

Abstract
Next-generation DNA sequencing (NGS) projects, such as the 1000 Genomes Project, are already revolutionizing our understanding of genetic variation among individuals. However, the massive data sets generated by NGS—the 1000 Genome pilot alone includes nearly five terabases—make writing feature-rich, efficient, and robust analysis tools difficult for even computationally sophisticated individuals. Indeed, many professionals are limited in the scope and the ease with which they can answer scientific questions by the complexity of accessing and manipulating the data produced by these machines. Here, we discuss our Genome Analysis Toolkit (GATK), a structured programming framework designed to ease the development of efficient and robust analysis tools for next-generation DNA sequencers using the functional programming philosophy of MapReduce. The GATK provides a small but rich set of data access patterns that encompass the majority of analysis tool needs. Separating specific analysis calculations from common data management infrastructure enables us to optimize the GATK framework for correctness, stability, and CPU and memory efficiency and to enable distributed and shared memory parallelization. We highlight the capabilities of the GATK by describing the implementation and application of robust, scale-tolerant tools like coverage calculators and single nucleotide polymorphism (SNP) calling. We conclude that the GATK programming framework enables developers and analysts to quickly and easily write efficient and robust NGS tools, many of which have already been incorporated into large-scale sequencing projects like the 1000 Genomes Project and The Cancer Genome Atlas.
In recent years, there has been a rapid expansion in the number of next-generation sequencing platforms, including Illumina (Bentley et al. 2008), the Applied Biosystems SOLiD System (McKernan et al. 2009), 454 Life Sciences (Roche) (Margulies et al. 2005), Helicos HeliScope (Shendure and Ji 2008), and most recently Complete Genomics (Drmanac et al. 2010). Many tools have been created to work with next-generation sequencer data, from read based aligners like MAQ (Li et al. 2008a), BWA (Li and Durbin 2009), and SOAP (Li et al. 2008b), to single nucleotide polymorphism and structural variation detection tools like BreakDancer (Chen et al. 2009), VarScan (Koboldt et al. 2009), and MAQ. Although these tools are highly effective in their problem domains, there still exists a large development gap between sequencing output and analysis results, in part because tailoring these analysis tools to answer specific scientific questions can be laborious and difficult. General frameworks are available for processing next-generation sequencing data but tend to focus on specific classes of analysis problems—like quality assessment of sequencing data, as in PIQA (Martinez-Alcantara et al. 2009)—or require specialized knowledge of an existing framework, as in BioConductor in the ShortRead toolset (Morgan et al. 2009). The lack of sophisticated and flexible programming frameworks that enable downstream analysts to access and manipulate the massive sequencing data sets in a programmatic way has been a hindrance to the rapid development of new tools and methods.
With the emergence of the SAM file specification (Li et al. 2009) as the standard format for storage of platform-independent next-generation sequencing data, we saw the opportunity to implement an analysis programming framework which takes advantage of this common input format to simplify the up-front coding costs for end users. Here, we present the Genome Analysis Toolkit (GATK), a structured programming framework designed to ease the development of efficient and robust analysis tools for next-generation DNA sequencers using the functional programming philosophy of MapReduce (Dean and Ghemawat 2008). By separating specific analysis calculations from common data management infrastructure, tools are easy to write while benefiting from ongoing improvements to the core GATK. The GATK engine is constantly being refined and optimized for correctness, stability, and CPU and memory efficiency; this well-structured software core allows the GATK to support advanced features such as distributed and automatic shared-memory parallelization. Here, we highlight the capabilities of the GATK, which has been used to implement a range of analysis methods for projects like The Cancer Genome Atlas (http://cancergenome.nih.gov) and the 1000 Genomes Project (http://www.1000genomes.org), by describing the implementation of depth of coverage analysis tools and a Bayesian single nucleotide polymorphism (SNP) genotyper, and show the application of these tools to the 1000 Genomes Project pilot data.
Methods
The GATK development environment is currently provided as a platform-independent Java 1.6 framework. The core system uses the nascent standard sequence alignment/map (SAM) format to represent reads using a production-quality SAM library, which is publicly available (http://picard.sourceforge.net). This SAM Java development kit handles parsing the sequencer reads, as well as providing ways to query for reads that span specific genomic regions. The binary alignment version of the SAM format, called binary alignment/map (BAM), is compressed and indexed, and is used by the GATK for performance reasons due to its smaller size and ability to be indexed for search. The core system can accommodate reads from any sequencing platform following conversion to BAM format and sorting on read coordinate order and has been extensively tested on Illumina (Bentley et al. 2008), Applied Biosystems SOLiD System (McKernan et al. 2009), 454 Life Sciences (Roche) (Margulies et al. 2005), and Complete Genomics (Drmanac et al. 2010). The GATK supports BAM files with alignments emitted from most next-generation sequence aligners and has been tested with many BAMs aligned using a variety of publicly available alignment tools. Many other forms of genomic information are supported as well; including common public database formats like the HapMap (International HapMap Consortium 2003) and dbSNP (Sherry et al. 2001) variation databases. A variety of genotyping and variation formats are also supported by the GATK, including common emerging SNP formats like GLF (http://samtools.sourceforge.net), VCF (http://www.1000genomes.org/wiki/doku.php?id=1000_genomes:analysis:vcfv3.2), and the GELI text format (http://www.broadinstitute.org/gsa/wiki/index.php/Single_sample_genotyper#The_GeliText_file_format). As the list of available variant and other reference associated metadata formats is constantly growing, the GATK allows end users to incorporate modules for new formats into the GATK; further information can be found on our website. The GATK is available as an open-source framework on The Broad Institute's website, http://www.broadinstitute.org/gsa/wiki/index.php/The_Genome_Analysis_Toolkit.
GATK architecture
The GATK was designed using the functional programming paradigm of MapReduce. This approach makes a contract with the developer, in which analysis tools are constructed so that the underlying framework can easily parallelize and distribute processing; this methodology has been used by companies like Google and Yahoo! (Bhandarkar 2009) to manage massive computing infrastructures in a scalable way. MapReduce divides computations into two separate steps; in the first, the larger problem is subdivided into many discrete independent pieces, which are fed to the map function; this is followed by the reduce function, joining the map results back into a final product. Calculations like SNP discovery and genotyping naturally operate at the map level of MapReduce, since they perform calculations at each locus of the genome independently. On the other hand, calculations that aggregate data over multiple points in the genome, such as peak calling in chromatin immunoprecipitation with massively parallel sequencing (ChIP-seq) experiments, would utilize the reduce function of MapReduce to integrate the heights of read pileups across loci to detect sites of transcriptional regulation (Pepke et al. 2009).
Consequently, the GATK is structured into traversals, which provide the division and preparation of data, and analysis modules (walkers), which provide the map and reduce methods that consume the data. In this contract, the traversal provides a succession of associated bundles of data to the analysis walker, and the analysis walker consumes these bundles of data, optionally emitting an output for each bundle to be reduced. Since many analysis methods for next-generation sequencing data have similar access patterns, the GATK can provide a small but nearly comprehensive set of traversal types that satisfy the data access needs of the majority of analysis tools. The small number of these traversal types, shared among many tools, enables the core GATK development team to optimize each traversal for correctness, stability, CPU performance, and memory footprint and in many cases allows them to automatically parallelize calculations.
Traversal types
As stated above, the GATK provides a collection of common data presentation schemes, called traversals, to walker developers (Table 1). For example, traversals “by each sequencer read” and “by every read covering each single base position in a genome” (Fig. 1) are the standard methods for accessing data for several analyses such as counting reads, building base quality histograms, reporting average coverage of sequencer reads over the genome, and calling SNPs. The “by every read covering each single base position in a genome” traversal, called a locus-based traversal, is the most commonly used traversal type. It presents the analysis walkers with all the associated genomic data, including all the reads that span the genomic location, all reference ordered data (which includes variation data, associated interval information, and other genetic features), and the reference base at the specific locus in the genome. Each of these single-base loci are passed to the walker's map function in succession. This traversal type accommodates common analysis methods that are concerned with computation over the sequencer-read pile up (the collection of nucleotides from each read at this location), like genotyping and depth of coverage calculations, along with methods that are concerned with variant analysis and concordance calculations.
Table 1.
Traversal types available in the Genome Analysis Toolkit

The GATK was designed in a modular way, which allows the addition of new traversal types that address users' analysis needs, in addition to providing established common traversal methods, as listed in the table.
Figure 1.

Read-based and locus-based traversals. Read-based traversals provide a sequencer read and its associated reference data during each iteration of the traversal. Locus-based traversals are provided with the reference base, associated reference ordered data, and the pileup of read bases at the given locus. These iterations are repeated respectively for each read or each reference base in the input BAM file.
The other common traversal type, a read based traversal, presents the analysis walker with each read individually, passing each read once and only once to the walker's map function. Along with the sequencer read, the walker is presented with the reference bases that the read overlaps (reads that do not align to reference do not have accompanying reference sequence). This type of traversal is useful for analyzing read quality scores, alignment scores, and merging reads from multiple bam files. Currently, traversals of overlapping or otherwise multilocus arrangements are not implemented, but the architecture is sufficiently general to enable such complex assess patterns with a concomitant increase in memory requirements. As an example, we are currently designing a traversal accessing read mate pairs together using additional computation and memory resources to look-ahead for the associated reads in the BAM file at every locus.
Sharding
One of the most challenging aspects of next-generation sequencing is managing the massive scale of sequencing data. Intelligently shattering this overwhelming collection of information into manageable pieces is critical for scalability, limiting memory consumption, and effective parallelization of tasks. In this respect the GATK has taken a novel approach by dividing up data into multikilobase-size pieces, which we have termed “shards.” Exact shard sizes are calculated by the GATK engine and are based on the underlying storage characteristics of the BAM and the demands on the system. These shards contain all the information for the associated subregion of the genome, including the reference bases, SNP information, and other reference ordered data, along with the reads taken from the SAM file. Each of these shards will be subdivided again by the traversal engine as it feeds data to the walker, but this shattering of data allows large chunks of data to be handed out in a manageable and controlled fashion. The sharding system is agnostic to the underlying file system or overarching execution manager, a design decision made to ensure compatibility with as many system configurations as possible. It would be possible to implement sharding systems that take advantage of data localizing file systems such as Hadoop (http://hadoop.apache.org), or parallel computing platforms such as LSF (http://www.platform.com) or Sun Grid Engine (http://gridengine.sunsource.net).
The GATK also handles associating reference-ordered data with specific loci in these shards. The engine matches user-supplied data, for instance, dbSNP or Hapmap variation information, to their specific locus. The GATK is not limited in the number of reference ordered data tracks that can be presented to the analysis modules. Multisite data are provided for each base in reads, and for each locus passed to the analysis module. Support for multilocus events, such as genomic translocations, is an active research area and is ongoing.
Interval processing
Many biological questions have context in only limited stretches of the genome; the GATK allows users to bracket the region or regions presented to the analysis walkers by specifying active intervals. These intervals can be specified either on the command line in common interval formats like the UCSC's BED format (Kent et al. 2002), or with a custom interval format that the GATK has defined. This provides end users with the ability to target regions of interest, like processing only HapMap called sites or determining coverage over all of the exons of a gene set.
Merging input files
Many analysis methods are concerned with computation over all data for a single individual, group, or population. For a variety of reasons, data produced from next-generation sequencing are not always organized and clustered in the same manner, making the management and collation of a single composite data source for an analysis tedious and error prone. To address this, the GATK is capable of merging multiple BAM files on the fly, allowing multiple sequencing runs or other input files to be clustered together seamlessly into a single analysis without altering the input files. Sequencer run information, including read-group information, is preserved in this process, which allows walkers to determine the original sequencing information if necessary. The merged sequencing data can also be written to disk; this is an effective means of merging data into meaningful groupings for later use.
Parallelization
The GATK provides multiple approaches for the parallelization of tasks. With interval processing, users can split tasks by genomic locations (i.e., dividing up a job by chromosome) and farm out each interval to a GATK instance on a distributed computing system, like Sun Grid Engine or LSF. A sample script is included in the supplemental material. The GATK also supports an automatic shared-memory parallelization, where the GATK manages multiple instances of the traversal engine and the given walker on a single machine. Walkers that wish to use this shared memory parallelization implement the TreeReducible interface, which enables the GATK to merge together two reduce results (Fig. 2). With these methods, the GATK is able to ensure correct serial reassembly of the results from multiple threads in reference-based order. The GATK also collects the output from the individual walkers, merging them in the correct reference based order, alleviating the tedious task of tracking output sources from tool developers.
Figure 2.

Shared memory parallel tree-reduction in the GATK. Each thread executes independent MapReduce calls on a single instance of the analysis walker, and the GATK uses the user specified tree-reduce function to merge together the reduce results of each thread in sequential order. The final in-order reduce result is returned.
Data collection and processing
The following analyses were conducted using publicly available pilot data from the 1000 Genomes Project. The data were collected from the Data Coordination Center (DCC) as BAM files aligned using MAQ (Li et al. 2008a) for Illumina reads, SSAHA2 (Ning et al. 2001) for 454 reads, and Corona (http://solidsoftwaretools.com/gf/project/corona/) for SOLiD reads. Sequencing data were stored on a cluster of five Isilon IQ 12000x network area storage devices and processed on a distributed blade farm using Platform Computing's LSF software. Shared memory parallel processing jobs were executed on an AMD Opteron 16 processor server with 128 gigabytes of RAM.
Results
Depth of coverage walker
Determining the depth of coverage (DoC) in the whole genome, whole exome, or in a targeted hybrid capture sequencing run is a computationally simple, but critical analysis tool. Depth-of-coverage calculations play an important role in accurate CNV discovery, SNP calling, and other downstream analysis methods (Campbell et al. 2008). Although computationally simple, the creation of this analysis tool is traditionally entangled with the tedious and often fragile task of loading and managing massive stores of sequence read-based information.
We have implemented a depth of coverage walker in the GATK to illustrate the power of the GATK framework, as well as to demonstrate the simplicity of coding to the toolkit. The DoC code contains 83 lines of code, extending the locus walker template. At each site the walker receives a list of the reads covering the reference base and emits the size of the pileup. The end user can optionally exclude reads of low mapping quality, reads indicated to be deletions at the current locus, and other read filtering criteria. Like all GATK-based tools, the DoC analysis can also be provided with a list of regions to calculate coverage, summing the average coverage over each region. This capability is particularly useful in quality control and assessment metrics for hybrid capture resequencing (Gnirke et al. 2009). This methodology can also be used to quantify sequencing results over complex or highly variable regions. One of these regions, the extended major histocompatibility complex (MHC), can have problematic sequencer-read alignments due to the high rate of genetic variability (Stewart et al. 2004). A simple test of an upstream sequencing pipeline is to analyze this region for effective mapping of reads using the depth of coverage walker, the output of which is shown in Figure 3. The figure shows the depth of coverage in the MHC region of all JPT samples from pilot 2 of the 1000 Genomes Project; high variability in read coverage is clearly visible, especially in regions that correspond to HLA regions of the MHC (de Bakker et al. 2006).
Figure 3.

MHC depth of coverage in JPT samples of the 1000 Genomes Project pilot 2, calculated using the GATK depth of coverage tool. Coverage is averaged over 2.5-kb regions, where lines represent a local polynomial regression of coverage. The track containing all known annotated genes from the UCSC Genome Browser is shown in gray, with HLA genes highlighted in red. Coverage drops near 32.1 M and 32.7 M correspond with increasing density of HLA genes.
Simple Bayesian genotyper
Bayesian estimation of the most likely genotype from next-generation DNA resequencing reads has already proven valuable (Li et al. 2008a,b; Li and Durbin 2009). Our final example GATK tool is a simple Bayesian genotyper. The genotyper, though naïve, provides a framework for implementing more advanced genotyping and variant discovery approaches that incorporate more realistic read-mapping error and base-miscall models. An improved framework could also handle samples or regions of the genome where the ploidy is not two, such as in tumor samples or regions of copy-number variation. This simple Bayesian genotyper serves both as the starting point for more advanced statistical inference tools and also as an ideal place to highlight the shared memory and distributed parallelization capabilities of the GATK core engine.
In brief, our example genotyper computes the posterior probability of each genotype, given the pileup of sequencer reads that cover the current locus, and expected heterozygosity of the sample. This computation is used to derive the prior probability each of the possible 10 diploid genotypes, using the Bayesian formulation (Shoemaker et al. 1999):
where D represents our data (the read base pileup at this reference base) and G represents the given genotype. The term p(G) is the prior probability of seeing this genotype, which is influenced by its identity as a homozygous reference, heterozygous, or homozygous nonreference genotype. The value p(D) is constant over all genotypes, and can be ignored, and
where b represents each base covering the target locus. The probability of each base given the genotype is defined as 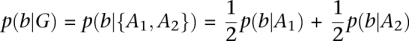 , when the genotype G = {Aa,A2} is decomposed into its two alleles. The probability of seeing a base given an allele is
, when the genotype G = {Aa,A2} is decomposed into its two alleles. The probability of seeing a base given an allele is
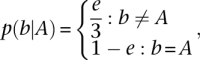 |
and the epsilon term e is the reversed phred scaled quality score at the base. Finally, the assigned genotype at each site is the genotype with the greatest posterior probability, which is emitted to disk if its log-odds score exceeds a set threshold.
The algorithm was implemented in the GATK as a locus based walker, in 57 lines of Java code (Fig. 4). Along with implementing the locus walker strategy, it also implements the Tree-Reducible interface, which allows the GATK to parallelize the MapReduce calls across processors. We applied the genotyping algorithm above to Pilot 2 deep coverage data for the CEU daughter, sample NA12878, on chromosome 1 of the 1000 Genomes Project data using Illumina sequencing technology. On a single processor, this calculation requires 863 min to process the 247,249,719 million loci of chromosome 1.
Figure 4.

Code sample for the simple genotyper walker. The map function uses a naïve Bayesian method to generate genotypes, given the pileup of reference bases at the current locus, and emits a call containing the likelihoods for each of the 10 possible genotypes (assuming a diploid organism). This is then output to disk. The implementation of the tree-reduce function provides directions to the GATK engine for reducing two in-order parallel reduce results, allowing parallelization of the genotyper.
Moreover, the GATK's built-in support for shared memory parallelization allows us to quickly add CPU resources to reduce the run-time of target analyses. The elapsed time to genotype NA12878s chromosome 1 drops nearly exponentially through the addition of only 11 additional processing nodes, with no change to the analysis code. The 12 processor version takes only slightly more than one-twelfth the time of the single processing version (Fig. 5). This flexibility allows end users to allocate CPU resources to a pool of analyses based on the priority of their completion, or to quickly complete an analysis by assigning large computing resources to a single run. Using the distributed parallelization scheme (see Supplemental material), the GATK can be partitioned to even large computational clusters, further reducing elapsed time for end user analyses.
Figure 5.

Parallelization of genotyping in the GATK. (A) 1000 Genomes Project sample NA12878s chromosome 1 was genotyped using both shared memory parallelization and distributed parallelization methods. Both methods follow a near exponential curve (B) as the processor count was increased, and using the distributed methodology it was possible to see elapsed time gains out to 50 processors.
Even this naïve genotyper performs reasonably well at identifying variants—315,202 variants were called on chromosome 1, with 81.70% in dbSNP and a concordance figure of 99.76%. This compares well against previous single individual genotyping efforts, which have seen concordance values for an individual of 86.4% and 99.6% (Wang et al. 2008; Wheeler et al. 2008). For HapMap sites the overall concordance figure is 99.84%, with 99.81% of homozygous variants correctly called. Given that the error rate in HapMap is 0.5% (International HapMap Consortium 2003), these values are remarkably good. However, this simple genotyper generates many false-positive SNP calls, as indicated by the 82% dbSNP rate given our expectation for 90% for a member of the CEPH population. Nevertheless, a better genotyper incorporating a more realistic model of machine error, and thereby reducing the false-rate, can as easily be implemented in the GATK, such as the production-grade multisample genotyper available from the GATK website (M DePristo, E Banks, R Poplin, K Garimella, J Maguire, C Hartl, A Philippakis, G del Angel, M Rivas, M Hanna, et al., in prep.).
Discussion
The Genome Analysis Toolkit (GATK) provides a structured Java programming framework for writing efficient and robust analysis tools for next-generation resequencing projects. The GATK's MapReduce architecture separates the complex infrastructure needed to access the massive next-generation sequencing data from the logic specific to each analysis tool. Since the GATK engine encapsulates the complexity of efficiently accessing the next-generation sequencing data, researchers and developers are free to focus on their specific analysis algorithms. This not only vastly improves the productivity of the developers, who can focus their effect on analysis methods, but also results in tools that are efficient and robust and can benefit from improvement to a common data management engine. Moreover, this separation has enabled us to optimize the GATK engine for correctness, stability, and CPU and memory efficiency and even to automatically parallelize most analysis tools on both shared memory machines and distributed clusters. Despite less than 1 yr of development, the GATK already underlies several critical tools in both the 1000 Genomes Project and The Cancer Genome Atlas, including quality-score recalibration, multiple-sequence realignment, HLA typing, multiple-sample SNP genotyping, and indel discovery and genotyping. The GATK's robustness and efficiency has enabled these tools to be easily and rapidly deployed in recent projects to routinely process terabases of Illumina, SOLiD, and 454 data, as well processing hundreds of lanes each week in the production resequencing facilities at the Broad Institute. In the near future, we intend to expand the GATK to support additional data access patterns to enable the implementation of local reference-guided assembly, copy-number variation detection, inversions, and general structural variation algorithms.
Acknowledgments
We thank our colleagues in the Medical and Population Genetics and Cancer Informatics programs at the Broad Institute, who have encouraged and supported us during the development the Genome Analysis Toolkit and have been such enthusiastic early adopters, in particular, Gad Getz, Anthony Philippakis, and Paul de Bakker. We also thank our reviewers for their valuable feedback on the manuscript. This work was supported by grants from the National Human Genome Research Institute, including the Large Scale Sequencing and Analysis of Genomes grant (54 HG003067) and the Joint SNP and CNV calling in 1000 Genomes sequence data grant (U01 HG005208).
Footnotes
[Supplemental material is available online at http://www.genome.org.]
Article published online before print. Article and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.107524.110.
References
- Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR, et al. 2008. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456: 53–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandarkar M 2009. Practical problem solving with hadoop and pig. In USENIX. The USENIX Association, San Diego, CA [Google Scholar]
- Campbell PJ, Stephens PJ, Pleasance ED, O'Meara S, Li H, Santarius T, Stebbings LA, Leroy C, Edkins S, Hardy C, et al. 2008. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat Genet 40: 722–729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean J, Ghemawat S 2008. MapReduce: Simplified data processing on large clusters. Commun ACM 51: 107–113 [Google Scholar]
- de Bakker PIW, McVean G, Sabeti PC, Miretti MM, Green T, Marchini J, Ke X, Monsuur AJ, Whittaker P, Delgado M, et al. 2006. A high-resolution HLA and SNP haplotype map for disease association studies in the extended human MHC. Nat Genet 38: 1166–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drmanac R, Sparks AB, Callow MJ, Halpern AL, Burns NL, Kermani BG, Carnevali P, Nazarenko I, Nilsen GB, Yeung G, et al. 2010. Human genome sequencing using unchained base reads on self-assembling DNA nanoarrays. Science 327: 78–81 [DOI] [PubMed] [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. 2009. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nat Biotechnol 27: 182–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap Consortium 2003. The International HapMap Project. Nature 426: 789–796 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The Human Genome Browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L 2009. VarScan: Variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25: 2283–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ruan J, Durbin R 2008a. Mapping short DNA sequencing reads and calling variants using mapping quality scores. Genome Res 18: 1851–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Li Y, Kristiansen K, Wang J 2008b. SOAP: Short oligonucleotide alignment program. Bioinformatics 24: 713–714 [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen Y-J, Chen Z, et al. 2005. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437: 376–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Alcantara A, Ballesteros E, Feng C, Rojas M, Koshinsky H, Fofanov V, Havlak P, Fofanov Y 2009. PIQA: Pipeline for Illumina G1 genome analyzer data quality assessment. Bioinformatics 25: 2438–2439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKernan KJ, Peckham HE, Costa GL, McLaughlin SF, Fu Y, Tsung EF, Clouser CR, Duncan C, Ichikawa JK, Lee CC, et al. 2009. Sequence and structural variation in a human genome uncovered by short-read, massively parallel ligation sequencing using two-base encoding. Genome Res 19: 1527–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan M, Anders S, Lawrence M, Aboyoun P, Pages H, Gentleman R 2009. ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25: 2607–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning Z, Cox AJ, Mullikin JC 2001. SSAHA: A fast search method for large DNA databases. Genome Res 11: 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepke S, Wold B, Mortazavi A 2009. Computation for ChIP-seq and RNA-seq studies. Nat Methods 6: S22–S32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Ji H 2008. Next-generation DNA sequencing. Nat Biotechnol 26: 1135–1145 [DOI] [PubMed] [Google Scholar]
- Sherry S, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski E, Sirotkin K 2001. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res 29: 308–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker JS, Painter IS, Weir BS 1999. Bayesian statistics in genetics: A guide for the uninitiated. Trends Genet 15: 354–358 [DOI] [PubMed] [Google Scholar]
- Stewart CA, Horton R, Allcock RJN, Ashurst JL, Atrazhev AM, Coggill P, Dunham I, Forbes S, Halls K, Howson JMM, et al. 2004. Complete MHC haplotype sequencing for common disease gene mapping. Genome Res 14: 1176–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang W, Li R, Li Y, Tian G, Goodman L, Fan W, Zhang J, Li J, Zhang J, et al. 2008. The diploid genome sequence of an Asian individual. Nature 456: 60–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler DA, Srinivasan M, Egholm M, Shen Y, Chen L, McGuire A, He W, Chen Y-J, Makhijani V, Roth GT, et al. 2008. The complete genome of an individual by massively parallel DNA sequencing. Nature 452: 872–876 [DOI] [PubMed] [Google Scholar]