

Abstract
Spina bifida, or failure of the vertebrae to close at the midline, is a common congenital malformation in humans that is often synonymous with neural tube defects (NTDs). However, it is likely that other etiologies exist. Genetic disruption of platelet-derived growth factor receptor (PDGFR) α results in spina bifida, but the underlying mechanism has not been identified. To elucidate the cause of this birth defect in PDGFRα mutant embryos, we examined the developmental processes involved in vertebrae formation. Exposure of chick embryos to the PDGFR inhibitor imatinib mesylate resulted in spina bifida in the absence of NTDs. We next examined embryos with a tissue-specific deletion of the receptor. We found that loss of the receptor from chondrocytes did not recapitulate the spina bifida phenotype. By contrast, loss of the receptor from all sclerotome and dermatome derivatives or disruption of PDGFRα-driven phosphatidylinositol 3′ kinase (PI3K) activity resulted in spina bifida. Furthermore, we identified a migration defect in the sclerotome as the cause of the abnormal vertebral development. We found that primary cells from these mice exhibited defects in PAK1 activation and paxillin localization. Taken together, these results indicate that PDGFRα downstream effectors, especially PI3K, are essential for cell migration of a somite-derived dorsal mesenchyme and disruption of receptor signaling in these cells leads to spina bifida.
Keywords: Spina bifida, PDGF, PI3 kinase, Cell migration, S6K1, PAK1, Mouse, Chick
INTRODUCTION
Spina bifida is a congenital birth defect where the vertebrae fail to protect the spinal cord (Stedman, 2000). Currently, there are several proposed etiologies for spina bifida. One category is attributed to failure of neural tube closure, also known as NTD (reviewed by Juriloff and Harris, 2000). Prenatal folic acid supplementation has been highly effective in reducing the occurrence of NTD, but analysis of several mouse models of spina bifida suggest that additional causes may exist for this birth defect (Helwig et al., 1995; Payne et al., 1997; Takahashi et al., 1992). These analyses point to defects in the mesoderm surrounding the neural tube as a potential cause of spina bifida. Previous analyses of PDGFRα and PDGF ligand null and mutant alleles have clearly demonstrated that loss of PDGFRα signaling results in spina bifida, but the cellular basis responsible for these defects has remained elusive (Ding et al., 2004; Helwig et al., 1995; Payne et al., 1997; Soriano, 1997). One reason is that PDGFRα is broadly expressed in mesenchymal populations during embryonic development, and complete loss of the receptor affects cells in many tissues and causes early embryonic lethality. Therefore, the reduced viability of later gestation embryos prohibits the study of spina bifida in these mutants.
More recently, analysis of a series of signaling point mutant alleles in the PDGFRα demonstrated that loss of PI3K signaling downstream of the receptor also resulted in spina bifida (Klinghoffer et al., 2002). These mice live until birth, providing a means to investigate the cause of spina bifida in a PDGFRα mutant background. In addition to vertebral defects, mislocalization of oligodendrocytes and melanocytes hinted that loss of PI3K activation downstream of PDGFRα may result in aberrant cell migration. Although stimulation of PDGF receptors can induce cytoskeletal rearrangements and cell migration in vitro, few studies have demonstrated a clear requirement for PDGF receptor-driven cell migration in vivo. In Xenopus embryos, PDGFRα directs mesoderm towards the blastocoel roof and disruption of this signaling pathway leads to randomized movement of cells (Nagel et al., 2004). In Drosophila, PVR, a receptor tyrosine kinase related to the mammalian PDGF and VEGF receptors, directs border cell migration to the oocyte (Duchek et al., 2001) and hemocyte migration (Wood et al., 2006).
In this report, we demonstrate that normal vertebral arch formation requires PDGFRα signal transduction. Surprisingly, the cells dependent on this receptor are not chondrocytes. Using mice defective in PDGFRα-initiated PI3K (PDGFRαPI3K/PI3K) signaling and lacking PDGFRα from a broad range of somite cells, we demonstrate that proliferation and survival of this somite-derived population are unaffected. Instead, cell migration is defective. We further show that cells derived from mutant embryos fail to activate pathways implicated in actin reorganization and migration. PDGFRαPI3K/PI3K mutant cells fail to activate Pak1, Rac1 and S6K1 in response to PDGF stimulation. These signaling disruptions result in loss of paxillin localization to focal contacts. These findings demonstrate that a somite-derived cell population requires PDGFRα-induced PI3K activity for migration in vivo and that the presence of other PDGFRα signaling pathways cannot bypass the requirement for PI3K activation.
MATERIALS AND METHODS
In ovo chick studies
Fertilized White Leghorn (Gallus gallus) eggs were obtained from the Texas A&M Poultry Science Department (College Station, TX), incubated at 37°C, then opened and staged at 2–3 days of development. Imatinib mesylate (a kind gift from R. Ilaria) or vehicle alone was applied by bath application of 500 μl to HH stage 14–17 embryos. Openings were sealed with tape and allowed to develop to HH stage 36. Embryos were frozen-embedded for histological sectioning. We analyzed two vehicle-treated embryos, and four each of 25 μM and 50 μM imatinib-treated embryos.
Mice
The mutant and Cre alleles used in these experiments were: PDGFRαPI3K/PI3K (Klinghoffer et al., 2002), PDGFRαGFP (Hamilton et al., 2003), PDGFRαfl (Tallquist and Soriano, 2003), Twist2Cre (Yu et al., 2003), and Col2a1-CreTg (Ovchinnikov et al., 2000). The Twist2Cre line was kindly provided by E. Olson. The Col2a1-CreTg mice were kindly provided by G. Karsenty. Embryos possessing a tissue-specific deletion of the PDGFRα were generated by crossing males (PDGFRαfl/+; Cre+) to females homozygous for the PDGFR αfl/fl.
Cell culture
Mesenchymal cells were isolated from the thoracic and lumbar regions of E13.5 embryos by removing surface ectoderm, neural tube, dorsal root ganglia and vertebral column components. Remaining tissue was rinsed extensively with PBS and dispase (Roche, Basel, Switzerland) treated (0.5 mg/ml for 10 minutes at 37°C) to form a single cell suspension. Single cell suspensions were rinsed with PBS then plated. Cultures containing PDGFRαGFP alleles were scored for GFP-expression as a marker of PDGFRα-expression. Passage 1 cultures were 90% (±3%) GFP+ (n=4). Passage 4 cultures were 89% (±4%) GFP+ (n=5). Marker analysis (Twist2 for dermatome and dermis; Col2a1 and scleraxis for sclerotome and chondrocytes) was performed by RT-PCR to confirm cell culture identity and purity. Cultures were positive for Twist2, negative for Col2a1 and scleraxis. RNA was extracted by Trizol (Invitrogen, Carlsbad, CA) and cDNA was generated using PowerScript Reverse Transcriptase (Clontech, Mountain View, CA).
Migration
Primary cells in single suspension were plated in triplicate at a density of 4×104 cells per well in a 96-well chemotaxis chamber (NeuroProbe), on a filter (8 μm pore size, PVP-free) pre-treated for 1 hour with 10 μg/ml rat tail collagen (Sigma, St Louis, MO). Growth factors were added to the bottom wells at the indicated concentrations. PDGFAA, PDGFBB and pTGFβ1 ligands were obtained from R&D Systems; bFGF was from Sigma. Long-term rapamycin treatment (22 μM, Sigma) was 48 hours before plating cells in the chemotaxis chamber at 1×104 cells per well. For short-term rapamycin treatment, cells were plated in the chemotaxis chamber at 2×104 cells per well in rapamycin (44 μM, Sigma) or vehicle. After addition of cells and growth factors, the migration chamber was incubated at 37°C for 6 hours. Each experiment was performed at least twice with independent cell lines, and each condition was assayed in triplicate.
Histological procedures and skeletal staining
For H&E and Safranin-O staining, embryos were fixed in 4% paraformaldehyde at 4°C overnight, embedded in paraffin, sectioned at a thickness of 7 μm, and stained according to standard procedures. Skeletal preparations were performed according to (Hogan, 1994). For green fluorescent protein (GFP) expression, embryos were fixed in 4% PFA overnight, saturated in 10% sucrose at 4°C overnight, embedded in OCT and sectioned at 10 μm. For BrdU analysis, pregnant females were injected with 10 μg BrdU (Sigma) per gram of body weight 1 hour prior to sacrifice. Embryos were fixed and sectioned as described above. BrdU was detected by anti-BrdU (Becton Dickinson, Franklin Lakes, NJ) primary antibody (1:50 dilution in block).
Cell number, proliferation and TUNEL index
Images of high-quality sections through the lumbar region were taken at 40×. For all quantification, the region that was quantified was demarcated by a box of consistent width that extended below the surface ectoderm to the perineural plexus) in three sections for each embryo. Cells were identified by the nuclear hematoxylin-QS counterstain (Vectastain). One embryo for each genotype at each time point was examined. Cells were counted in the region dorsal to the neural tube. Proliferation index was calculated by dividing the number of BrdU-positive cells by the total number of cells within the boxed area and multiplying by 100. TUNEL index was performed by standard procedures using biotinylated-14-dATP and detected using the streptavidin-HRP and DAB (Vectastain) kits. The TUNEL index was calculated by dividing the number of TUNEL-positive cells by the total number of cells in the demarcated region. No TUNEL-positive cells were detected in this region although TUNEL positive cells could be found in other tissues of the embryo. We also performed the assay on a positive control (DNAse I treated section) to verify the detection of fragmented DNA.
Micromass culture analysis
Limb buds isolated from E11.5 embryos were rinsed extensively with PBS, dispase treated (1 U/ml for 30 minutes at 37°C), rinsed and dispersed to make a single cell suspension. Cells were plated in 25 μl droplets at a density of 1×107 cells/ml of basal media (DMEM with 2% serum) and incubated at 37°C for 1 hour. Wells were flooded with basal media. After 24 hours, conditioned media (1:1) and growth factors (concentrations as indicated) were added. Growth factor and conditioned media treatments were performed in triplicate. Conditioned media was harvested from cells cultured in 10% serum at density of 5×105 cells per well after 24 hours. Micromass cultures were fixed in ethanol after 5 or 7 days of culture, and chondrocyte nodules were stained with 1% Alcian Blue. Stained cultures were rinsed with 3% acetic acid to remove excess stain. Alcian Blue was extracted with 300 μl 4 M guanidine-HCl. Extracted Alcian Blue was measured by OD600 (Amersham Biosciences GeneQuant pro spectrophotometer). The 4- to 5-day culture was repeated three times, and the 7-day culture was repeated twice.
Western blot
For western blot analysis, cells were starved (in DMEM with 0.1% serum) for 48 hours, then stimulated with 10 ng/ml PDGF-AA or 10% serum for 5 minutes. Whole cell lysates were run on 7.5% SDS-PAGE and transferred to PVD membranes. Rac1 pull downs were performed as previously described (del Pozo et al., 2000). Cells were plated at 1×106 cells per 10 cm dish and then starved for 24 hours in 0.1% serum. After starvation, cells were stimulated with either 10% serum or 25 ng/ml PDGFAA for 2–20 minutes. Following stimulation, cells were washed with ice cold PBS prior to lysis. Lysis buffer (400 μl) plus aprotinin (Sigma, A6279-5ML) and PMSF were added. GTP-bound Rac1 was then isolated using the Rac1 activation kit (Upstate, Billerica, MA, 17–283).
Immunocytochemistry
GFP-expressing cells were plated for 24 hours in 1% FBS in DMEM. Cells were stimulated for 10 minutes with either 10% serum containing media or 10 ng/ml PDGFAA. PDGFRαPI3K/PI3K and control cells were plated overnight and then starved in 0.1% serum for 6 hours. After starvation cells were stimulated for 10 minutes with either 10% serum-containing media or 25 ng/ml PDGFAA. Cells were then fixed in 3% paraformaldehyde, permeablized, blocked and stained for paxillin (BD Transduction Laboratories, Franklin Lakes, NJ, 51-9002034) at a dilution of 1:400. Secondary antibody was donkey anti-mouse 594 (Molecular Bioprobes, Carlsbad, CA). Cells were imaged on a Zeiss Axiovert 200 microscope.
Antibodies
Cytoskeletal actin loading control (Novus, Littleton, CO 1:5000), AKT-p(Ser473) (Cell Signaling, Danvers, MA 1:1000), AKT (Cell Signaling, 1:1,000), FAK (Cell Signaling, 1:1000), FAK-p(Tyr576/577) (Cell Signaling, 1:1000), MAPK-p(P44,42) (Cell Signaling, 1:5000), MAPK (Cell Signaling, 1:2500), PAK1 (Cell Signaling, 1:1000), PAK1-p(Ser199/204) (Cell Signaling, 1:1000), PAK1-p(Thr423) (Cell Signaling, 1:1000), PDGFRα (Santa Cruz, Santa Cruz, CA 1:333), p70S6K-p(Thr389) (Cell Signaling, 1:1000) and p70S6K (Cell Signaling, 1:1000).
In situ hybridization
Embryos were fixed in 4% PFA overnight, saturated in 10% sucrose at 4°C overnight, embedded in OCT, and sectioned at 14 μm. In situ analyses were performed as previously described (Wilkinson, 1992).
RT-PCR primers
Twist2: forwards, CAGAGCGACGAGATGGACAATAAG; reverse GGTTGGTCTTGTGTTTCCTCAGG. Col2a1: forwards, TATGGAAGC-CCTCATCTTGCCG; reverse, TCTTTTCTCCCTTGTCACCACG. Scleraxis: forwards, TTCTCACCTGGGCAATGTGC; reverse AGT-GTTCGGCTGCTTAGAGTCAAG.
RESULTS
Treatment with imatinib mesylate recapitulates PDGFRα spina bifida phenotype
One of the most widely accepted causes of spina bifida is failure of neural tube closure (Juriloff and Harris, 2000). Several PDGF receptor mutant alleles exhibit spina bifida and wavy neural tubes, but no neural tube defects have been reported in these embryos. To determine whether disruption of PDGF receptor activity can cause spina bifida after neural tube closure has occurred, we used a pharmacological inhibitor of PDGF receptor tyrosine kinase activity (imatinib mesylate) in developing embryos (Buchdunger et al., 2002). Using the avian system, we treated HH stage 14–17 embryos with imatinib mesylate. This stage is just after neural tube closure and compartmentalization of the rostral somites occurs (equivalent to an E11.5–12.5 mouse embryo). We then examined vertebral formation at stage 36 after chondrogenesis of the lumbar vertebrae is completed (reviewed by Romanoff, 1960). Comparing imatinib mesylate-treated and untreated embryos, we observed no differences in embryo size. In the majority of drug-treated embryos, their outward appearance was normal, including the craniofacial region and limbs. However, in two embryos treated with 50 μM of imatinib mesylate, we observed failure of ventral closure, which has also been reported in PDGFRα-null embryos (Soriano, 1997). Serial sections of the embryos revealed that all drug-treated embryos exhibited spina bifida. In each of these embryos, we observed failure of the spinal lamina (plates of bone that form the dorsal wall of the vertebrae) to extend to the dorsal midline throughout the lumbar and lumbar-sacral regions. All vehicle-treated embryos possessed normal vertebral development (Fig. 1). In addition, the neural tubes of drug-treated embryos were comparable with vehicle-treated embryos in that they were closed and exhibited no obvious defects. In the slightly older drug-treated embryos, we also observed feather primordia. These data suggest that inhibition of PDGF receptor signaling after neural tube closure leads to spina bifida.
Fig. 1. Induction of spina bifida by imatinib-mesylate.
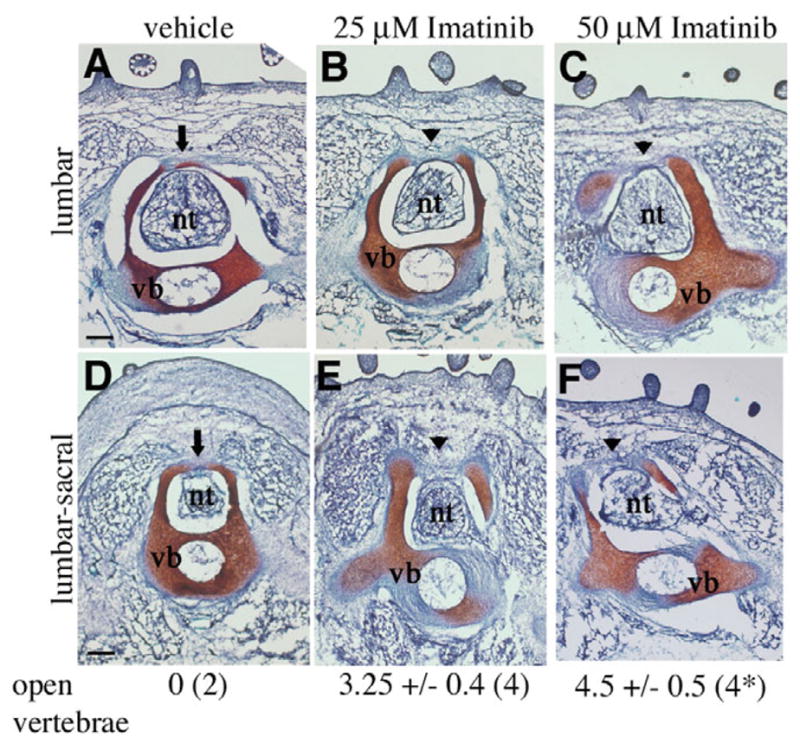
(A-F) Representative Safranin-O stained transverse sections through the lumbar and lumbar-sacral regions of chick embryos (HH stage 36). (A,D) Vehicle treated, and (B,E) 25 μM and (C,F) 50 μM imatinib-mesylate treated. The lamina (indicated by an arrow in A,D) failed to form across the dorsal midline in imatinib-treated embryos (indicated by arrowheads in B,C,E,F), resulting in spina bifida. Images show the most advanced arch formation found in serial sections of each treatment group. Scale bars: 200 μm. nt, neural tube; vb, vertebral body. Values listed below each column indicate the number of open vertebrae in all serial sections through the lower thoracic, lumbar and sacral region in each embryo. The number of embryos examined is indicated in parentheses. *Two out of four embryos treated with 50 μM imatinib-mesylate also exhibited failure of ventral closure.
Loss of PDGFRα from cartilage does not result in spina bifida
The lumbar vertebrae form by extension of the somite-derived sclerotome around the neural tube. PDGFRα is expressed in the epithelial somite but becomes restricted to a subset of cells, including the perichondrium, the condensing mesenchyme surrounding the vertebral arch and the dermis (Hamilton et al., 2003; Orr-Urtreger and Lonai, 1992; Payne et al., 1997; Takakura et al., 1997). To gain a better understanding of the cellular disruption leading to spina bifida, we examined three independent mouse lines with defective PDGFRα signaling. Using a PDGFRα allele that is flanked by loxP sites (Tallquist and Soriano, 2003) and tissue-specific Cre-expressing mouse lines, we generated mice that lacked PDGFRα in distinct somitic cell populations. Embryos deficient in PDGFRα in vertebral cartilage populations were generated using Col2a1-CreTg mice (Ovchinnikov et al., 2000) (referred to as PDGFRαCKO). In these embryos, Cre is expressed in the sclerotome as early as E9.5 and results in recombination of loxP-flanked alleles in all cartilage and perichondrium of the axial skeleton (Day et al., 2005; Yoon et al., 2005). A broader deletion in condensed mesenchyme was generated using mice that express Cre from the Twist2 locus (Yu et al., 2003) (referred to as PDGFRαTKO). In this mouse line, Cre is expressed in the sclerotome condensing mesenchyme, the dermatome and osteoblasts. Finally, we also examined vertebral development in embryos expressing a PDGFRα that was incapable of signaling through PI3K (PDGFRαPI3K/PI3K) and had been previously reported to exhibit spina bifida (Klinghoffer et al., 2002). This allele contains engineered point mutations in the domain of the PDGFRα that normally binds PI3K, resulting in a loss of this signaling pathway downstream of the receptor.
Examination of skeletal preparations at E18.5 demonstrated that loss of PDGFRα in chondrocytes resulted in normal formation and closure of the vertebrae (Fig. 2B) when compared with controls (Fig. 2A), although 100% of these mice died at birth from a clefting of the secondary palate similar to mice with neural crest cell deletion of the receptor (Tallquist and Soriano, 2003). This suggests that deletion of PDGFRα occurred in chondrocyte populations, but that loss of the receptor in vertebral chondrocytes and perichondrium did not affect its development. By contrast, defects were observed in the dorsal-most sclerotome derivatives in PDGFRαTKO and PDGFRαPI3K/PI3K embryos. In the lumbar region, the progression of the lamina was significantly impeded, and the spinous process was absent (Fig. 2C,D). This phenotype was specific to the lumbar vertebrae (L1-L6) and sometimes included the immediate surrounding thoracic (T10–13) and sacral (S1-2) vertebrae. In all embryos examined, the vertebral bodies, vertebral arches, pedicles, ribs and appendicular skeleton developed normally (Fig. 2 and data not shown). When postnatal day 1 (P1) PDGFRαTKO mice were recovered, we observed protrusion of the spinal cord through the opening in the vertebrae (data not shown). This suggested that during the process of birth, constriction of the embryo caused the spinal cord to emerge between the open vertebrae, resulting in subcutaneous myelomeningocele (or spina bifida aperta). Because of the distinct phenotypes of the PDGFRαCKO and the PDGFRαTKO embryos, we conclude that the defect in lamina development does not result from a primary abnormality in perichondrium or chondrocytes. Instead, the defect was probably caused by a loss of PDGFRα signaling in a somite cell population other than chondrocytes.
Fig. 2. Spina bifida in PDGFRα mutant embryos.
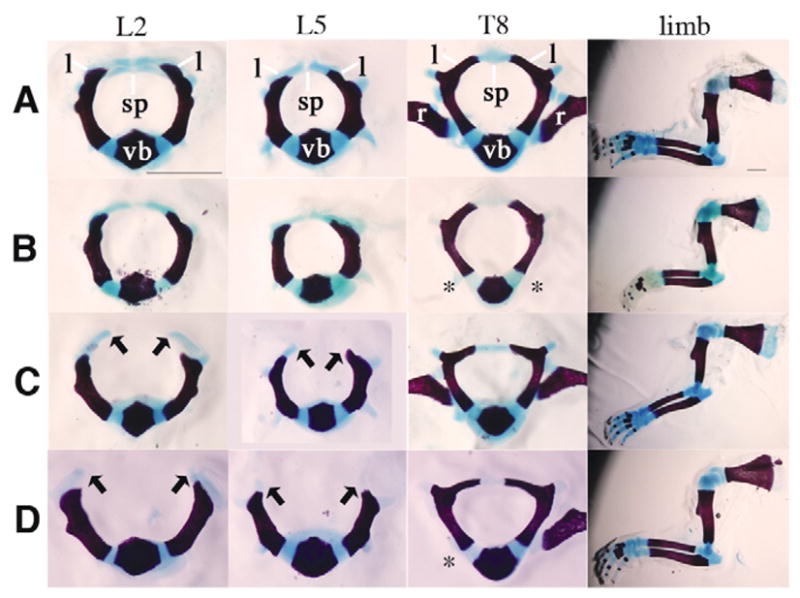
Skeletal preparations of E18.5 embryos display defects in lumbar vertebrae formation. (A) Control, (B) PDGFRαCKO (C) PDGFRαPI3K/PI3K and (D) PDGFRαTKO. Headings at the top of each column indicate the bone isolated. L, lumbar; T, thoracic. The lumbar vertebral arches form normally in A and B but fail to progress in C and D. Vertebral arch development of the thoracic vertebrae and limbs was normal in all embryos. Arrows indicate the extent that Alcian Blue cartilage can be detected. Scale bars: 1 mm. Asterisks indicate location of bones lost during dissection. sp, spinous process; l, lamina; vb, vertebral body; r, rib. Data representative of skeletal preparations performed on E18.5 and P1 embryos: wild type (n=10); PDGFRαCKO (n=4); PDGFRαPI3K/PI3K (n=10); and PDGFRαTKO (n=9).
Loss of progression of the dorsal vertebrae
Because spina bifida is commonly associated with NTD, we examined the PDGFRαTKO and PDGFRαPI3K/PI3K embryos throughout gestation for neural tube closure and found that the neural tube had a normal morphology and closed at the expected time point (Fig. 3B and data not shown). To identify when the defect in vertebral development appeared, we examined the lumbar region by H&E and Safranin O (cartilage) staining between E14.5 and E16.5 (Fig. 3A-J and data not shown). At E14.5, control, PDGFRαPI3K/PI3K and PDGFRαTKO embryos appeared similar (Fig. 3A,B), but by E15.5, defects in vertebral development were observed (Fig. 3E,F,I–L). In wild-type embryos, the chondrocytes expanded along the dorsal aspect of the neural tube, but in mutant embryos small condensations of unstained mesenchyme were observed (Fig. 3E,F). By E16.5, mesenchymal condensations could be identified, but the area was dramatically reduced in size and did not stain with Safranin O (Fig. 3I-J). Using collagen type 2a1 as an earlier marker for chondrocyte progenitors and chondrocytes, we found that the precursors of the lamina were present, but they failed to extend towards the dorsal midline (see Fig. S1A in the supplementary material). Examination of cells dorsal to the neural tube revealed an increase in cell density between E14.5 to E15.5 in the wild type but not in the mutant (Fig. 3C,D,G,H; see Table S1 in the supplementary material). When we examined the lumbar region of E18.5 PDGFRαTKO embryos, we found a similar result. Although the skin was similar to control sections with regards to epidermis, dermis and subcutis formation, the lamina and the adjacent dense mesenchyme were absent (Fig. 3K,L). These data demonstrate that the defect in the vertebral arch formation in the PDGFRαPI3K/PI3K and PDGFRαTKO embryos occurred after E14.5 and specifically affected the lamina of the lumbar vertebrae.
Fig. 3. Timing of vertebral arch defect.
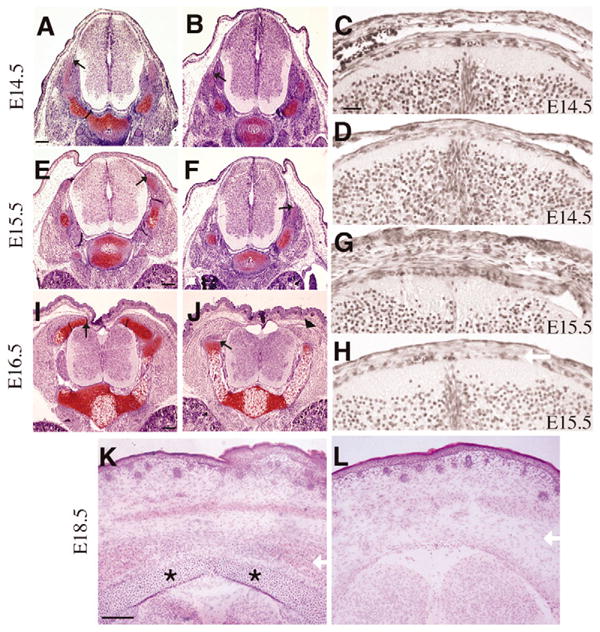
(A,B,E,F,I,J) Safranin-O stained transverse sections through lumbar vertebrae at the indicated ages. (A,E,I) are control and (B,F,J) are PDGFRαPI3K/PI3K embryos. Safranin-O sections are representative of the furthest progression of the vertebral arch in the lumbar region at each time-point. By E15.5 the development of the vertebral arch in the mutant lags behind that of the control littermate, and failure of the vertebral arch to fully form was observed at E16.5. Arrowhead indicates condensing cartilage that fails to stain with Safranin-O. Black arrows denote furthest progression of vertebral arch. (C,D,G,H) H&E (in black and white) of (C,G) control and (D,H) PDGFRαPI3K/PI3K embryos at the indicated ages. (K,L) Hematoxylin and Eosin staining of E18.5 (K) control and (L) PDGFRαTKO. Asterisks indicate the lamina of the vertebral arch. White arrows indicate region of mesenchymal cells dorsal to the neural tube and their absence in the mutant embryos. Scale bars: 100 μm, except C,D,G,H (20 μm).
Mesenchymal cell-secreted factors promote chondrogenesis
Previous reports suggest that growth factor secretion from adjacent tissues may affect the development of the dorsal vertebrae (Boyd et al., 2004). To investigate the possibility that loss of PDGFRα signaling negatively affected expression of chondrocyte growth factors, we isolated primary cells from the lumbar region of E13.5 control, PDGFRαPI3K/PI3K and PDGFRαTKO embryos (see Materials and methods) and compared their abilities to promote chondrogenic growth and differentiation in a limb bud micromass culture system. In this assay, undifferentiated limb bud mesenchyme is stimulated with conditioned media from primary cells. Because the limb bud mesenchyme is capable of cartilage differentiation in vitro, one can determine whether secreted factors, such as growth factors or extracellular matrix, in conditioned media are capable of enhancing the chondrocyte differentiation (Stott and Chuong, 2000). We found that conditioned media from both wild-type and PDGFRα mutant cells (TKO and PI3K/PI3K) were capable of inducing the production of cartilage proteoglycans, similar to stimulation with TGFβ1, as measured by Alcian Blue binding (see Fig. S2 in the supplementary material). To determine whether chondrocyte induction was specific to primary mesenchymal cells, we also tested conditioned media from mouse embryonic fibroblasts (MEF) and HEK293T epithelial cell lines. MEF-conditioned media promoted chondrocyte differentiation, whereas HEK293T conditioned media induction was similar to control media. These data indicate that disruption of PDGFRα signaling does not alter the secretion of factors enhancing chondrocyte differentiation.
Disrupted sclerotome migration in PDGFRα mutant embryos
A major function of the PDGFRα during development is to promote the proliferation of progenitor cell populations (Betsholtz, 2003). To determine whether a proliferation defect was the cause of the aberrant vertebral formation, we compared proliferation of control, PDGFRαTKO and PDGFRαPI3K/PI3K embryos. All exhibited similar rates of proliferation in comparable regions of the neural arch, as well as the loose sclerotome cells adjacent to the developing vertebrae and the dermis (see Fig. S3 in the supplementary material). We also investigated the proliferation of primary cells from E13.5 embryos and found that whereas control and PDGFRαPI3K/PI3K cells proliferated in response to media containing 10% serum, neither cell type proliferated in response to PDGFAA stimulation (data not shown). Thus, in contrast to many other cell populations that rely upon PDGFRα signal transduction for proliferation, sclerotome is unaffected by loss of the receptor.
Because we had noted a specific decrease in the cell population dorsal to the neural tube, we examined the expansion of this cell population in the lumbar region during vertebral development. Between E14.5 and E15.5 control embryos demonstrated an expansion in the total number of cells present, whereas PDGFRαPI3K/PI3K cells showed little increase (see Table S1 in the supplementary material). To determine whether the failure in expansion of this cell population was caused by reduced proliferation or increased apoptosis, we quantified the number of cells incorporating BrdU and the number staining with TUNEL labeling. We found that there was no significant difference in the number of TUNEL-positive cells or in the proliferation index of cells dorsal to the neural tube (see Table S1 in the supplementary material), suggesting that cell survival and proliferation in this region are normal in the PDGFRαPI3K/PI3K embryos.
To determine which cell populations in these regions expressed the PDGFRα, we examined embryos that expressed GFP from the PDGFRα locus (PDGFRαGFP) (Hamilton et al., 2003). Sections of E14.5 control embryos demonstrated the location of PDGFRα-expressing cells in the lumbar region. The most abundant receptor expression was in the mesenchyme surrounding developing vertebrae and in the dermis. Receptor expression was also present in the perichondrium, but only a low expression level was observed in the developing chondrocytes (Fig. 4A). A similar expression pattern has been reported previously for PDGFRα transcripts (Payne et al., 1997). At E16.5, PDGFRα (GFP+) cells had migrated to the dorsal neural tube. The outer cell population was probably dermis, whereas the cells adjacent to the neural tube were connective mesenchyme derived from somites (see Fig. S1D in the supplementary material).
Fig. 4. Loss of PDGFRα-PI3kinase signaling results in defective migration.
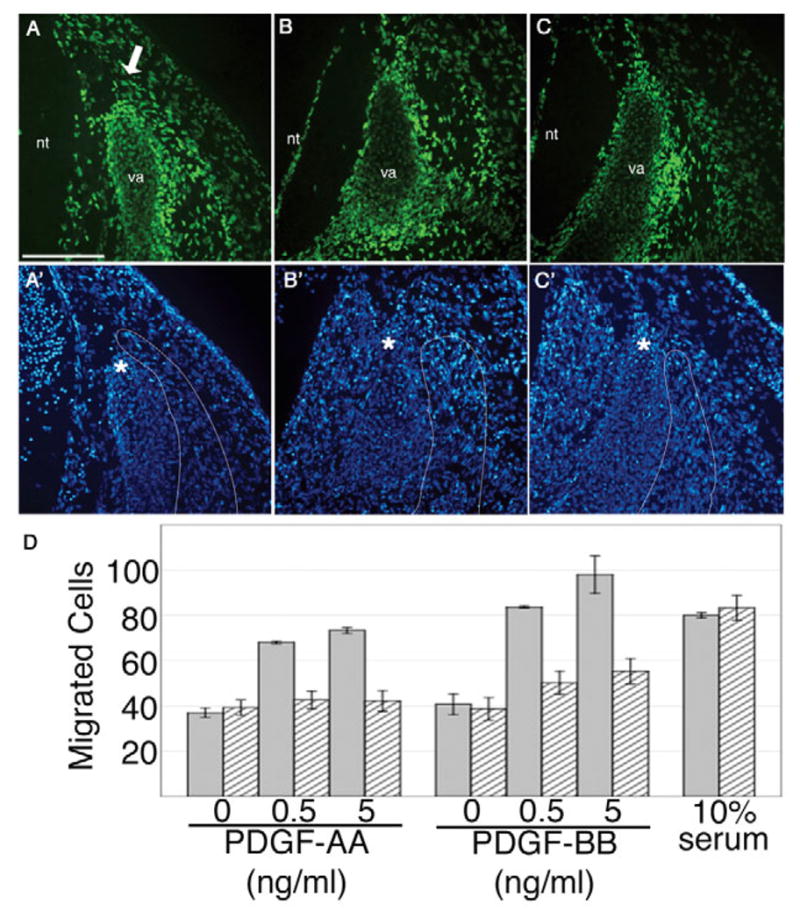
(A,A′) PDGFRαGFP/+, (B,B′) PDGFRαPI3K/GFP and (C,C′) PDGFRαTKO/GFP. PDGFRα-expressing cells (as detected by PDGFRαGFP) are present in the perichondrium and in the mesenchyme surrounding the vertebral arch. (A,A′) Heterozygote control section is at the level of the perichondrium and therefore displays more PDGFRα-positive chondrocytes. The PDGFRα-positive mesenchyme adjacent to the vertebral arch extends just beyond the tip of the arch in the heterozygote control, but has failed to advance in the (B,C) mutants. Sections are representative of furthest advancement of the vertebral arch and adjacent mesenchyme population in two embryos of each genotype. (A′-C′) DAPI fluorescence of the sections in A-C. Adjacent mesenchyme is outlined. Asterisks denote the tip of the vertebral arch. Arrow indicates mesenchyme. va, vertebral arch; nt, neural tube. Scale bar: 100 μm. (D) In vitro migration assay. Heterozygous cells (grey bars) migrated in a dose-dependent manner in response to PDGF-AA and PDGF-BB ligands, whereas mutant cells (striped bars) were unable to migrate in response to PDGF-AA and showed a decreased response to PDGF-BB. Both genotypes were able to migrate in response to 10% serum. This experiment is representative of three independent experiments.
These observations suggested that failure in migration of PDGFRα-expressing cells might lead to the observed reduction in cells in the dorsal region of the embryo. Comparison of sections through control PDGFRαGFP/+ embryos and PDGFRα mutant embryos further supported this possibility. In control embryos, the mesenchyme immediately adjacent to the forming arch had advanced to the extent of the tip of the vertebral arch. By contrast, in PDGFRαGFP/PI3K embryos GFP+ cells were present lateral to the vertebral arch, but the majority of GFP+ cells had not advanced even half the distance of the vertebral arch (Fig. 4B). This effect was even more pronounced when we examined GFP+ cells in PDGFRαTKO/GFP embryos (Fig. 4C). At E16.5, a stage when vertebral arch development is halted in the mutant animals, we find a cluster of GFP+ cells dorsal to the developing vertebral arches in the lumbar region of PDGFRαTKO/GFP embryos (see Fig. S1D,E in the supplementary material). These results suggest that PDGFRα signaling is likely to be required for migration of this somite cell population. Furthermore, because these cells are capable of secreting chondrogenic factors (as demonstrated in the micromass assay), failure of these cells to migrate could result in disrupted vertebral development.
To examine the ability of PDGF receptor stimulation to induce migration, we tested primary cells from embryos whose genotypes were either PDGFRαPI3K/GFP or PDGFRαGFP/+ in a modified Boyden chamber assay. Cells isolated from wild-type embryos migrated towards PDGF ligands and serum. By contrast, mutant cells had a reduced ability to migrate towards PDGFAA and PDGFBB (Fig. 4D) but were still capable of migration induced by other stimuli. These results indicate that loss of PI3K signals downstream of the PDGFRα lead to a defect in cell motility.
Normal Ras/MAPK and FAK activation but aberrant Akt and S6K1 phosphorylation in PDGFRαPI3K/PI3K cells
Generation of PtdIns(3,4,5)P3 by PDGFRα-initiated PI3K activity leads to activation of multiple downstream pathways. We examined how efficiently these pathways were disrupted in PDGFRαPI3K/PI3K cells by western blot. Both mutant and wild-type cells expressed similar levels of PDGFRα (Fig. 5A). However, we found that phosphorylation of Akt on S473 and p70S6 kinase (S6K1) on T389 was drastically reduced in the PDGFRαPI3K/PI3K cells in response to PDGFAA stimulation (Fig. 5B). Loss of phosphorylation of both of these effectors confirmed that the PI3K pathway was inactive in PDGF stimulated PDGFRαPI3K/PI3K cells. By contrast, MAPK phosphorylation of S6K1 (T421/S424) was similar in control and PDGFRαPI3K/PI3K cells in response to PDGFAA and 10% serum (data not shown). We then examined the activation status of other key motility pathways downstream of the PDGFRα. The Ras effectors ERK1/2 have been associated with establishment of pseudopodia and focal adhesions during chemotaxis (Brahmbhatt and Klemke, 2003; Fincham et al., 2000). Therefore, we determined whether ERK1/2 activation was disrupted in the PDGFRαPI3K/PI3K cells. We found that phosphorylation of these proteins was comparable in mutant and control samples (Fig. 5A). These data are in agreement with previous analysis of MEF cells isolated from the PDGFRαPI3K/PI3K embryos that suggest the PI3K mutation does not disrupt other signaling pathways downstream of the PDGFRα (Klinghoffer et al., 2002). Activation of focal adhesion kinase (FAK) in response to Src phosphorylation has also been demonstrated to be important for migration (reviewed by Hanks et al., 2003; Parsons, 2003). Similar to ERK1/2 activation, we observed no differences in FAK phosphorylation between wild-type and PDGFRαPI3K/PI3K cells (Fig. 5A). These data support the idea that disruption of pathways directly downstream of PI3K activity is the cause of the motility defect and that other downstream pathways, such as Src and MAPK, remain intact.
Fig. 5. Disruption of PI3K and PAK pathway activation in PDGFRαPI3K/PI3K cells.

(A-C) Western blot analysis of whole cell lysates from primary mesenchymal cells. (A) PDGFRα is expressed and ERK1/2 is phosphorylated in PDGFAA and serum-stimulated control and PDGFRαPI3K/PI3K cells. (B) Akt and S6K1 are not phosphorylated in PDGFAA-stimulated PDGFRαPI3K/PI3K cells. (C) PAK1 is not phosphorylated in PDGFAA stimulated PDGFRαPI3K/PI3K cells. Loading controls (immediately beneath the sample) were either actin (for the PDGFRα) or stripped blots that were reblotted for the unphosphorylated form of the protein. Stimulation conditions: st, starved; AA, 10 ng/ml PDGF-AA; se, 10% serum. Blots are representative of three independent experiments.
Recent data have suggested that rapamycin affects both mTOR complexes (mTORC1 and mTORC2) and that this effect varies depending on the cell type and the duration of rapamycin treatment (Sabatini, 2006; Sarbassov et al., 2006; Zeng et al., 2007). Because we observed a failure in S6K1 phosphorylation on the mTOR site in PDGFRαPI3K/PI3K cells, we next examined the consequence of disrupting the mTOR pathway during PDGFRα-stimulated migration. To accomplish this we treated wild-type primary, somite-derived mesoderm cells with rapamycin and determined their ability to migrate towards PDGFAA. We observed a loss of motility in response to PDGFAA, bFGF and TGFβ1 stimulation (see Fig. S4A in the supplementary material). By contrast, rapamycin had no effect on the migration of these cells towards serum. Interestingly, migration was inhibited regardless of the treatment time, either 24 hours prior to migration or during the migration assay (see Fig. S4B in the supplementary material).
PI3K activity at the cell membrane leads to translocation and activation of a variety of proteins at the cell surface, including phosphoinositide-dependent kinase 1 (PDK1), Akt and mTOR. One of the downstream effectors of these signals is Rac1, which is important for cytoskeletal organization. We therefore tested the ability of PDGFRαPI3K/PI3K cells to activate Rac1. We found that Rac1 activation occurred as early as 1 minute after stimulation and persisted up to 20 minutes in PDGFAA stimulated wild-type cells (Fig. 6A). By contrast, the amount of active Rac1 did not increase above background when we stimulated PDGFRαPI3K/PI3K cells with PDGFAA (Fig. 6A), but we were able to recover activated Rac1 in response to stimulation by serum (Fig. 6A). We next investigated a downstream effector of activated Rac1: p21-activated kinase (Pak1). Pak proteins are serine/threonine kinase effectors of Rho family GTPases (Dharmawardhane et al., 1997; Sells et al., 1997). Upon Rac1 binding, Pak1 becomes autophosphorylated on S199 and T204. Using a phosphospecific antibody to these sites, we found that Pak1 autophosphorylation did not occur in PDGFRαPI3K/PI3K cells stimulated with PDGFAA (Fig. 5C). Because Pak1 can also be directly phosphorylated on T423 by PDK1 downstream of PI3K activity, we investigated this event via western blot and observed a lack of phosphorylation on T423 in response to PDGF stimulation in PDGFRαPI3K/PI3K cells (Fig. 5C). By contrast, when these same cells are stimulated with serum containing media, we observe phosphorylation of Pak1 at both the autophosphorylation site and the PDK1 site, demonstrating that other signaling pathways could still activate these pathways. These data demonstrate that PDGFRα association with PI3K leads to Pak1 phosphorylation, and suggest that failure of this activation may result in defective migration.
Fig. 6. Effects of PDGFRαPI3K/PI3K mutation on Rac1 activation and paxillin localization.
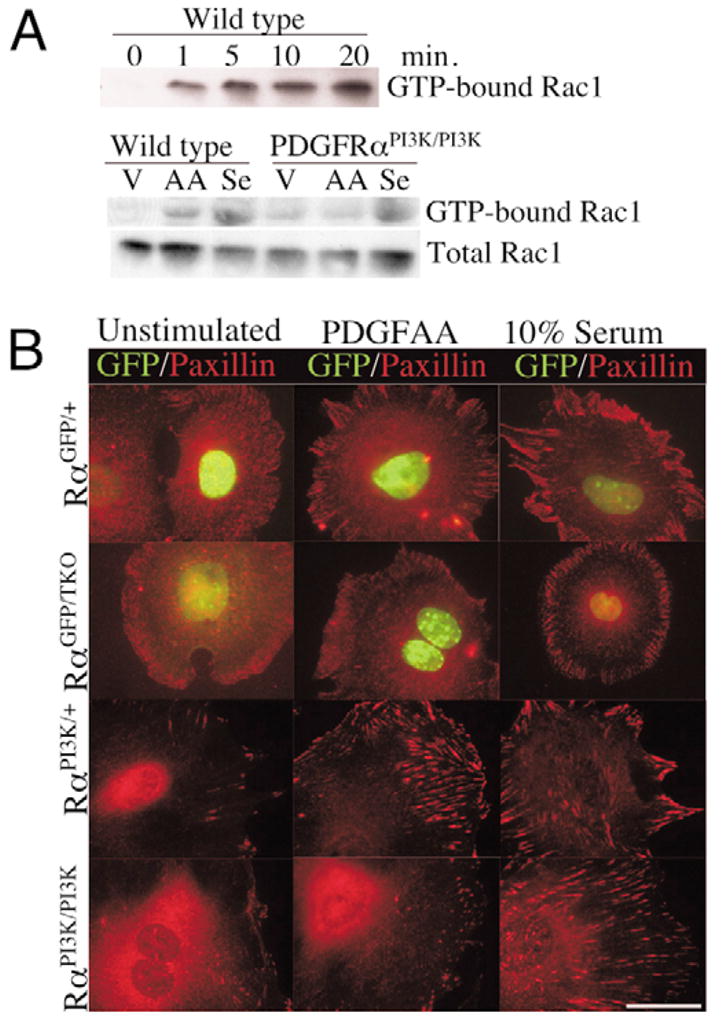
(A) Western blot for GTP-bound Rac1 and total Rac1 in control and PDGFRαPI3K/PI3K cells. Upper blot demonstrates the time course of Rac1 activation in control cells. Lower blot illustrates the failure of PDGFRαPI3K/PI3K cells to stimulate Rac1 activation when stimulated with PDGFAA. Cells were stimulated for 10 minutes. Results are representative of three independent experiments.
(B) Immunocytochemistry for paxillin localization in control and PDGFRα mutant cells. Genotypes are indicated on the left. Cells were plated for two hours, starved for 24 hours, and then stimulated for 30 minutes with the indicated stimulation. Images are representative of five independent experiments and represent the majority of cells within the culture for each stimulation. Scale bar: 50 μm.
Finally, because we observed profound motility defects in the mutant cells, we examined the ability of the PDGFRα mutant cells to form focal contacts and reorganize actin in response to PDGFAA stimulation. Paxillin is a multidomain protein that localizes to focal contacts and is a docking site for many regulators of actin dynamics (Brown and Turner, 2004). Using cells derived from PDGFRαTKO that were heterozygous for the PDGFRαGFP allele, we found that control and mutant cells localized paxillin to the cellular periphery when stimulated with media containing 10% serum (Fig. 6B). In cells stimulated with PDGFAA, we found control cells possessed localized regions of intense paxillin staining, but mutant cells appeared similar to unstimulated cells. We next examined paxillin localization in PDGFRαPI3K/PI3K cells. Analogous to what we observed in the PDGFRα-deficient cells, paxillin localized to cellular protrusions in control cells upon PDGF stimulation, but failed to localize in the PDGFRαPI3K/PI3K cells (Fig. 6B). Taken together, these data indicate the PDGFRα-mediated migration and cytoskeletal rearrangement is dependent on PI3K signal transduction.
DISCUSSION
Failure of neural tube closure is commonly believed to be the major cause of spina bifida. In this report, we demonstrated that a likely cause of spina bifida in PDGFRα mutant mice was a defect in a non-sclerotome somite cell population and that neural tube defects did not precede the spina bifida. The failure of this cell population to migrate towards the dorsal neural tube halted further development of vertebral elements in the lumbar region. We also showed that migration of these cells was directed by PI3K signaling downstream of the PDGFRα. Similar to a requirement for PI3K signaling downstream of the PDGFRβ (Aoki et al., 2001; Franke et al., 1995), stimulation of PDGFRαPI3K/PI3K cells resulted in failure in activation of Akt (this report) (Klinghoffer et al., 2002) and S6K1. These signaling disruptions resulted in loss of Rac1 and Pak1 activation, and a failure in generation of focal contacts. Thus, PI3K is an essential signaling pathway for cytoskeletal reorganization downstream of the PDGFRα, and signaling through Src and MAPK pathways does not compensate for loss of PI3K.
Until now, the cause of spina bifida in PDGFRα mutant mice has been unclear. The difficulty in identifying the etiology of this defect previously has been the requirement of PDGFRα in many tissues in the developing embryo. Using chick embryos, as well as conditional and hypomorphic PDGFRα mouse lines, we have been able to define the cell population responsible for spina bifida, as well as identifying a potential cellular function mediated by the receptor. The imatinib-treated chick embryos most closely resembled spina bifida occulta, whereas defects in the mutant mouse embryos were similar to spina bifida aperta. We attribute this difference to the fact that, in the mouse, a genetic lesion exists, whereas, in the chick, a single treatment of the PDGF receptor inhibitor was the inducing factor. One surprising outcome of our data is the limited region of vertebrae affected by disruption of PDGFRα signaling. In both the imatinib-treated embryos and the genetic disruption of the receptor, defects were limited to the lumbar region and the immediately adjacent thoracic and sacral vertebrae. This is in striking contrast to the vertebral defects reported for PDGFRα-null alleles (Morrison-Graham et al., 1992; Payne et al., 1997; Soriano, 1997). In these mouse lines, defects occur along the entire length of the vertebral column. One explanation for the disparity in the phenotypes is that the PDGFRα-deficient mutants may have mesodermal hypoplasia and exhibit surface ectoderm detachment (Morrison-Graham et al., 1992; Orr-Urtreger et al., 1992; Soriano, 1997). These disruptions may cause a more global defect in cell organization, survival and growth factor production. Therefore, complete loss of PDGFRα signaling during embryogenesis may affect many cell populations leading to more severe disruption of somite morphogenesis, vasculature or even neural tube closure.
Our observations are in agreement with studies in the avian system that investigated the formation of the dorsal vertebrae. These experiments suggested that signaling of sonic hedgehog and BMP4 from the dorsal ectoderm and roof plate of the neural tube directed a superficial somite cell population to form the dorsal vertebrae (Monsoro-Burq et al., 1994; Monsoro-Burq et al., 1996; Monsoro-Burq and Le Douarin, 2000; Watanabe et al., 1998). Although our data support the idea that a somite-derived dorsal mesenchyme is required for chondrocyte development, the lack of phenotype in the PDGFRαCKO suggests an indirect role for PDGFRα signaling in vertebrae development. Chick/quail chimeras have indicated that both the dorsal chondrocyte and mesenchymal cell population arise from the ventral half of the somite (Christ et al., 2004). Therefore, the dorsal mesenchyme population is derived from the same area of the somite as the vertebral derivatives, and is also required in this signaling paradigm. The ability of soluble factors from non-chondrocyte dorsal mesenchyme to stimulate chondrogenesis supports the possibility that coordinated migration of mesenchymal cells alongside the developing vertebral arches plays a role in the development of the vertebral arch. There are multiple growth factors that could be involved in regulating the chondrocyte growth and differentiation. It is clear from a number of studies that normal bone development often requires a balance of growth factor signals (Naski and Ornitz, 1998). In many circumstances, either an excess or a deficiency in growth factor signaling can lead to impaired development. The factor(s) secreted by the mesenchymal cell population could be either a growth factor or a growth factor inhibitor (Day et al., 2005; Hung et al., 2007). Although it is possible that the secreted molecule is a growth factor, we cannot rule out the possibility that these cells may also be depositing extracellular matrix components such as collagens, matrix proteoglycans and matrix metalloproteases that also have a demonstrated role in bone growth and morphogenesis (Blair et al., 2002; Lee, 2006; Malemud, 2006).
In vitro, PDGF receptors direct multiple cellular activities including proliferation, survival, actin reorganization and motility (reviewed by Heldin and Westermark, 1999), and in Xenopus and Drosophila, PDGF receptor homologs are required for migration of several cell populations (Cho et al., 2002; Duchek et al., 2001; McDonald et al., 2003; Nagel et al., 2004). Although migration signals downstream of PDGF receptors have been implicated in the mouse (Abramsson et al., 2007; Klinghoffer et al., 2002; Richarte et al., 2007), it is often difficult to separate proliferation from migration defects (reviewed by Hoch and Soriano, 2003). Although the PDGFRβ seems to consistently promote cell motility in multiple cell types, the ability of PDGFRα to regulate chemotaxis has proven to be a cell type-dependent phenomenon (reviewed by Ronnstrand and Heldin, 2001). In some cells, such as NIH and Swiss 3T3 cells, PDGFRα stimulation induces cell motility (Hosang et al., 1989; Rosenkranz et al., 1999), whereas in endothelial and vascular smooth muscle cells, PDGFRα activation represses migratory signals from other receptors (Koyama et al., 1992; Yokote et al., 1996). Our results suggest a primary role for the PDGFRα in promoting migration of this somite-derived population. By contrast, cells that are destined to become dermis that also express PDGFRα migrated appropriately in the PDGFRα mutants investigated. Therefore, during development PDGF receptors are likely to play distinct roles in promoting cellular functions, and each cell type must be examined individually to define the role of the receptor.
In mammals, PI3K signaling has been linked to many cellular outcomes, including growth, metabolism, survival and migration (Fruman et al., 1998). Loss of all PI3K p85 regulatory subunits results in embryonic lethality and a phenotype strikingly similar to complete loss of PDGFRα, and cells from these p85 mutant embryos failed to form membrane ruffles in response to PDGF (Brachmann et al., 2005). Now, we have shown that the loss of this signaling pathway renders a specific population of cells incapable of migration during embryogenesis, even though PDGFRαPI3K/PI3K mutant cells retained the ability to signal through Src and MAPK. The cellular outcome of PI3K signaling not only depends on the receptor promoting it, but also depends on the cell type responding to it. For example, disruption of PI3K activation downstream of the Kit receptor resulted in a block in differentiation of gametes, and mast cell and melanocyte migration were unperturbed (Blume-Jensen et al., 2000; Kissel et al., 2000). In Drosophila the PDGF-related receptor PVR guides border cell migration in a PI3K-independent manner, suggesting that PDGF receptors may use different signals depending on the developmental context (Bianco et al., 2007). Mice deficient in PI3K signaling downstream of the PDGFRβ displayed reduced interstitial fluid homeostasis but no apparent defect in pericyte migration (Heuchel et al., 1999; Tallquist et al., 2003). Our data clearly demonstrate a requirement for PI3K signaling downstream of the PDGFRα to initiate migration, whereas cell survival and proliferation are not dependent on this signaling.
The promotion of cell motility requires the coordination of many intracellular signaling molecules. Although PI3K activation is important for both Akt and Rac1 activation (Nobes and Hall, 1995; Wennstrom et al., 1994a), it is not the only pathway that has been implicated in cytoskeletal reorganization driven by PDGF stimulation. For example, PLCγ and Src signaling have been implicated in actin ruffle formation and cell motility driven by PDGFRβ stimulation in vitro (Boyle et al., 2007; Kundra et al., 1994; Wennstrom et al., 1994b). Nonetheless, in our mesenchymal cells, these pathways were not sufficient to stimulate motility in vivo or in vitro.
The hierarchy of signaling is yet to be established for many of these interactions. It has been shown that paxillin phosphorylation by PAK1 leads to increased cell motility (Nayal et al., 2006), and paxillin can also modulate Rac1 activity (Chen et al., 2005). Finally, it has also been suggested that S6K1 could be involved in cell motility via interactions with Rac1 (Berven et al., 2004; Liu et al., 2006). Thus, loss of PDGFRα-PI3K activation results in a failure in multiple components associated with focal adhesions and cell motility. The complex interactions of these proteins suggest multiple levels of regulation, and it will be interesting to use these cells to further determine the essential actions of each of these components.
Although Rac1 activation is an important component of cell motility, and this pathway is clearly disrupted in our cells, it is unlikely that loss of Rac1 is the only disruption leading to a failure in migration. Recent evidence in Rac1-null mouse embryonic fibroblasts has shown that these cells are capable of migrating towards PDGF (Vidali et al., 2006). This suggests that other signaling pathways downstream of PDGF receptors can also induce migration. Our data suggest that the mTORC1 or potentially mTORC2 complexes are also important in PDGF stimulated chemotaxis. We not only see a loss of phosphorylation of Akt on the mTORC2 site (S473), but rapamycin also inhibits migration of cells towards PDGF. This result is in contrast to the lack of inhibition of migration to serum, suggesting that the rapamycin does not render cells incapable of migration because of a generalized mechanism. Studies of neuronal migration in response to HGF stimulation have demonstrated that rapamycin does not affect migration (Segarra et al., 2006). These results emphasize that different cell types rely on distinct signaling pathways for induction of cell motility.
The role of cell polarity and cytoskeletal reorganization has been appreciated in the morphogenesis of the neural tube. Multiple lines of evidence indicate that defects in these processes can result in neural tube defects (Harris and Juriloff, 2007; Wallingford, 2006). Now, we have shown that spina bifida can be caused by defects unrelated to neural tube closure. In addition, disrupted cytoskeletal rearrangement and cell movement are involved in this second etiology. In these studies, the importance of PDGFRα signaling, especially through PI3K, in somite-derived tissues suggests that cases of human spina bifida resistant to folate supplementation may not be directly related to failure in neural tube closure. Second, because disruption of PDGF receptor signaling has been proposed for treatment of several human pathologies, concern should be placed on effects that PDGF receptor inhibition could have on fetuses.
Supplementary Material
Acknowledgments
We thank our laboratory colleagues, Jeff Hildebrand and Ondine Cleaver for their review of the manuscript. We also especially thank Stacy Glasgow and Ondine Cleaver with assistance in establishing the chick culture system. This research was supported in part by grants from NIH/NHLBI (HL74257) and the March of Dimes (FY05-116) to M.D.T.
Footnotes
Supplementary material for this article is available at http://dev.biologists.org/cgi/content/full/135/3/589/DC1
References
- Abramsson A, Kurup S, Busse M, Yamada S, Lindblom P, Schallmeiner E, Stenzel D, Sauvaget D, Ledin J, Ringvall M, et al. Defective N-sulfation of heparan sulfate proteoglycans limits PDGF-BB binding and pericyte recruitment in vascular development. Genes Dev. 2007;21:316–331. doi: 10.1101/gad.398207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki M, Blazek E, Vogt PK. A role of the kinase mTOR in cellular transformation induced by the oncoproteins P3k and Akt. Proc Natl Acad Sci USA. 2001;98:136–141. doi: 10.1073/pnas.011528498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berven LA, Willard FS, Crouch MF. Role of the p70(S6K) pathway in regulating the actin cytoskeleton and cell migration. Exp Cell Res. 2004;296:183–195. doi: 10.1016/j.yexcr.2003.12.032. [DOI] [PubMed] [Google Scholar]
- Betsholtz C. Biology of platelet-derived growth factors in development. Birth Defects Res C Embryo Today. 2003;69:272–285. doi: 10.1002/bdrc.10030. [DOI] [PubMed] [Google Scholar]
- Bianco A, Poukkula M, Cliffe A, Mathieu J, Luque CM, Fulga TA, Rorth P. Two distinct modes of guidance signalling during collective migration of border cells. Nature. 2007;448:362–365. doi: 10.1038/nature05965. [DOI] [PubMed] [Google Scholar]
- Blair HC, Zaidi M, Schlesinger PH. Mechanisms balancing skeletal matrix synthesis and degradation. Biochem J. 2002;364:329–341. doi: 10.1042/BJ20020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blume-Jensen P, Jiang G, Hyman R, Lee KF, O’Gorman S, Hunter T. Kit/stem cell factor receptor-induced activation of phosphatidylinositol 3′-kinase is essential for male fertility. Nat Genet. 2000;24:157–162. doi: 10.1038/72814. [DOI] [PubMed] [Google Scholar]
- Boyd LM, Chen J, Kraus VB, Setton LA. Conditioned medium differentially regulates matrix protein gene expression in cells of the intervertebral disc. Spine. 2004;29:2217–2222. doi: 10.1097/01.brs.0000142747.90488.1d. [DOI] [PubMed] [Google Scholar]
- Boyle SN, Michaud GA, Schweitzer B, Predki PF, Koleske AJ. A critical role for cortactin phosphorylation by Abl-family kinases in PDGF-induced dorsal-wave formation. Curr Biol. 2007;17:445–451. doi: 10.1016/j.cub.2007.01.057. [DOI] [PubMed] [Google Scholar]
- Brachmann SM, Yballe CM, Innocenti M, Deane JA, Fruman DA, Thomas SM, Cantley LC. Role of phosphoinositide 3-kinase regulatory isoforms in development and actin rearrangement. Mol Cell Biol. 2005;25:2593–2606. doi: 10.1128/MCB.25.7.2593-2606.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmbhatt AA, Klemke RL. ERK and RhoA differentially regulate pseudopodia growth and retraction during chemotaxis. J Biol Chem. 2003;278:13016–13025. doi: 10.1074/jbc.M211873200. [DOI] [PubMed] [Google Scholar]
- Brown MC, Turner CE. Paxillin: adapting to change. Physiol Rev. 2004;84:1315–1339. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- Buchdunger E, O’Reilly T, Wood J. Pharmacology of imatinib (STI571) Eur J Cancer. 2002;38(Suppl 5):S28–S36. doi: 10.1016/s0959-8049(02)80600-1. [DOI] [PubMed] [Google Scholar]
- Chen GC, Turano B, Ruest PJ, Hagel M, Settleman J, Thomas SM. Regulation of Rho and Rac signaling to the actin cytoskeleton by paxillin during Drosophila development. Mol Cell Biol. 2005;25:979–987. doi: 10.1128/MCB.25.3.979-987.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho NK, Keyes L, Johnson E, Heller J, Ryner L, Karim F, Krasnow MA. Developmental control of blood cell migration by the Drosophila VEGF pathway. Cell. 2002;108:865–876. doi: 10.1016/s0092-8674(02)00676-1. [DOI] [PubMed] [Google Scholar]
- Christ B, Huang R, Scaal M. Formation and differentiation of the avian sclerotome. Anat Embryol. 2004;208:333–350. doi: 10.1007/s00429-004-0408-z. [DOI] [PubMed] [Google Scholar]
- Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–750. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. EMBO J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharmawardhane S, Sanders LC, Martin SS, Daniels RH, Bokoch GM. Localization of p21-activated kinase 1 (PAK1) to pinocytic vesicles and cortical actin structures in stimulated cells. J Cell Biol. 1997;138:1265–1278. doi: 10.1083/jcb.138.6.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding H, Wu X, Bostrom H, Kim I, Wong N, Tsoi B, O’Rourke M, Koh GY, Soriano P, Betsholtz C, et al. A specific requirement for PDGF-C in palate formation and PDGFR-alpha signaling. Nat Genet. 2004;36:1111–1116. doi: 10.1038/ng1415. [DOI] [PubMed] [Google Scholar]
- Duchek P, Somogyi K, Jekely G, Beccari S, Rorth P. Guidance of cell migration by the Drosophila PDGF/VEGF receptor. Cell. 2001;107:17–26. doi: 10.1016/s0092-8674(01)00502-5. [DOI] [PubMed] [Google Scholar]
- Fincham VJ, James M, Frame MC, Winder SJ. Active ERK/MAP kinase is targeted to newly forming cell-matrix adhesions by integrin engagement and v-Src. EMBO J. 2000;19:2911–2923. doi: 10.1093/emboj/19.12.2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481–507. doi: 10.1146/annurev.biochem.67.1.481. [DOI] [PubMed] [Google Scholar]
- Hamilton TG, Klinghoffer RA, Corrin PD, Soriano P. Evolutionary divergence of platelet-derived growth factor alpha receptor signaling mechanisms. Mol Cell Biol. 2003;23:4013–4025. doi: 10.1128/MCB.23.11.4013-4025.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanks SK, Ryzhova L, Shin NY, Brabek J. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front Biosci. 2003;8:d982–d996. doi: 10.2741/1114. [DOI] [PubMed] [Google Scholar]
- Harris MJ, Juriloff DM. Mouse mutants with neural tube closure defects and their role in understanding human neural tube defects. Birth Defects Res A Clin Mol Teratol. 2007;79:187–210. doi: 10.1002/bdra.20333. [DOI] [PubMed] [Google Scholar]
- Heldin CH, Westermark B. Mechanism of action and in vivo role of platelet-derived growth factor. Physiol Rev. 1999;79:1283–1316. doi: 10.1152/physrev.1999.79.4.1283. [DOI] [PubMed] [Google Scholar]
- Helwig U, Imai K, Schmahl W, Thomas BE, Varnum DS, Nadeau JH, Balling R. Interaction between undulated and Patch leads to an extreme form of spina bifida in double-mutant mice. Nat Genet. 1995;11:60–63. doi: 10.1038/ng0995-60. [DOI] [PubMed] [Google Scholar]
- Heuchel R, Berg A, Tallquist M, Ahlen K, Reed RK, Rubin K, Claesson-Welsh L, Heldin CH, Soriano P. Platelet-derived growth factor beta receptor regulates interstitial fluid homeostasis through phosphatidylinositol-3′ kinase signaling. Proc Natl Acad Sci USA. 1999;96:11410–11415. doi: 10.1073/pnas.96.20.11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch RV, Soriano P. Roles of PDGF in animal development. Development. 2003;130:4769–4784. doi: 10.1242/dev.00721. [DOI] [PubMed] [Google Scholar]
- Hogan BL. Manipulating the Mouse Embryo. Cold Spring Harbor, NY: Cold Spring Harbor Press; 1994. [Google Scholar]
- Hosang M, Rouge M, Wipf B, Eggimann B, Kaufmann F, Hunziker W. Both homodimeric isoforms of PDGF (AA and BB) have mitogenic and chemotactic activity and stimulate phosphoinositol turnover. J Cell Physiol. 1989;140:558–564. doi: 10.1002/jcp.1041400322. [DOI] [PubMed] [Google Scholar]
- Hung IH, Yu K, Lavine KJ, Ornitz DM. FGF9 regulates early hypertrophic chondrocyte differentiation and skeletal vascularization in the developing stylopod. Dev Biol. 2007;307:300–313. doi: 10.1016/j.ydbio.2007.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juriloff DM, Harris MJ. Mouse models for neural tube closure defects. Hum Mol Genet. 2000;9:993–1000. doi: 10.1093/hmg/9.6.993. [DOI] [PubMed] [Google Scholar]
- Kissel H, Timokhina I, Hardy MP, Rothschild G, Tajima Y, Soares V, Angeles M, Whitlow SR, Manova K, Besmer P. Point mutation in kit receptor tyrosine kinase reveals essential roles for kit signaling in spermatogenesis and oogenesis without affecting other kit responses. EMBO J. 2000;19:1312–1326. doi: 10.1093/emboj/19.6.1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinghoffer RA, Hamilton TG, Hoch R, Soriano P. An allelic series at the PDGFalphaR locus indicates unequal contributions of distinct signaling pathways during development. Dev Cell. 2002;2:103–113. doi: 10.1016/s1534-5807(01)00103-4. [DOI] [PubMed] [Google Scholar]
- Koyama N, Morisaki N, Saito Y, Yoshida S. Regulatory effects of platelet-derived growth factor-AA homodimer on migration of vascular smooth muscle cells. J Biol Chem. 1992;267:22806–22812. [PubMed] [Google Scholar]
- Kundra V, Escobedo JA, Kazlauskas A, Kim HK, Rhee SG, Williams LT, Zetter BR. Regulation of chemotaxis by the platelet-derived growth factor receptor-beta. Nature. 1994;367:474–476. doi: 10.1038/367474a0. [DOI] [PubMed] [Google Scholar]
- Lee ER. Proteolytic enzymes in skeletal development: histochemical methods adapted to the study of matrix lysis during the transformation of a “cartilage model” into bone. Front Biosci. 2006;11:2538–2553. doi: 10.2741/1989. [DOI] [PubMed] [Google Scholar]
- Liu L, Li F, Cardelli JA, Martin KA, Blenis J, Huang S. Rapamycin inhibits cell motility by suppression of mTOR-mediated S6K1 and 4E-BP1 pathways. Oncogene. 2006;25:7029–7040. doi: 10.1038/sj.onc.1209691. [DOI] [PubMed] [Google Scholar]
- Malemud CJ. Matrix metalloproteinases: role in skeletal development and growth plate disorders. Front Biosci. 2006;11:1702–1715. doi: 10.2741/1916. [DOI] [PubMed] [Google Scholar]
- McDonald JA, Pinheiro EM, Montell DJ. PVF1, a PDGF/VEGF homolog, is sufficient to guide border cells and interacts genetically with Taiman. Development. 2003;130:3469–3478. doi: 10.1242/dev.00574. [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Le Douarin N. Duality of molecular signaling involved in vertebral chondrogenesis. Curr Top Dev Biol. 2000;48:43–75. doi: 10.1016/s0070-2153(08)60754-1. [DOI] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Bontoux M, Teillet MA, Le Douarin NM. Heterogeneity in the development of the vertebra. Proc Natl Acad Sci USA. 1994;91:10435–10439. doi: 10.1073/pnas.91.22.10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monsoro-Burq AH, Duprez D, Watanabe Y, Bontoux M, Vincent C, Brickell P, Le Douarin N. The role of bone morphogenetic proteins in vertebral development. Development. 1996;122:3607–3616. doi: 10.1242/dev.122.11.3607. [DOI] [PubMed] [Google Scholar]
- Morrison-Graham K, Schatteman GC, Bork T, Bowen-Pope DF, Weston JA. A PDGF receptor mutation in the mouse (Patch) perturbs the development of a non-neuronal subset of neural crest-derived cells. Development. 1992;115:133–142. doi: 10.1242/dev.115.1.133. [DOI] [PubMed] [Google Scholar]
- Nagel M, Tahinci E, Symes K, Winklbauer R. Guidance of mesoderm cell migration in the Xenopus gastrula requires PDGF signaling. Development. 2004;131:2727–2736. doi: 10.1242/dev.01141. [DOI] [PubMed] [Google Scholar]
- Naski MC, Ornitz DM. FGF signaling in skeletal development. Front Biosci. 1998;3:d781–d794. doi: 10.2741/a321. [DOI] [PubMed] [Google Scholar]
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente-Manzanares M, Horwitz AR. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol. 2006;173:587–589. doi: 10.1083/jcb.200509075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Lonai P. Platelet-derived growth factor-A and its receptor are expressed in separate, but adjacent cell layers of the mouse embryo. Development. 1992;115:1045–1058. doi: 10.1242/dev.115.4.1045. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Bedford MT, Do MS, Eisenbach L, Lonai P. Developmental expression of the alpha receptor for platelet-derived growth factor, which is deleted in the embryonic lethal Patch mutation. Development. 1992;115:289–303. doi: 10.1242/dev.115.1.289. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov DA, Deng JM, Ogunrinu G, Behringer RR. Col2a1-directed expression of Cre recombinase in differentiating chondrocytes in transgenic mice. Genesis. 2000;26:145–146. [PubMed] [Google Scholar]
- Parsons JT. Focal adhesion kinase: the first ten years. J Cell Sci. 2003;116:1409–1416. doi: 10.1242/jcs.00373. [DOI] [PubMed] [Google Scholar]
- Payne J, Shibasaki F, Mercola M. Spina bifida occulta in homozygous Patch mouse embryos. Dev Dyn. 1997;209:105–116. doi: 10.1002/(SICI)1097-0177(199705)209:1<105::AID-AJA10>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Richarte AM, Mead HB, Tallquist MD. Cooperation between the PDGF receptors in cardiac neural crest cell migration. Dev Biol. 2007;306:785–796. doi: 10.1016/j.ydbio.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanoff A. The Avian Embryo: Structural and Functional Development. New York: Macmillan Co; 1960. [Google Scholar]
- Ronnstrand L, Heldin CH. Mechanisms of platelet-derived growth factor-induced chemotaxis. Int J Cancer. 2001;91:757–762. doi: 10.1002/1097-0215(200002)9999:9999<::aid-ijc1136>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Rosenkranz S, DeMali KA, Gelderloos JA, Bazenet C, Kazlauskas A. Identification of the receptor-associated signaling enzymes that are required for platelet-derived growth factor-AA-dependent chemotaxis and DNA synthesis. J Biol Chem. 1999;274:28335–28343. doi: 10.1074/jbc.274.40.28335. [DOI] [PubMed] [Google Scholar]
- Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Segarra J, Balenci L, Drenth T, Maina F, Lamballe F. Combined signaling through ERK, PI3K/AKT, and RAC1/p38 is required for met-triggered cortical neuron migration. J Biol Chem. 2006;281:4771–4778. doi: 10.1074/jbc.M508298200. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Soriano P. The PDGF alpha receptor is required for neural crest cell development and for normal patterning of the somites. Development. 1997;124:2691–2700. doi: 10.1242/dev.124.14.2691. [DOI] [PubMed] [Google Scholar]
- Stedman TL. Stedman’s Medical Dictionary. Philadelphia: Lippincott Williams & Wlikins; 2000. [Google Scholar]
- Stott NS, Chuong CM. Retroviral gene transduction in limb bud micromass cultures. Methods Mol Biol. 2000;135:509–513. doi: 10.1385/1-59259-685-1:509. [DOI] [PubMed] [Google Scholar]
- Takahashi Y, Monsoro-Burq AH, Bontoux M, Le Douarin NM. A role for Quox-8 in the establishment of the dorsoventral pattern during vertebrate development. Proc Natl Acad Sci USA. 1992;89:10237–10241. doi: 10.1073/pnas.89.21.10237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura N, Yoshida H, Ogura Y, Kataoka H, Nishikawa S, Nishikawa S. PDGFR alpha expression during mouse embryogenesis: immunolocalization analyzed by whole-mount immunohistostaining using the monoclonal anti-mouse PDGFR alpha antibody APA5. J Histochem Cytochem. 1997;45:883–893. doi: 10.1177/002215549704500613. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, Soriano P. Cell autonomous requirement for PDGFRalpha in populations of cranial and cardiac neural crest cells. Development. 2003;130:507–518. doi: 10.1242/dev.00241. [DOI] [PubMed] [Google Scholar]
- Tallquist MD, French WJ, Soriano P. Additive effects of PDGF receptor beta signaling pathways in vascular smooth muscle cell development. PLoS Biol. 2003;1:E52. doi: 10.1371/journal.pbio.0000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidali L, Chen F, Cicchetti G, Ohta Y, Kwiatkowski DJ. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol Biol Cell. 2006;17:2377–2390. doi: 10.1091/mbc.E05-10-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallingford JB. Planar cell polarity, ciliogenesis and neural tube defects. Hum Mol Genet. 2006;15(Suppl 2):R227–R234. doi: 10.1093/hmg/ddl216. [DOI] [PubMed] [Google Scholar]
- Watanabe Y, Duprez D, Monsoro-Burq AH, Vincent C, Le Douarin NM. Two domains in vertebral development: antagonistic regulation by SHH and BMP4 proteins. Development. 1998;125:2631–2639. doi: 10.1242/dev.125.14.2631. [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Hawkins P, Cooke F, Hara K, Yonezawa K, Kasuga M, Jackson T, Claesson-Welsh L, Stephens L. Activation of phosphoinositide 3-kinase is required for PDGF-stimulated membrane ruffling. Curr Biol. 1994a;4:385–393. doi: 10.1016/s0960-9822(00)00087-7. [DOI] [PubMed] [Google Scholar]
- Wennstrom S, Siegbahn A, Yokote K, Arvidsson AK, Heldin CH, Mori S, Claesson-Welsh L. Membrane ruffling and chemotaxis transduced by the PDGF beta-receptor require the binding site for phosphatidylinositol 3′ kinase. Oncogene. 1994b;9:651–660. [PubMed] [Google Scholar]
- Wilkinson DG. Whole mount in situ hybridization to vertebrate embryos. Oxford: IRL Press; 1992. [Google Scholar]
- Wood W, Faria C, Jacinto A. Distinct mechanisms regulate hemocyte chemotaxis during development and wound healing in Drosophila melanogaster. J Cell Biol. 2006;173:405–416. doi: 10.1083/jcb.200508161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokote K, Mori S, Siegbahn A, Ronnstrand L, Wernstedt C, Heldin CH, Claesson-Welsh L. Structural determinants in the platelet-derived growth factor alpha-receptor implicated in modulation of chemotaxis. J Biol Chem. 1996;271:5101–5111. doi: 10.1074/jbc.271.9.5101. [DOI] [PubMed] [Google Scholar]
- Yoon BS, Ovchinnikov DA, Yoshii I, Mishina Y, Behringer RR, Lyons KM. Bmpr1a and Bmpr1b have overlapping functions and are essential for chondrogenesis in vivo. Proc Natl Acad Sci USA. 2005;102:5062–5067. doi: 10.1073/pnas.0500031102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Zeng Z, Sarbassov DD, Samudio IJ, Yee KWL, Munsell MF, Jackson CE, Giles FJ, Sabatini DM, Andreeff M, Konopleva M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–3512. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.