrium that can be used for detection of selection signatures. It is, however, only applicable for studies like the IBHM that involve large numbers of genetically diverse populations, such as breeds of livestock.
Validation with known loci:
The ability of this method to identify signatures of selection was tested by applying it to two subsets of breeds with common phenotypes, black coat color and lack of horns, both of which are controlled by genes in well-defined genomic locations. Matukumalli et al. (2009) used groups of breeds with the same pair of traits for characterizing and evaluating the accuracy of a high-density SNP typing assay for cattle.
Black coat color:
Coat color in cattle is largely determined by polymorphism in the melanocortin 1 receptor (MC1R) gene on BTA18. At least three major alleles exist at this locus, the E+ wild type, ED dominant black locus, and e recessive red locus (Klungland et al. 1995). MC1R is located between bp 13,776,888 and 13,778,639 (Btau_4.0 build). Among the breeds in the IBHM, Holsteins and Angus have the characteristic black phenotype resulting from presence of ED. Therefore, a subset was made using the data from these two breeds and CLL18j were calculated for BTA18 and compared to CCLL18,0.01 based on random samples of 48 cattle. No SNPs in MC1R were included in the IBHM analysis panel; the two closest SNPs flanked MC1R, at bp 13,497,415 and 14,111,894.
Absence of horns:
Cattle are naturally horned and most of the breeds included in the IBHM share this phenotype. However, a dominant mutation can cause cattle to be hornless, or polled. This condition is generally considered to be desirable in most production environments. Therefore, some breeds have been selected to be 100% polled, including the Angus and Red Angus in the IBHM, and others such as the Hereford and Limousin breeds in the IBHM have a majority of polled animals. The gene responsible for horns has not yet been characterized, but the causative mutation has been localized to a region of ∼1 Mbp on the proximal end of BTA1 (Brenneman et al. 1996; Drögemüller et al. 2005). The most recent data indicate that the polled gene lies between bp 600,000 and 1,600,000 (Drögemüller et al. 2005).
CLL1j were therefore calculated for a subset of the four breeds with significant numbers of polled animals (i.e., Angus, Red Angus, Hereford, and Limousin). To gauge significance, the CLL1j were compared to CCLL1,0.01 generated with random groups of 96 individuals.
Search for selection signatures for dairy production:
The method was then applied to all chromosomes, by using the B. taurus breeds selected primarily for milk production. This subset comprised five breeds, Brown Swiss, Guernsey, Holstein, Jersey, and Norwegian Red. CLLij were calculated for a subset of these five breeds and compared to CCLLi,0.01 of randomly sampled groups of 120 (i.e., 5 × 24) individuals. Following this procedure, the SNP windows with the greatest CLL were identified for each chromosome and the number of distinct selection signatures was counted. Adjacent signatures were considered “distinct” if they were separated by at least three consecutive windows with nonsignificant CLL (P > 0.05, genome-wide).
The approach described above would tend to detect putative signatures of selection that were associated with mutations creating alleles with positive influences on dairy production that occurred prior to divergence of the B. taurus into specialized breeds. However, in some instances, recombination might have occurred in these regions after the radiation of founder populations of specific breeds. When this happens, each single breed of the subset could be expected to have significant differences in SNP allele frequencies from the entire IBHM, but the direction of the difference may differ from breed to breed. In such a case, averaging allele frequencies across the subset would tend to “cancel out” the significant differences in the individual breeds, precluding detection of a signature of selection.
Therefore, the test was also applied separately to each of the five breeds, by comparing CLLij to CCLLiα created through random sampling of 24 individuals. Regions where statistically significant CLL was observed in multiple breeds were then identified, and assumed to represent signatures of selection for dairy traits, even if no signature was observed in the combined data from all five dairy breeds.
Test of ascertainment bias:
The approach used to select genetic markers can introduce ascertainment bias in population genetics studies (Nielsen 2004). No specific adjustments were made in this study to account for possible sources of ascertainment bias. However, several features of the analysis applied herein were assumed to render it relatively robust against ascertainment bias. First, the basis for the study was a large group of very diverse breeds (Brunelle et al. 2008; Bovine HapMap Consortium et al. 2009; Seabury et al. 2010), including breeds that did and did not contribute significantly to the SNP ascertainment process. Also, the test sets always included multiple breeds, decreasing the influence of any single breed. As noted earlier, the method described and applied here is only applicable to studies of multiple breeds, such as would be available in a HapMap study. Second, windows of SNP were used, limiting the influence of any single SNP for which ascertainment bias may be present. Finally, a certain proportion of any ascertainment bias that may have been present would have contributed to greater variability in the permutation test as well as the actual tests for selection signatures.
Nevertheless, a specific investigation of one possible source of ascertainment bias was undertaken. As noted earlier, the IBHM included a wide group of breeds, including B. taurus, B. indicus, and hybrid breeds. Given their diverse domestication history and documented genomic differences (e.g., Brunelle et al. 2008; Bovine HapMap Consortium et al. 2009; Seabury et al. 2010), including both taurine and indicine breeds in the study had the potential to introduce ascertainment bias. A parallel study was thus done to examine this possibility. Specifically, the tests for selection signatures in dairy breeds were also performed by using a subset of the IBHM from which the indicine and hybrid breeds (Beefmaster, Brahman, Gir, Nelore, Santa Gertrudis, and Sheko) had been removed. The parallel study was initially performed for the first 10 chromosomes. Results with and without the indicine breeds were quite similar. The correlation of CLL from the two analyses was ∼0.70. Perhaps more importantly, the extreme values of CLL generally fell in the same genomic regions in both analyses. However, exclusion of the indicine breeds greatly decreased significance of the results. First, historical selection for milk production in the indicine breeds has been weak or indirect, or both, decreasing the potential for allelic differences between the five dairy breeds and the overall population. Second, removing these breeds decreased the precision of the test. For these reasons, inclusion of both taurine and indicine breeds was deemed the best strategy and only those results will be discussed further.
RESULTS AND DISCUSSION
Table 2 shows CCLLi,0.01 significance threshold values for each chromosome for subpopulations consisting of a single breed and of five breeds for a sliding window of nine SNPs. In general, the location of selection signatures was similar for all three window lengths tested, so only results obtained with a sliding window of nine SNPs are presented and discussed.
Trends observed in CCLL with respect to size of the subpopulation and number and density of SNPs were as expected. Namely, CCLL decreased as the size of the subpopulation increased (from one to five breeds), because sampling variation decreased. The CCLL increased as the number of SNPs per chromosome increased, as this increased the number of trials (i.e., sliding windows) for which CCLLs were generated, and as SNP density increased, because of greater linkage disequilibrium within the shorter windows of SNP and, in turn, greater codependency (i.e., greater covariance) of allelic frequencies of the SNP within each window. The largest CCLLs were observed for chromosomes 6, 14, and 25 for which SNP density was greatest.
Signatures of selection for known genes:
Black coat color:
Figure 1 shows the CLL for windows of nine SNPs along chromosome 18 for the subset of the breeds with black coat color. As mentioned previously, the MC1R locus controlling this phenotype is located between bp 13,776,888 and 13,778,639. A very clear signature for selection is indicated, with extremely large CLL for the windows that include the region surrounding MC1R. The maximum CLL was 299.49, for the nine-SNP window from bp 12,600,188 to 14,155,202. For comparison, the CCLL for P < 0.01 genome-wide significance was 95.95 and the greatest CLL observed among all 50,000 permutations was 130.69. The pattern of allelic frequencies around the MC1R locus in these breeds was extremely unlikely to have occurred by chance, thus supporting the notion that the parametric CLL has the ability to detect signatures of selection.
Figure 1.—

The pattern of (negative) composite log likelihood (CLL) (——) on BTA18 for the subset of breeds with black coat color. MC1R is located between bp 13,776,888 and 13,778,639. (—–) = P < 0.01 genome-wide threshold.
Several other putative signatures of selection are observed on BTA18 (Figure 1). These signatures were usually the result of a large deviation from the IBHM for one of the breeds, usually Holstein, rather than for both breeds, indicating that the parametric CLL approach may lack robustness if the number of breeds is small. The exception was for the region between ∼1 and 2 Mbp, where significant (P < 0.01, genome-wide) deviation was present for both breeds. Identification of any single gene that was likely to be responsible for this result was problematic, however. This region is gene rich, with 22 putative or provisional genes, none of which had an obvious effect on phenotypes of the Holstein and Angus that differs from the rest of the breeds in the IBHM. In addition, this region included an interval of >0.5 Mbp (from bp 1,300,489 to 1,822,486) that was not represented by any SNP.
Absence of horns:
Figure 2 shows the CLL for the first 40 Mbp of BTA1 for the subset of breeds with a majority of polled animals. A clear and statistically significant divergence from the IBHM is observable in the area of the location of the yet-unidentified horned locus in cattle. The maximum CLL was 204.98, observed for the window centered at the SNP for bp 772,511 and comprising the region from bp 487,590 to 1,338,205, which agrees very closely with the results of Drögemüller et al. (2005). This region includes 11 putative genes (Table 3). The single SNP with the largest difference in allelic frequency from the IBHM was at bp 1,202,223, where the selected breeds had major allele frequency of 0.80 vs. only 0.53 for the IBHM. This SNP is within IFNGR2, interferon gamma receptor 2. All sliding windows between bp 224,076 and 2,199,642 had significant departures from the IBHM (Figure 2).
Figure 2.—

The pattern of (negative) composite log likelihood (CLL) (——) on BTA1 for the subset of breeds with large proportions of polled animals. The polled gene lies between bp 600,000 and 1,600,000 (Drögemüller et al. 2005). (—–) = P < 0.01 genome-wide threshold.
TABLE 3.
Putative genes located within the window of nine SNP with the greatest composite log likelihood for a subset of hornless breeds and their position on BTA1
| Gene | First bp | Last bp |
|---|---|---|
| ATP5O, ATP synthase, H+ transporting, mitochondrial F1 complex, O subunit | 720,699 | 728,057 |
| ITSN1, intersectin 1 (SH3 domain protein) | 742,353 | 987,633 |
| CRYZL1, crystallin, zeta (quinone reductase)-like 1 | 988,462 | 1,021,991 |
| DONSON, downstream neighbor of SON | 1,022,856 | 1,029,638 |
| DONSON, downstream neighbor of SON | 1,032,585 | 1,042,626 |
| SON, SON DNA binding protein | 1,042,883 | 1,073,741 |
| GART, phosphoribosylglycinamide formyltransferase, phosphoribosylglycinamide synthetase, phosphoribosylaminoimidazole synthetase | 1,074,722 | 1,101,013 |
| LOC784171 similar to chromosome 21 open reading frame 55, isoform 2 | 1,115,574 | 1,120,609 |
| TMEM50B, transmembrane protein 50B | 1,135,359 | 1,172,197 |
| IFNGR2, interferon gamma receptor 2 | 1,186,973 | 1,219,051 |
| IFNAR1, interferon alpha receptor 1 | 1,278,530 | 1,306,982 |
Numerous other departures from the IBHM frequencies are observable in Figure 2 and present in the remainder of BTA1, not shown in Figure 2. This result is not surprising, considering that breeds in this subset are all selected for beef production, and two of them, the Angus and Red Angus, are essentially the same breed, differing primarily in coat color.
Signatures of selection for dairy production:
Multiple regions with statistically significant (P < 0.01, genome-wide) departures of allelic frequencies of the subset of five dairy breeds from the mean frequencies of the entire IBHM set were observed. Supporting information (Figure S1, Figure S2, Figure S3, Figure S4, Figure S5, Figure S6, Figure S7, Figure S8, Figure S9, Figure S10, Figure S11, Figure S12, Figure S13, Figure S14, Figure S15, Figure S16, Figure S17, Figure S18, Figure S19, Figure S20, Figure S21, Figure S22, Figure S23, Figure S24, Figure S25, Figure S26, Figure S27, Figure S28, Figure S29, Figure S30) shows graphically the CLL for windows of nine SNPs for all 29 autosomes and the X chromosome. Table 4 has the number of statistically significant signatures for each chromosome. Nearly 700 (699) different putative signatures of selection were observed. This result is consistent with the hypothesis that milk production is a complex trait controlled by many genes. Moreover, the phenotype of dairy breeds differs from other breeds not only for increased milk yield, but also for various other morphometric and physiological traits.
TABLE 4.
Information regarding number and locations of signature of selection for dairy production for each chromosome (BTA)
| BTA | Significant signaturesa | Maximum CLL | Location (bp) of maximum | Gene closest to maximum |
|---|---|---|---|---|
| 1 | 40 | 337.63 | 117,157,118 | RAP2B, member of RAS oncogene family |
| 2 | 32 | 232.67 | 80,018,456 | CNTNAP5, contactin associated protein-like 5 |
| 3 | 32 | 288.31 | 44,433,721 | OLFM3, olfactomedin 3 |
| 4 | 36 | 199.77 | 54,727,829 | TFEC, transcription factor EC |
| 5 | 28 | 315.09 | 27,358,607 | TMCC3, transmembrane and coiled-coil domain family 3 |
| 6 | 23 | 461.07 | 72,801,968 | KIT, v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog |
| 7 | 27 | 250.74 | 50,991,839 | HBEGF, heparin-binding EGF-like growth factor |
| 8 | 43 | 230.77 | 104,735,298 | PALM2-AKAP2, PALM2-AKAP2 read-through transcript |
| 9 | 33 | 208.46 | 25,735,072 | LOC787103, similar to Vimentin |
| 10 | 26 | 196.13 | 76,532,634 | SYT16, synaptotagmin XVI |
| 11 | 30 | 329.70 | 39,293,698 | RTN4, reticulon 4 |
| 12 | 18 | 193.22 | 29,092,788 | LOC507053, similar to INSL3 receptor; INSL3R; GREAT |
| 13 | 26 | 242.32 | 10,218,293 | CAMK1D, calcium/calmodulin-dependent protein kinase ID |
| 14 | 18 | 259.31 | 52,239,353 | KCNV1, potassium channel, subfamily V, member 1 |
| 15 | 26 | 244.97 | 45,768,306 | CCKBR, cholecystokinin B receptor |
| 16 | 19 | 231.20 | 66,232,833 | KCNK2, potassium channel, subfamily K, member 2 |
| 17 | 25 | 274.09 | 36,388,685 | SPRY1, sprouty homolog 1, antagonist of FGF signaling (Drosophila) |
| 18 | 18 | 247.76 | 14,857,880 | ITFG1, integrin alpha FG-GAP repeat containing 1 |
| 19 | 18 | 220.49 | 48,165,022 | ITGB3, integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) |
| 20 | 22 | 243.85 | 14,123,694 | SFRS12, splicing factor, arginine/serine-rich 12 |
| 21 | 16 | 217.56 | 43,696,780 | AKAP6, A kinase (PRKA) anchor protein 6 |
| 22 | 22 | 206.77 | 29,231,882 | PPP4R2, protein phosphatase 4, regulatory subunit 2 |
| 23 | 16 | 188.77 | 10,842,876 | SFRS3, splicing factor, arginine/serine-rich 3 |
| 24 | 21 | 194.29 | 38,708,428 | SMCHD1, structural maintenance of chromosomes lexible hinge domain containing |
| 25 | 15 | 272.42 | 2,650,732 | KCTD5, potassium channel tetramerization domain containing 5 |
| 26 | 14 | 189.59 | 36,779,948 | GFRA1, GDNF family receptor alpha 1 |
| 27 | 15 | 152.50 | 45,238,475 | UBE2E2, ubiquitin-conjugating enzyme E2E 2 |
| 28 | 14 | 224.95 | 34,176,363 | ZMIZ1, zinc finger, MIZ-type containing 1 |
| 29 | 15 | 191.99 | 17,109,850 | No gene |
| X | 11 | 272.94 | 42,127,052 | CHM, choroideremia (Rab escort protein 1) |
For each chromosome (BTA), the length in base pairs (Mbp), the number of evaluated genotypes (SNP), SNP density statistics, and the critical values of the negative composite log likelihood (CCLL) above which significance was declared as P < 0.01 on a genome-wide level for single- and 5-breed subpopulations.
P < 0.01 on a genome-wide basis.
The largest numbers of putative selection signatures were observed on BTA1 and BTA8. BTA8 also had the greatest density of selection signatures (0.37/Mbp), followed by BTA22 (0.35/Mbp) and BTA25 (0.34/Mbp). The X chromosome had the fewest putative signatures of selection both overall and per base pair. Among the autosomes, BTA6 had the smallest density of selection signatures (0.19/Mbp), around half that of BTA8. Relatively few significant signatures of selection were also observed on BTA14. Both of these chromosomes had a large SNP density and large CCLL, perhaps decreasing the power for detection. These results were in contrast, however, from BTA25, which had a large density of significant signatures despite having a high SNP density and the third largest CCLL after BTA6 and -14.
Table 3 also shows the largest CLL observed for nine SNP windows on each chromosome, the location of the central SNP of the sliding window with the largest CLL and the gene closest to midpoint of the window. Although the sliding window regions with the greatest CLL for each chromosome often included more than a single gene, several of the results based on the central gene of the window were quite intriguing.
For example, instances were observed where genes from the same general family were at the center of the multi-SNP window with the largest CLL on more than one chromosome. Specifically, potassium channel genes were associated with the largest CLL on BTA14, -16, and -25; integrin genes were observed at the points of maximum CLL on BTA18 and -19; and arginine/serine-rich splicing factors were at the points of largest CLL on BTA20 and -23.
Among these three groups of genes, the potassium channel genes may be the most interesting. From a purely statistical point of view, the likelihood of observing three potassium channel genes among the 30 SNP windows with maximum CLL by pure chance is quite small. As of October 2009, the National Center for Biotechnology Information reported 116 potassium channel genes among the putative 24,500 bovine genes, according to the Btau_4.0 build, for a relative proportion of P = 0.004735. Making a rough calculation on the basis of the binomial distribution, the likelihood of three potassium channel genes appearing at random among the points of maximum CLL among the 30 bovine chromosomes is <0.0004. Although this simple calculation is not exact, as it ignores the differential distribution of genes across chromosomes, it is unlikely to grossly underestimate the true likelihood.
The potassium channel results are also interesting from a biological perspective. A number of relationships between potassium channels and mammary function have been reported in the literature. Potassium is the primary cation in milk (Underwood and Suttle 1999) and blocking of potassium channels has been demonstrated to downregulate milk secretion (Silanikove et al. 2000, 2009). Czarnecki et al. (2003) reported a function of potassium channels in the growth of mammasomatotroph cell lines. Mammasomatotroph cells are responsible for the production of prolactin, a key hormone for milk production.
Hayes et al. (2008) cited another potassium channel gene, potassium channel tetramerization domain containing 8 (KCTD8), as the most-likely explanation for a selection signature on BTA6 in Norwegian Red cattle, one of the breeds involved in this analysis. A putative signature of selection was also observed in the same region (bp 65,880,230 to 65,617,020 of BTA6) in this study.
To investigate this issue further, the CLL of the genomic locations of the first 40 (out of 116) unique results obtained on Entrez Gene when searching the bovine genome for “potassium channel” were compared to the CCLL for their respective chromosomes. Among these 40 results, 33 were within SNP windows with significant CLL at a P < 0.01 genome-wide, three more were significant at P < 0.05, and an final potassium channel gene was in a window with significant CLL (P < 0.01) for the Holstein breed (see Table S1 for more details).
Integrins are involved in the interaction and attachment of cells to surrounding tissue, as well as in signaling pathways. Among the integrins noted in Table 3, ITGB3 is particularly interesting, especially in the context of the previously discussed results, as it has been reported to play a role in regulation of endothelial cells and the extracellular matrix, particularly in calcium-activated potassium channels (Kawasaki et al. 2004). Both ITGB3 (integrin, beta 3) and ITFG1, (integrin alpha FG-GAP repeat containing 1) are expressed in the mammary gland (Lemay et al. 2009). Subunits of ITGB3 have been identified on the bovine oocyte vitelline membrane (Pate et al. 2007) and have been reported to be involved in receptors for foot-and-mouth disease (Duque et al. 2004).
SFRS3 (BTA23) and SFRS12 (BTA20) are arginine/serine-rich splicing factors 3 and 12, respectively, and play roles in processing of mRNA, which could clearly have an influence on dairy production, although no such particular role has been reported. SFRS12 has been reported to be expressed in the virgin mammary gland (Lemay et al. 2009). Another arginine/serine-rich splicing factor, SFRS8, was reported to be differentially expressed over time in the liver of high-producing periparturient Holstein dairy cattle (Loor et al. 2005).
KIT was at the position of the most significant CLL on BTA6, as well as the largest CLL in the entire genome, which is not surprising, given the phenotypes with which KIT is associated. In particular, KIT is responsible for the “Piebald” spotted coat-color pattern in cattle and other species (Grosz and Macneil 1999). This phenotype is present in four of the breeds (Guernsey, Holstein, Jersey, and Norwegian Red) included in the dairy subset. Interestingly, a strong selection signature was also observed at this location in the Brown Swiss breed, which does not show the Piebald phenotype. However, KIT is known to play roles other than in determining coat color, including reproduction (Koch et al. 2009) and is expressed in the lactating bovine mammary gland (Lemay et al. 2009). Flori et al. (2009) also reported a selection signature in this region among dairy cattle breeds, but ascribed it to PDGFRA, platelet-derived growth factor receptor alpha polypeptide.
Other genes in Table 4 are expressed in the mammary gland (Lemay et al. 2009): TMCC3 and PPP4R2 in the lactating gland, GFRA1, HBGEF, and SPRY1 in the virgin gland, ITFG1 and PPP4R2 in the mastitic gland, and KCTD5 in both the virgin and involuted glands.
Table 5 lists other 25 chromosomal regions that were not associated with the greatest CLL on their respective chromosomes in the across-breed analysis, but had highly significant CLL (P < 0.01, genome-wide) for at least four of the breeds included in the study. The 8 regions denoted with an asterisk (*) had highly significant (P < 0.01, genome-wide) CLL for all five breeds. In addition to the data in Table 5, 78 other regions had highly significant (P < 0.01, genome-wide) CLL for at least three of the five breeds (Table S2) and 44 additional regions had CLL giving at least an indication of a selection signature (P < 0.25, genome-wide) in all five breeds (Table S3).
TABLE 5.
Information regarding selection signatures that were observed in at least four of the five dairy breeds
| BTA | Start | End | Maximum | Gene closest to maximum | Annotated | Non-Annotated |
|---|---|---|---|---|---|---|
| 5* | 26,178,047 | 27,291,073 | 26,708,796 | PLXNC1, plexin C1 | 3 | 1 |
| 5 | 49,933,340 | 51,644,704 | 51,399,583 | HELB, helicase (DNA) B | 6 | 1 |
| 7* | 9,602,447 | 13,106,374 | 10,876,588 | GADD45GIP1, growth arrest and DNA-damage-inducible, gamma interacting protein 1 | 17 | 12 |
| 8 | 47,900,176 | 49,116,897 | 48,473,800 | MAMDC2, MAM domain containing 2 | 4 | 1 |
| 8* | 52,301,026 | 53,991,682 | 53,113,746 | RORB, RAR-related orphan receptor B | 3 | 4 |
| 9 | 38,191,173 | 39,369,494 | 39,369,494 | MARCKS, myristoylated alanine-rich protein kinase C substrate lac | 2 | 0 |
| 11* | 29,156,927 | 30,507,890 | 29,775,412 | EPAS1, endothelial PAS domain protein 1 | 8 | 2 |
| 11* | 67,359,659 | 68,382,926 | 68,274,305 | ETAA1, Ewing tumor-associated antigen 1 | 1 | 1 |
| 12* | 84,307,073 | 85,109,167 | 84,711,550 | CUL4A, cullin 4A | 19 | 3 |
| 13NS | 32,892,499 | 33,741,173 | 33,073,175 | EPC1, enhancer of polycomb homolog 1 | 6 | 0 |
| 15* | 45,666,739 | 47,416,476 | 45,768,306 | PRKCDBP, protein kinase C, delta binding protein | 5 | 22 |
| 16 | 38,698,090 | 41,234,968 | 38,698,447 | AGTRAP, angiotensin II receptor-associated protein | 24 | 3 |
| 17 | 808,318 | 2,265,823 | 1,590,263 | TLL1, tolloid-like 1 | 3 | 0 |
| 19 | 50,972,227 | 51,660,321 | 50,972,227 | PSMD12, proteasome (prosome, macropain) 26S subunit, non-ATPase, 12 | 16 | 3 |
| 20 | 43,989,371 | 44,669,003 | 44,195,229 | PDZD2, PDZ domain containing 2 | 3 | 2 |
| 21 | 4,805,808 | 6,846,501 | 5,896,027 | MEF2A, myocyte enhancer factor 2A | 8 | 2 |
| 21 | 23,905,745 | 24,660,803 | 24,409,362 | BNC1, basonuclin 1 | 2 | 4 |
| 21 | 28,945,159 | 29,649,208 | 29,561,180 | CHRNA7, cholinergic receptor, nicotinic, alpha 7 | 3 | 2 |
| 23 | 4,019,318 | 6,608,019 | 6,210,051 | LOC100141197 similar to Uncharacterized calcium-binding protein KIAA0494 | 2 | 7 |
| 23* | 30,661,700 | 31,663,669 | 31,374,887 | HMGN4, high mobility group nucleosomal binding domain 4 | 22 | 34 |
| 26 | 22,389,526 | 23,604,859 | 23,099,901 | CRISP1, cysteine-rich secretory protein 1 | 7 | 3 |
| 28 | 5,303,070 | 6,437,329 | 5,781,123 | TARBP1, TAR (HIV-1) RNA binding protein 1 | 5 | 2 |
| 28 | 26,871,628 | 27,364,767 | 27,041,597 | CDH23, cadherin-like 23 | 4 | 0 |
| 28 | 33,551,444 | 34,691,301 | 34,176,363 | LOC786412, similar to laminin receptor | 2 | 2 |
Chromosome (BTA) and starting, ending, and maximum base pairs of regions with a significant (P < 0.01, genome-wide) signature in at least four of the five breeds, the gene closest to the maximum and the number of annotated and nonannotated (genes found within the signature. *Highly significant (P < 0.01, genome-wide) selection signature observed in all five dairy breeds; NS, nonsignificant across breeds.
Because the selection signatures reported in Table 5 were based on results from individual breeds and thus had smaller numbers of animals in each significance test, the identification of a specific SNP window with the greatest CLL was less precise than with the across breed analysis (Table 4). Table 5 thus shows the intervals of SNP encompassing the windows of SNP with the maximum CLL for each of the four (or five) breeds with significant CLL. The SNP at the center of the nine-SNP window with the greatest CLL in the across-breed analysis is also given, along with the gene closest to the across-breed maximum. Finally, the number of annotated and nonannotated genes located in the interval of significant SNP windows is presented. In some cases, large numbers of genes are concentrated in the region of the putative selection signature, most notably on BTA7, -12, -16, -19, and -23, with no particularly sharp peak in CLL. These results could indicate signatures of selection for multiple genes.
Unlike with the results based on the maximum CLL across breeds (Table 4), no groups of similar genes were identified on multiple chromosomes. Nevertheless, Table 5 has some interesting candidate genes. TLL1 and CUL4A are expressed in the lactating bovine mammary gland (Lemay et al. 2009) and CUL4A was upregulated in breast carcinomas (Binghui et al. 2002). CUL4A is also expressed in the virgin mammary gland, along with MARCKS, EPAS1, PRKCDBP, AGTRAP, and MEF2A (Lemay et al. 2009), whereas HELB is expressed in the mammary gland of pregnant cattle. EPAS1 is involved in angiogenesis and Bionaz et al. (2008) reported a much greater expression (22.3 times) of EPAS1 in liver cells of periparturient cattle than in Madin–Darby bovine kidney cells from the same animals.
Mutations in CDH23 are associated with hearing loss in humans, including through the condition called Usher syndrome (Wagatsuma et al. 2007). While this relationship may not seem relevant to dairy production, Lanier et al. (2000) reported a significant difference in sensitivity to sound between Holstein dairy cattle and beef cattle. Moreover, CDH23 is believed to exert its effect through the formation of a transmembrane complex with the PDZ domains of the protein harmonin (Siemens et al. 2002). PDZD2, PDZ domain containing 2, was at the center of a strong selection signature observed in four breeds (Table 5).
Finally, like the potassium channel genes in Table 3, CHRNA7 (BTA21) is also involved in the function of voltage-gated ion channels. In humans, this gene is believed to be associated with schizophrenia and other psychological disorders (e.g., Leonard and Freedman 2006). Although schizophrenia is not a widely diagnosed problem in cattle, it is plausible that this gene influences behavior. Gutierrez-Gil et al. (2008) reported overlap between quantitative trail loci for behavior for cattle and genes associated with schizophrenia and anxiety in humans. Dairy cattle have likely been more severely selected for tameness (or in any case, for a different behavior) than cattle bred for other purposes. Dairy cattle interact with humans on a much more frequent basis than do beef cattle, due to twice-daily milking, and culling of animals with unruly temperament is likely to be more common.
Comparison to other studies:
Several other studies have detected signatures of selection in cattle, using different data and methods. As already mentioned, Hayes et al. (2008) reported a selection signature on BTA6 that was also observed in this study. Flori et al. (2009) highlighted 13 significant signatures, all of which were observed in this study (see Figure S3, Figure S4, Figure S5, Figure S6, Figure S14, Figure S18, Figure S20, and Figure S26). MacEachern et al. (2009) compared differences in allelic frequencies of Australian Angus and Holstein cattle at >7,500 SNPs. They reported three regions with large differences among breeds, at bp 61,300,000 to 62,500,000 on BTA8; bp 3,210,000 to 3,400,000 on BTA20; and bp 21,600,000 to 22,200,000 on BTA24. Significantly large CLLs were observed in the same locations on BTA8 and -24 from the across-breed data in this study (see Figure S8 and Figure S24) and on BTA20 within the Holstein and Jersey breeds. Quantitative trait loci influencing beef production have been previously reported in these regions.
Prasad et al. (2008) examined allelic frequencies of Holsteins and Angus for 355 and 175 SNPs on BTA19 and -29, respectively. They reported 14 regions with large differences between the two breeds. Their work was based on the Btau_3.1 build of the genome and regions spanned from 0.7 to 3.4 Mbp, so precise direct comparisons were not possible, but some interesting similarities with the results of this study were observed. Among the 14 regions, only 2 were not associated with regions of significantly large (P < 0.01, genome-wide) CLLs in this study and strong agreement was shown at 10 regions. The most interesting result was for a signature corresponding to the region between ∼33.7 and 34.5 Mbp (Btau_4.0). This region includes two potassium channel genes (KCNJ1 and KCNJ5) and a locus resembling Rho GTPase-activating protein. A similarly annotated locus on BTA21 was associated with highly significant CLLs in four of the five breeds in this study (Table 5).
The IBHM study (Bovine Hapmap Consortium 2009) reported selection signatures based on extreme FST across all breeds. The seven regions with elevated FST on BTA2 (∼64.8 Mbp), 5 (∼53.0 Mbp, 7 (∼47.7 Mbp), 19 (46.0 Mbp), and X (41–44 Mbp and 49–50 Mbp) all had significantly large CLLs across the five dairy breeds. However, not surprisingly, none of the seven regions with extremely low FST among all breeds had significantly large CLLs among the dairy breeds. Barendse et al. (2009), using the data from the IBHM as well as from 189 animals from 13 beef breeds in Australia, identified regions on BTA2 (∼64.7–64.8 Mbp), 5 (∼51.1 Mbp), and 28 (∼24.5 Mbp) with large FST in both samples (IBHM and Australia) and associations with feed efficiency (residual feed intake). All three of these regions showed strong signatures for selection in this subset of five dairy breeds from the IBHM (see Figures S1–S30 and Table 5). Biological differences among beef and dairy breeds in feed efficiency and utilization have been demonstrated by various researchers, usually from the perspective of beef production. Pfuhl et al. (2007) found that Charolais bulls were more efficient in protein accretion than Holsteins, which directed more energy to producing fat. Robelin and Geay (1984) also reported leaner carcasses in the Charolais (and Limousin), relative to the Holstein, although other beef breeds had fatter carcasses.
In summary, the use of a parametric composite log likelihood (CLL) to compare differences in allelic frequencies within a window of SNP between a subset of phenotypically similar subpopulations (breeds) and the general population seems to be a valid approach to detect putative signatures of selection relevant to the common phenotype of the subpopulations. The robustness of this approach increases as the subset includes more different breeds.
The known locations for genes controlling black coat color and horns in cattle were clearly observed by application of this method. Approximately 700 putative signatures of selection were observed when applying this approach to a group of five dairy cattle breeds. The genes located closest to the locations with the greatest CLL were identified and several have hypothetical relationships with milk production.
This study was, however, largely an exercise in hypothesis generation, rather than hypothesis testing. Phenomena other than selection, such as genetic drift or genotyping anomalies could also be responsible for some of the results observed, but these causes are less likely as the number of breeds increases. Additional biological studies are necessary to verify the hypothesized relationships between genes identified in putative selection signatures and differences between dairy breeds and other cattle.
Acknowledgments
The authors thank the various cattle breeding associations and “Breed Champions” for making their animals and data available for this study. As noted previously, the data used in this study were from the IBHM (Bovine HapMap Consortium 2009).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.110.116111/DC1.
References
- Barendse, W., B. E. Harrison, R. J. Bunch, M. B. Thomas and L. B. Turner, 2009. Genome wide signatures of positive selection: the comparison of independent samples and the identification of regions associated to traits. BMC Genomics 10 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binghui, L., J. C. Ruiz and K. T. Chun, 2002. CUL-4A is critical for early embryonic development. Mol. Cell Biol. 22 4997–5005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bionaz, M., Everts, R. E., Lewin, H. A., Drackley, J. K., and J. J. Loor, 2008. Madin-Darby bovine kidney (MDBK) cells and liver tissue of periparturient cows share remarkable similarity in gene expression profile. J. Dairy Sci. 86(E-Suppl 1): 61. [Google Scholar]
- Bovine HapMap Consortium, 2009. Genome-wide survey of SNP variation uncovers the genetic structure of cattle breeds. Science 324 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenneman, R. A., S. K. Davis, J. O. Sanders, B. M. Burns, T. C. Wheeler et al., 1996. The polled locus maps to BTA1 in a Bos indicus Bos taurus cross. J. Hered. 87 156–161. [DOI] [PubMed] [Google Scholar]
- Brunelle, B. W., J. J. Greenlee, C. M. Seabury, C. E. Brown, 2nd and E. M. Nicholson, 2008. Frequencies of polymorphisms associated with BSE resistance differ significantly between Bos taurus, Bos indicus, and composite cattle. BMC Vet. Res. 4 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill, G.A., and R.W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czarnecki, A., L. Dufy-Barbe, S. Huet, M. F. Odessa and L. Bresson-Bepoldin, 2003. Potassium channel expression level is dependent on the proliferation state in the GH3 pituitary cell line. Am. J. Physiol. Cell Physiol. 284 C1054–C1064. [DOI] [PubMed] [Google Scholar]
- Drögemüller, C., A. Wöhlke, S. Mömke and O. Distl, 2005. Fine mapping of the polled locus to a 1-Mb region on bovine chromosome 1q12. Mamm. Genome 16 613–620. [DOI] [PubMed] [Google Scholar]
- Duque, H., M. LaRocco, W. T. Golde and B. Baxt, 2004. Interactions of foot-and-mouth disease virus with soluble bovine alphaVbeta3 and alphaVbeta6 integrins. J. Virol. 78 9773–9781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis, S., 2007. Linkage disequilibrium as a tool for detecting signatures of natural selection. Methods Mol. Biol. 376 59–70. [DOI] [PubMed] [Google Scholar]
- Fay, J. C., and C. I. Wu, 2000. Hitchhiking under positive Darwinian selection. Genetics 155 1405–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flori, L., S. Fritz, F. Jaffrézic, M. Boussaha, I. Gut et al., 2009. The genome response to artificial selection: a case study in dairy cattle. PLoS ONE 4 e6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.X., and W. H. Li, 1993. Statistical test of neutrality of mutations. Genetics 133 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosz, M. D., and M. D. MacNeil, 1999. Brief communication. The ‘spotted’ locus maps to bovine chromosome 6 in Hereford-cross population. J. Hered. 90 233–236. [DOI] [PubMed] [Google Scholar]
- Gutiérrez-Gil, B., N. Ball, D. Burton, M. Haskell, J. L. Williams et al., 2008. Identification of quantitative trait loci affecting cattle temperament. J. Hered. 99 629–638. [DOI] [PubMed] [Google Scholar]
- Hayes, B. J., S. Lien, H. Nilsen, H. G. Olsen, P. Berg et al., 2008. The origin of selection signatures on bovine chromosome 6. Anim. Genet. 39 105–111. [DOI] [PubMed] [Google Scholar]
- Kawasaki, J., G. E. Davis and M. J. Davis, 2004. Regulation of Ca2+-dependent K+ current by v3 integrin engagement in vascular endothelium. J. Biol. Chem. 279 12959–12966. [DOI] [PubMed] [Google Scholar]
- Khatkar, M. S., P. C. Thomson, I. Tammen and H. W. Raadsma, 2004. Quantitative trait loci mapping in dairy cattle: review and meta-analysis. Genet. Sel. Evol. 36 163–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and R. Nielsen, 2004. Linkage disequilibrium as a signature of selective sweeps. Genetics 167 1513–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y., and W. Stephan, 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160 765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klungland, H., D. I. Vage, L. Gomez-Raya, S. Adalsteinsson and S. Lien, 1995. The role of melanocyte-stimulating hormone (MSH) receptor in bovine coat color determination. Mamm. Genome 6 636–639. [DOI] [PubMed] [Google Scholar]
- Koch, D., M. Sakurai, K. Hummitzsch, T. Hermsdorf, S. Erdmann et al., 2009. KIT variants in bovine ovarian cells and corpus luteum. Growth Factors 27 100–113. [DOI] [PubMed] [Google Scholar]
- Lanier, J. L., T. Grandin, R. D. Green, D. Avery and K. McGee, 2000. The relationship between reaction to sudden, intermittent movements and sounds and temperament. J. Anim. Sci. 78 1467–1474. [DOI] [PubMed] [Google Scholar]
- Lemay, D. G., D. J. Lynn, W. F. Martin, T. M. Casey, E. V. Kriventseva et al., 2009. The bovine lactation genome: insights into the evolution of mammalian milk. Genome Biol. 10 R43 Epub. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard, S., and R. Freedman, 2006. Genetics of chromosome 15q13-q14 in schizophrenia. Biol. Psych. 60 115–122. [DOI] [PubMed] [Google Scholar]
- Loor, J. J., H. M. Dann, R. E. Everts, R. Oliveira, C. A. Green et al., 2005. Gene expression profiling of liver from periparturient dairy cows reveals complex adaptive mechanisms in hepatic function. Physiol. Genomics 23 217–226. [DOI] [PubMed] [Google Scholar]
- MacEachern, S., B. Hayes, J. McEwan and M. Goddard, 2009. An examination of positive selection and changing effective population size in Angus and Holstein cattle populations (Bos taurus) using a high density SNP genotyping platform and the contribution of ancient polymorphism to genomic diversity in Domestic cattle. BMC Genomics 10 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matukumalli, L. K., C. T. Lawley, R. D. Schnabel, J. F. Taylor, M. F. Allan et al., 2009. Development and characterization of a high density SNP genotyping assay for cattle. PLoS ONE 4 e5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith, J., and J. Haigh, 1974. The hitch-hiking effect of a favourable gene. Genet. Res. 23 23–35. [PubMed] [Google Scholar]
- Nielsen, R., 2004. Population genetic analysis of ascertained SNP data. Hum. Genomics 1 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., S. Williamson, Y. Kim, M. J. Hubisz, A.G. Clark et al., 2005. Genomic scans for selective sweeps using SNP data. Genome Res. 15 1566–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsch, J., C. D. Meiklejohn and D. L. Hartl, 2001. Patterns of sequence variation suggest the recent action of positive selection in the janus-ocnus region of Drosophila simulans. Genetics 159 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pate, B. J., K. L. White, Q. A. Winger, L. F. Rickords, K. I. Aston et al., 2007. Specific integrin subunits in bovine oocytes, including novel sequences for alpha 6 and beta 3 subunits. Mol. Reprod. Dev. 74 600–607. [DOI] [PubMed] [Google Scholar]
- Pfuhl, R., O. Bellman, C. Kühn, F. Teuscher, K. Ender et al., 2007. Beef versus dairy cattle: a comparison of feed conversion, carcass composition and meat quality. Arch. Anim. Breed. 50 59–70. [Google Scholar]
- Prasad, A., R. D. Schnabel, S. D. McKay, B. Murdoch, P. Stothard et al., 2008. Linkage disequilibrium and signatures of selection on chromosomes 19 and 29 in beef and dairy cattle. Anim. Genet. 39 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robelin, J., and Y. Geay, 1984. Body composition as effected by physiological status, breed, sex and diet, pp. 525–548 in Herbivore Nutrition in the Subtropics, edited by F. M. C. Goldchrist and R. I. Mackie. The Science Press, Craighall, South Africa.
- Seabury, C. M., P. M. Seabury, J. E. Decker, R. D. Schnabel, J. F. Taylor et al., 2010. Diversity and evolution of 11 innate immune genes in Bos taurus taurus and Bos taurus indicus cattle. Proc. Natl. Acad. Sci. USA 107 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siemens, J., P. Kazmierczak, A. Reynolds, M. Sticker, A. Littlewood-Evans et al., 2002. The Usher syndrome proteins cadherin 23 and harmonin form a complex by means of PDZ-domain interactions. Proc. Natl. Acad. Sci. USA 99 14946–14951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silanikove, N., A. Shamay, D. Shinder and A. Moran, 2000. Stress down-regulates milk yield in cows by plasmin induced beta-casein product that blocks K+ channels on the apical membranes. Life Sci. 67 2201–2212. [DOI] [PubMed] [Google Scholar]
- Silanikove, N., F. Shapiro and D. Shinder, 2009. Acute heat stress brings down milk secretion in dairy cows by up-regulating the activity of the milk-borne negative feedback regulatory system. BMC Physiol. 9 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood, E. J., and N. F. Suttle, 1999. The Mineral Nutrition of Livestock. CABI, Oxfordshire, UK.
- Voight, B.F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4 e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagatsuma, M., R. Kitoh, H. Suzuki, H. Fukuoka, Y. Takumi et al., 2007. Distribution and frequencies of CDH23 mutations in Japanese patients with non-syndromic hearing loss. Clin. Genet. 72 339–344. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., L. R. Cardon, A. D. Anderson, D. M. Nielsen and W. G. Hill, 2006. Measures of human population structure show heterogeneity among genomic regions. Genome Res. 15 1468–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]