

Abstract
ATP is rapidly released from osteoblasts in response to mechanical load. We examined the mechanisms involved in this release and established that shear-induced ATP release was mediated through vesicular fusion and was dependent on Ca2+ entry into the cell via L-type voltage-sensitive Ca2+ channels. Degradation of secreted ATP by apyrase prevented shear-induced PGE2 release.
Introduction
Fluid shear induces a rapid rise in intracellular calcium ([Ca2+]i) in osteoblasts that mediates many of the cellular responses associated with mechanotransduction in bone. A potential mechanism for this increase in [Ca2+]i is the activation of purinergic (P2) receptors resulting from shear-induced extracellular release of ATP. This study was designed to determine the effects of fluid shear on ATP release and the possible mechanisms associated with this release.
Methods
MC3T3-E1 preosteoblasts were plated on type I collagen, allowed to proliferate to 90% confluency, then subjected to 12 dynes/cm2 laminar fluid flow using a parallel plate flow chamber. ATP release into the flow media was measured using a luciferin/luciferase assay. Inhibitors of channels, gap junctional intercellular communication (GJIC) and vesicular formation were added prior to shear and maintained in the flow medium for the duration of the experiment.
Results and Conclusions
Fluid shear produced a transient increase in ATP release compared to static MC3T3-E1 cells (59.8±15.7nM vs. 6.2±1.8nM, respectively), peaking within 1 min of onset. Inhibition of calcium entry through the L-type voltage-sensitive Ca2+ channel (L-VSCC) with nifedipine or verapamil significantly attenuated shear-induced ATP release. Channel inhibition had no effect on basal ATP release in static cells. Ca2+ -dependent ATP release in response to shear appeared to result from vesicular release, and not through gap hemichannels, since vesicle disruption with N-ethylmaleimide, brefeldin A, or monensin prevented increases in flow-induced ATP release, whereas inhibition of gap hemichannels with either 18α-glycyrrhetinic acid or 18β-glycyrrhetinic acid did not. Degradation of extracellular ATP with apyrase prevented shear-induced increases in PGE2 release. These data suggest a time line of mechanotransduction wherein fluid shear activates L-VSCC's to promote Ca2+ entry that, in turn, stimulates vesicular ATP release. Further, these data suggest that P2 receptor activation by secreted ATP mediates flow-induced prostaglandin release.
Keywords: ATP release, mechanotransduction, Ca2+ signaling, osteoblasts, fluid shear
Introduction
Bone is a dynamic organ, with its architecture constantly changing in accordance with the mechanical usage required of it. As external forces placed upon bone decrease, as occurs in prolonged bed rest, immobilization or microgravity, the skeleton undergoes net resorption resulting in significant bone loss (1). Conversely, increased external forces on the skeleton can produce net bone accumulation (2). Various in vitro loading techniques, including hypotonic swelling, substrate strain, and fluid shear stress (FSS), have been developed to study the cellular responses and mechanisms involved in the perception of mechanical stimuli by bone cells. While none of these models completely replicate the stresses endured by bone, most produce osteoblastic responses that are considered anabolic in vivo. These responses include transients in intracellular calcium levels ([Ca2+]i) (3), changes in gene expression (4-6), and prostaglandin release (7,8). We have focused this study on the effects of fluid shear since we have shown that fluid shear, and not physiologic levels of mechanical strain, increases the expression of osteopontin, c-fos, cyclooxygenase 2, and TGFβ (4,9)
Osteoblasts respond to FSS with a rapid increase in intracellular Ca2+ that is dependent on both extracellular Ca2+ entry and intracellular Ca2+ release (3). While we have shown that FSS-induced increases in gene expression are mediated by intracellular Ca2+ release in osteoblasts (10), Ca2+ entry in response to shear has been shown to be required for release of prostaglandins (11), nitric oxide (12), and TGFβ (13). Rapid Ca2+ entry must occur through ion channels and two potential candidates for mediation of FSS-induced Ca2+ entry in osteoblasts are the mechanosensitive, cation-selective channel (MSCC) and the L-type, voltage-sensitive Ca2+ channel (L-VSCC) (for review, see (14). The MSCC has been shown to be important in the release of prostaglandins (11) and TGFβ (13), while inhibition of the L-type VSCC has been shown to inhibit nitric oxide release in bone organ cultures (15) and reduce loading-induced bone formation in vivo (16).
There is a significant body of evidence demonstrating that ATP in the extracellular milieu induces a host of physiologic responses upon activation of ATP-binding purinergic (P2) receptors. These receptors are found in a wide variety of cell types and tissues and have been shown to alter Ca2+ signaling in numerous cell types. P2 receptors can be divided into two families of receptors: metabotropic P2Y receptors that induce intracellular Ca2+ release through activation of G proteins and ionotropic P2X receptors that are ligand-gated channels. Osteoblasts express a variety of P2Y and P2X receptors (17) and activation of these receptors have been shown to increase [Ca2+]i, propagate calcium waves (18), induce c-fos (17) and increase proliferation (19,20). Release of ATP from the cytosol to the pericellular environment is a regulated process and its extracellular availability for P2 receptor binding is limited by the presence of membrane-bound nucleotidases (21). The mechanism(s) of ATP release are unclear, yet chloride-conducting channels (22,23), gap junctional hemichannels (24,25), and vesicular mechanisms (26,27) have been implicated in the controlled release of ATP.
In this study, we examined the effects of fluid shear stress on ATP release in MC3T3-E1 osteoblasts. We demonstrate that shear transiently increases ATP and that this release is Ca2+-dependent. We further show that the shear-induced release of ATP is blocked by inhibition of the L-type voltage-sensitive calcium channel (L-VSCC) mediated by vesicular fusion. Most significant is the observation that ATP activation of P2 receptors is important for shear-induced PGE2 release.
Materials and Methods
Cell culture
MC3T3-E1 cells, a murine osteoblast-like cell line (a gift from Dr. Mary C. Farach-Carson, University of Delaware) were grown in minimal essential medium, α-modification containing 10% fetal bovine serum (Gibco, New York, NY), 100 U/ml penicillin G and 100 μg/ml streptomycin. Cells were maintained in a humidified incubator at 37°C with 5% CO2/95% air and subcultured every 72 hours. For shear studies, 80,000 cells were seeded onto rat-tail type I collagen-coated (100 μg/ml; BD, Franklin Lakes, NJ) glass slides. Fluid shear experiments were performed two days later, when the cells were 80-85% confluent. Flow media consisted of minimal essential medium, α-modification containing 0.5% fetal bovine serum, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 20 mM HEPES, pH 7.4
Fluid flow experiments
Fluid flow was applied to cells in a parallel plate flow chamber using a closed flow loop, as described previously (28) (Cytodyne, San Diego, CA). This system uses a constant hydrostatic pressure head to drive media through the channel of the flow chamber to subject the cell monolayer to steady laminar flow resulting in a well-defined fluid shear stress of 12 dynes/cm2. The apparatus was maintained at 37°C throughout the duration of experimentation. The correlation between shear and flow rate was calculated using the equation
| (Equation 1) |
where Q is the flow rate (cm3/s); μ is the viscosity of the flow media (0.01 dyne·s/cm2); h is the height of the channel (0.022cm); b is the slit width (3.2cm); and τ is the wall shear stress (dyne/cm2). For time course studies of ATP release, a programmable Harvard Syringe Pump (PHD programmable, Harvard Apparatus, Hollison, MA) was used to perfuse the flow chamber with fresh media at the aforementioned shear rate of 12 dynes/cm2.
Luciferin/Luciferase-dependent detection of ATP
An ATP bioluminescence assay containing luciferin/luciferase reagent was used to detect ATP (ATP Bioluminescence Assay kit HS II, Roche, Indianapolis, IN). This assay utilizes the conversion of D-luciferin by luciferase into oxyluciferin and light that requires ATP as a co-factor. The resultant luminescence, measured using a Monolight 3010 (BD Biosciences Pharmingen, San Diego, CA), reflects ATP concentration. Conditioned media samples were acquired in two separate protocols. To determine the time course of ATP release, media samples were taken at each time point and immediately frozen at −80 °C for further analysis. For studies using the closed flow loop, a known volume of media was added to the flow loop prior to fluid shear exposure for 5 minutes. After 5 minutes of fluid shear, 1mL of media was removed and stored as above. Controls were performed with each drug solution to ensure that the added drugs had no effect on luciferase activity. Results were normalized to cellular protein concentration, as determined by the amido black method.
Pharmacologic agents
All drugs tested were purchased from Sigma Chemical (St. Louis, MO) and dissolved into distilled water unless otherwise specified. Drugs were added 45 minutes prior to the onset of flow and remained present for the duration of the experiment. The following concentrations were used: 5μM nifedipine (from 5mM stock in ethanol), 10μM verapamil, 5μM 18α-glycyrrhetinic acid (AGA; from 15mM stock in DMSO), 5μM 18β-glycyrrhetinic acid (BGA; from 15mM stock in DMSO), 100μM monensin (from 100mM stock in MeOH), 10μM; brefeldin A (BFA; 35mM stock in EtOH), 100μM N-ethylmaleimide (NEM; from 100mM stock).
Assessment of plasma membrane integrity
Cell damage was assessed quantitatively by measuring the samples of recovered media for lactate dehydrogenase (LDH). Analysis of LDH levels was performed using the CytoTox96 Non-radioactive Cytotoxicity Assay (Promega, Madison, WI). This assay uses NADH, generated by oxidation of lactate into pyruvate, with the conversion of iodonitrotetrazolium into a red formazan product in the presence of diaphorase. The absorbance at 490nm is proportional to the amount of LDH in the media sample. For a positive control, cells were disrupted by lysing in 0.1% TritonX-100. Serial dilutions of this positive control were compared to LDH levels from media samples.
Gap Junctional Intercellular Communication Assays
GJIC assays were performed using double labeling and immunofluorescence as described previously (29,30). In these experiments, cells were loaded with the dyes calcein-AM (Molecular Probes, Eugene, OR) and 1,19-dioctadecyl-3,3,39,39-tetramethylindocarbocyanine perchlorate (DiI; Molecular Probes). Because of its small molecular weight (< 1 kDa), calcein is gap junction-permeable and able to transfer to neighboring cells in the presence of functional gap junctions, while DiI, a larger, lipophilic dye, incorporates into the membrane and is unable to pass through functional gap junctions. These double-labeled (donor) cells were dropped onto non-labeled (acceptor) cells in monolayer. If functional GJIC existed, calcein would be transferred to the acceptor cells, whereas DiI would stay in the donor cell. Donor cells can be distinguished from acceptor cells through double exposure with fluorescein and rhodamine filters: donor cells fluoresce yellow because of the presence of both calcein (green) and DiI (red), whereas acceptor cells only fluoresce green.
Two days prior to experimentation, MC3T3-E1 cells were seeded onto 35mm glass cover slips in 6-well plates at a density of 60,000 cells per well (acceptor cells). On the day of the experiments, the pre-confluent donor cells were removed from the incubator and washed with PBS followed by aspiration. The donor cells were labeled in a solution composed of 2mL HBSS, 2% BSA, 7μL DiI, 20μL calcein-AM, and 20μL pluronic acid (Molecular Probes) and incubated at 37°C for 30 minutes. After 30 minutes, the dye was aspirated and the donor cells were detached by trypsinization and resuspended in fresh growth medium. A cell pellet was obtained by 5 minutes of centrifugation at 200g. Five hundred double-labeled donor cells were then dropped onto the acceptor cells and incubated for 2 hours at 37°C. Following incubation, the cover slips were removed from the dishes, washed twice in HBSS, and mounted onto a glass slide using Fluoromount-G (Fischer Scientific). The slides were placed on a Nikon fluorescent microscope (Nikon EFD-3; Optical Apparatus, Ardmore, PA) and visualized using fluorescein and rhodamine to locate the calcein- and DiI-loaded cells, respectively. For studies using AGA or BGA to inhibit GJIC, 5μM AGA or BGA was added to acceptor cells 45 minutes prior to the addition of donor cells and maintained in the incubation media until the cover slips were mounted.
Quinacrine staining of intracellular ATP
Osteoblasts were seeded onto type I collagen-coated glass slides as described above. Two days later, when the slides were approximately 80-85% confluent, the cells were incubated in 25μM quinacrine for 30 minutes, rinsed twice in HBSS, and mounted with Fluoromount G. The slides were then immediately examined using the same Nikon fluorescent microscope as for GJIC assays.
Prostaglandin measurement
For measurement of prostaglandin E2 in sheared cells, experiments were conducted as described above but exposed to fluid shear for 60 minutes instead of 5 minutes. After the 60 minute shear treatment, slides of cells were overlaid with 1mL of fresh flow media (with or without drug, as appropriate) and incubated for 30 additional minutes at 37°C with 5% CO2. The media was then collected and PGE2 was measured using commercially available ELISA kits (Amersham Biosciences, Piscataway, NJ) according to the manufacturers instructions. Results were normalized to cellular protein levels. The effect of exogenous ATP addition to static cells was also addressed. Experiments were performed as above for PGE2 release, but overlaid with 1mL of ATP (at concentrations of 100nM – 1mM) in flow media for 30 minutes, after which time the media was collected and analyzed by ELISA.
Statistical analysis
A minimum of two slides per treatment were examined each day on at least three different days. Two way ANOVA analyses were used to compare ATP release from MC3T3-E1 cells treated with pharmacological agents. When a significant difference was found between samples, a Fisher's PLSD was performed to localize the significant difference. Statistical significance was considered at p < 0.05, and samples are presented as mean ± standard error of mean (SEM).
Results
Fluid shear stress induces ATP release
MC3T3-E1 cells exhibited a basal release of ATP (6.2±1.8 nM) that was significantly increased ∼10-fold (59.8±15.7 nM; p<0.001) when cells were subjected to 12 dynes/cm2 FSS (Fig. 1A). Since cytosolic ATP concentrations are in the millimolar range (31), it was necessary to determine whether the changes in extracellular ATP levels resulted from an active release rather than shear-induced cell lysis. To ensure that membrane damage did not contribute to shear-mediated ATP release, we analyzed the conditioned media from sheared cells for the presence of the cytosolic enzyme, lactate dehydrogenase (LDH): if ATP release resulted from cellular damage, LDH would be found in the conditioned media. We consistently found that fluid shear-induced ATP release occurred in the absence of significant plasmalemmal damage compared to static controls (data not shown).
Figure 1.
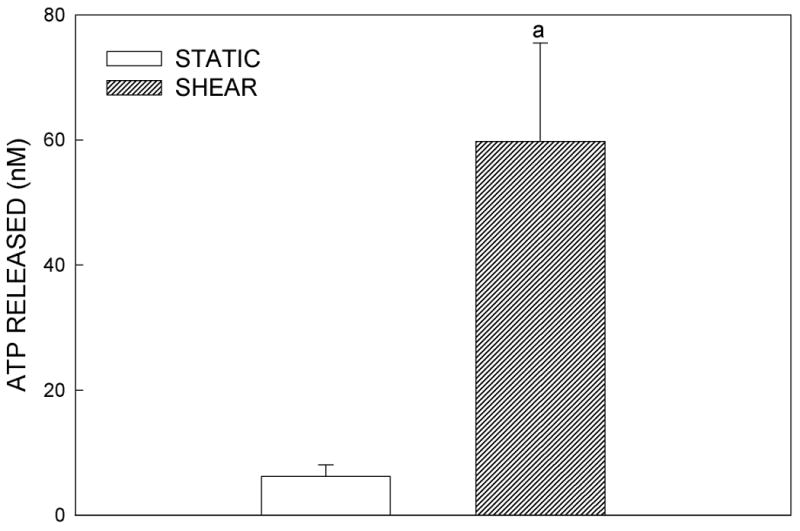
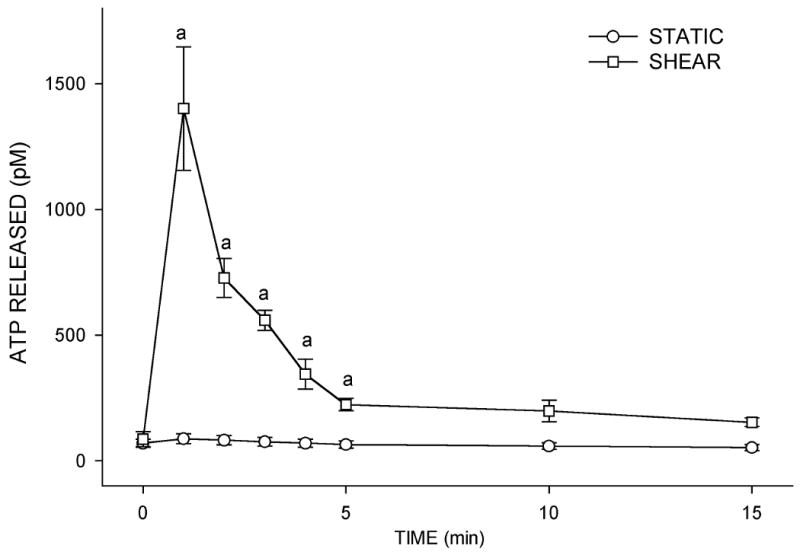
(A) Effects of fluid shear stress at 12 dynes/cm2 on ATP release in MC3T3-E1 osteoblasts. Following 5 minutes of flow, the flow media was removed for analysis of ATP content using an ATP-dependent luciferin-luciferase reaction. Fluid shear increased ATP release approximately 5-fold compared to static controls (a: p < .01 compared to static cells). Each bar represents the mean±SE of 10 experiments. (B) Time course of ATP release in response to shear. ATP was released rapidly from MC3T3-E1 cells, peaking within 1 min of shear application. ATP release returned to near, but elevated, baseline levels with sustained shear (a: p < 0.01 vs. static cells at same time point).
ATP release in response to a variety of stimuli has been shown to be transient, occurring rapidly after the stimulus and decreasing to basal levels over a period of 30-60 minutes (32). To determine if a similar pattern occurs in osteoblastic MC3T3-E1 cells in response to FSS, we examined ATP release at time points before and after 5 minutes using a programmable Harvard Syringe Pump to produce 12 dynes/cm2 shear. We found that FSS induced a rapid release of ATP within one minute of the onset of FSS that returned to pre-flow levels with prolonged fluid shear (Fig. 1B).
Shear induced ATP release requires calcium entry
To determine whether ATP release was Ca2+-dependent, we exposed static MC3T3-E1 cells to the Ca2+ ionophore, ionomycin (1 μM), for 10 min, then removed the bathing medium for ATP analysis. Addition of ionomycin produced a 3-fold (p<0.05) increase in ATP release compared to untreated controls (Fig. 2A). L-VSCC and MSCC channels have been implicated in the intracellular Ca2+ response to mechanical load (3,4,15,33). To test whether these channels are involved in shear-induced ATP release, we blocked the MSCC with GdCl3 (10 μM) and the L-VSCC with nifedipine (5 μM) and verapamil (10 μM). Nifedipine significantly attenuated the shear-induced ATP release (Fig. 2B) in a manner similar to verapamil inhibition (data not shown). Neither inhibitor altered basal ATP release. Block of the MSCC with GdCl3 did not alter either basal or shear-induced ATP release.
Figure 2.
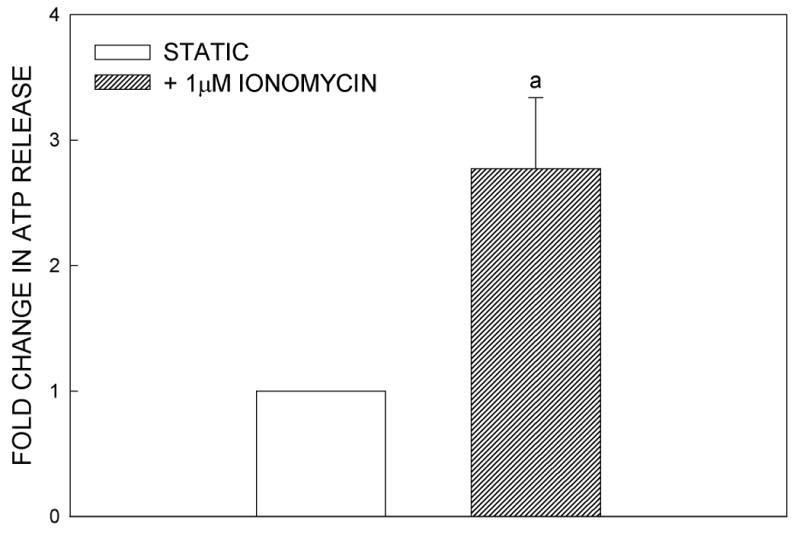
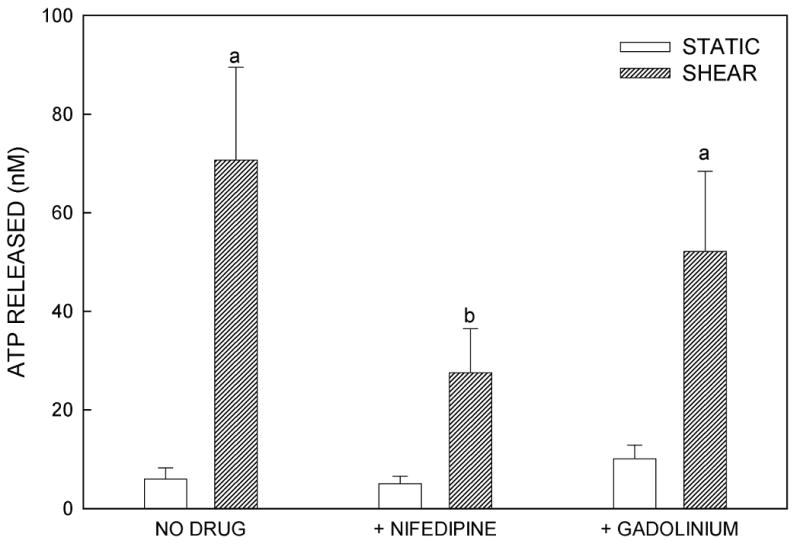
ATP release is dependent on extracellular Ca2+ entry. (A) MC3T3-E1 cells were treated with Ionomycin (1μM), a calcium ionophore, for 10 minutes. The media was then collected for ATP analysis. The addition of ionomycin significantly increased ATP release compared to control cells (a: p < .02 vs. vehicle control). (B) Effects of channel blockers on ATP release. MSCC inhibition with GdCl3 (10μM) had no effect on either basal or FSS-induced ATP release, while L-type VSCC inhibition with nifedipine (5μM) or verapamil (10μM) attenuated FSS-induced ATP release but not basal release (a: p < .05 vs. static cells in same conditions; b: p > .05 vs. static cells in same conditions).
Shear-induced ATP release does not require GJIC
Gap junctions and GJIC have been implicated in the mechanism through which other cell types release ATP in response to a mechanical signals (24,34). Because serum proteins can bind to AGA and titrate the effective AGA concentration (35), it was first necessary to demonstrate that GJIC was inhibited under the flow conditions used. GJIC was evaluated using the double-labeling technique as described previously. In Fig. 3A, the green (calcein) fluorescence indicates the coupled cells in the monolayer while the yellow (calcein and DiI) fluorescence indicates the donor cells. We found that 5μM AGA pre-treatment in flow media (containing 0.5% FBS) effectively inhibited GJIC in MC3T3-E1 osteoblasts. Under these conditions, we found that osteoblasts released ATP in the presence of 5μM AGA when exposed to fluid shear stress (Fig. 3B). Similar results were found when cells were sheared in the presence of another GJ inhibitor, BGA (5μM). These results suggest that shear-induced ATP release in murine osteoblasts does not require GJIC or hemichannels.
Figure 3.
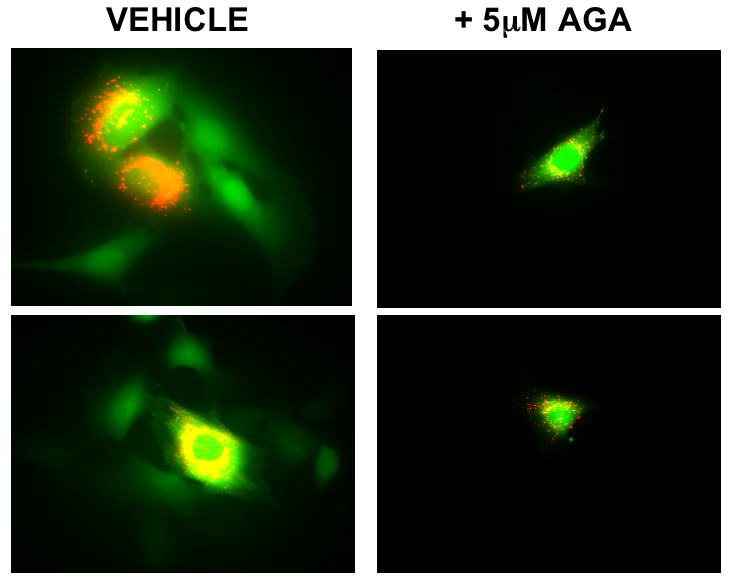
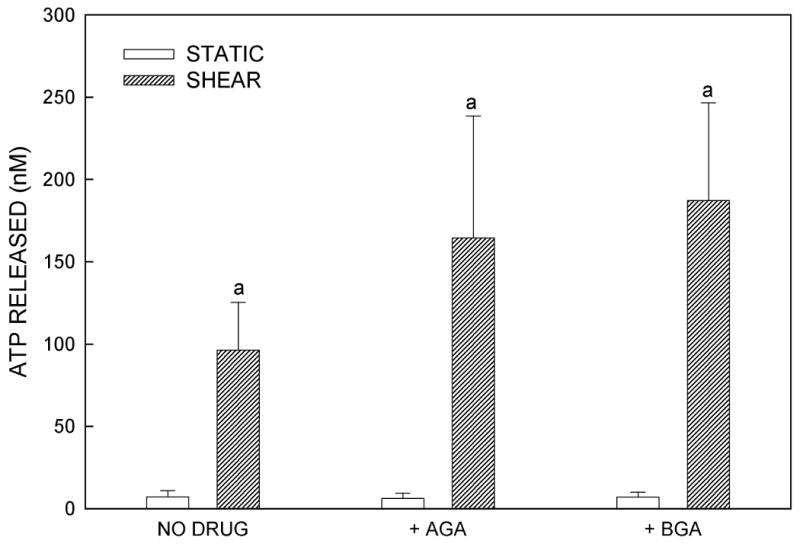
The role of GJIC in ATP release. (A) GJIC was assessed in MC3T3-E1 cells by dual label dye transfer. Donor cells double labeled with the fluorescent dyes calcein and 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) were placed in contact with unloaded cells in the monolayer. Dye transfer was evaluated after 2h. In the dual-exposure photographs, calcein has transferred to acceptor cells which are green, demonstrating functional GJIC; cells fluorescing yellow contain both calcein and DiI and are the dual-labeled donor cells. Pre-incubation with 5μM 18α-glycyrrhetinic acid inhibited GJIC. (B) MC3T3-E1 cells pretreated with vehicle control (DMSO) or GJ inhibitors, 18α-glycyrrhetinic acid or 18β-glycyrrhetinic acid, demonstrated that inhibition of GJIC had no significant effect on FSS-induced ATP release (a: p < .01 vs. static cells in same conditions).
Localization of intracellular ATP stores
We examined the localization of intracellular ATP stores in MC3T3-E1 osteoblasts using quinacrine, a cell-permeant fluorophore that binds to ATP. After 30 min incubation with quinacrine, a high level of punctuated fluorescence was seen, localized primarily within the cytosol of the cells (Fig. 4A). To assess the role of vesicular exocytosis in ATP release, we utilized three pharmacologic agents: BFA, which causes disruption of the Golgi apparatus (26); monensin, which prevents vesicle formation from the Golgi apparatus (26,27); and NEM, which prevents vesicle fusion to the plasma membrane by interfering with vesicle-associated NSF proteins (26,27). Fluid shear in the presence of each of these antagonists significantly attenuated ATP release compared to untreated controls (Fig. 4B-D). These data, combined with the highly granular and punctate intracellular localization of ATP as visualized by quinacrine staining, suggest that ATP is released from murine osteoblasts in a vesicular manner.
Figure 4.
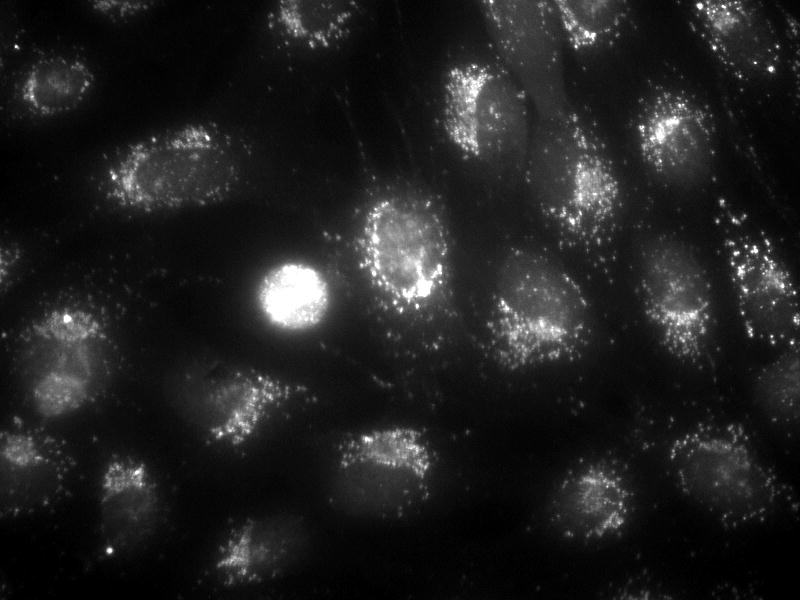
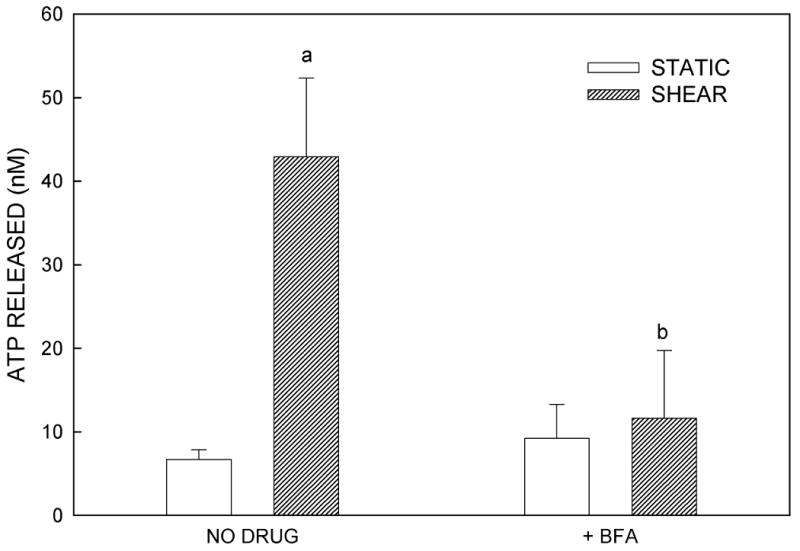
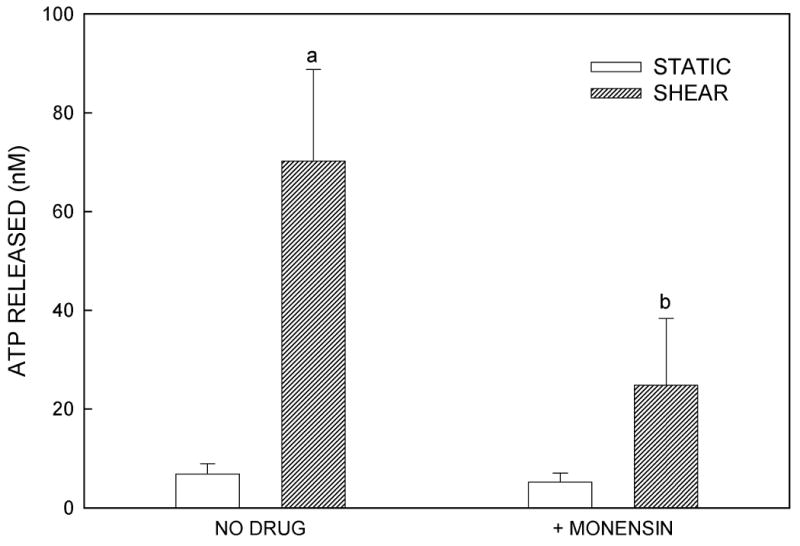
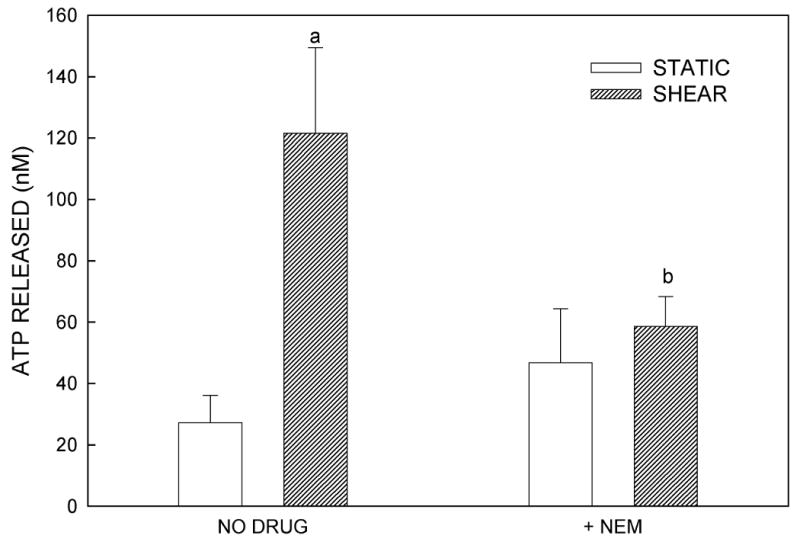
(A) Quinacrine staining of MC3T3-E1 cells demonstrates grainy, punctate localization of ATP, suggesting that ATP is contained in vesicles. (B) Brefeldin A, an agent that disrupts the Golgi and thereby prevents vesicle formation, attenuated FSS-induced increases in ATP release but had no effect on basal ATP release. (C) Monensin, which maintains the Golgi structure but prevents vesicle budding from the Golgi, similarly attenuated FSS-induced ATP release without affecting static ATP release. (D) N-ethylmaleimide prevents vesicular exocytosis by inhibiting NSF proteins. Addition of NEM also inhibited FSS-induced ATP release. (a: p < 0.01 vs. static cells in same conditions)
Extracellular ATP is required for flow-induced increases in PGE2
Prostaglandins are rapidly released in response to shear in osteoblasts (8,11) and their formation is required for load-induced bone formation (36). To determine whether shear-induced PGE2 release was mediated by extracellular ATP, we added apyrase (5U/ml), a nucleotidase that degrades nucleotide triphosphates into nucleotide monophosphates, to the flow medium. Apyrase attenuated flow-induced increases in PGE2 release (Fig. 5A). Similar results were obtained from experiments performed with the non-specific P2 antagonist PPADS, and additional experiments with heat-inactivated apyrase confirmed that the enzymatic activity of apyrase, and not some secondary, non-specific effect, was responsible for attenuating PGE2 release (data not shown); Further, when exogenous ATP was added to static cells, we observed significant increases in PGE2 release (Fig. 5B) without induction of Cox-2 (data not shown). These data suggest that shear-induced ATP secretion mediates the release of prostaglandins, paracrine factors that have been implicated in the anabolic response to exogenous mechanical load.
Figure 5.
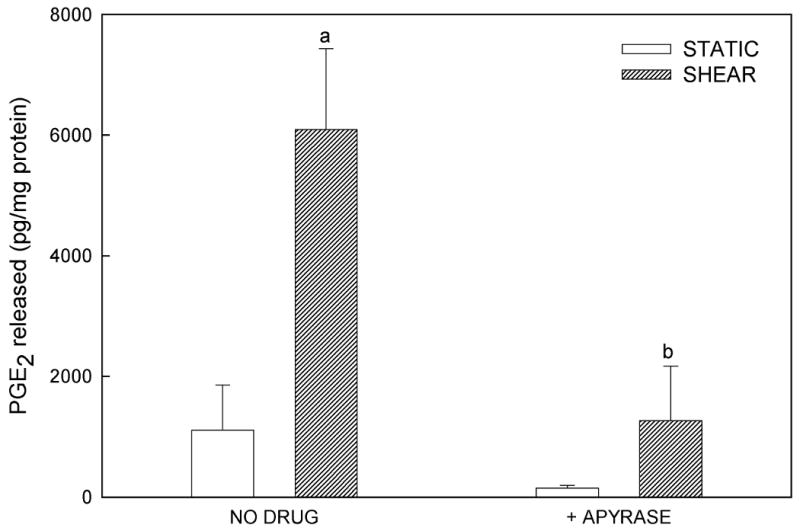
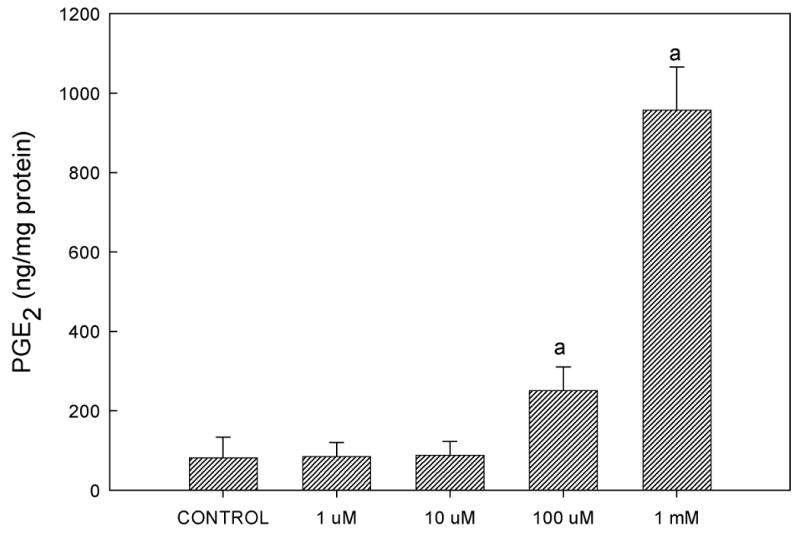
(A) Addition of apyrase (5 U/mL), which hydrolyzes extracellular ATP, significantly decreased PGE2 release from sheared osteoblasts, suggesting that FSS-induced ATP secretion mediates PGE2 release. (B) Addition of exogenous ATP to static, non-sheared cells dose-dependently increased PGE2 release (a: p < 0.01 to static control; b: p < 0.01 to shear in the absence of apyrase).
Discussion
Several mechanisms have been proposed for stimuli-induced ATP release from various cell types. These proposed mechanisms include release through a chloride conducting pathway (37), via gap junctional hemichannels (24,34) and Ca2+ dependent vesicular exocytosis (26). Our data indicate that shear-induced ATP release results from Ca2+ dependent vesicular release. However, we did observe that the general chloride channel blocker, 4,4′-diisothiocyanostilbene-2,2′-disulfonic acid (DIDS), inhibits both basal and shear-induced ATP release (data not shown). While this is a focus of another study, previous reports suggest that DIDS does not block ATP release through inhibition of a chloride conductance, but, rather, by binding to a sulfonylurea receptor on the vesicle to prevent fusion of the vesicle to the membrane (38). Another potential pathway, ATP conductance through gap junctional hemichannels, appears to be involved in other cell types (24,39), but not in the MC3T3-E1 cell model. We demonstrate that gap junction inhibition with either AGA or BGA prevented dye movement through the junctional complex, but had no effect on FSS-induced ATP release. These data support a previous report that found no difference in mechanically-stimulated ATP release in human osteoblast-like cells overexpressing the Cx43 connexin compared to wild type controls (25). Quinacrine, a ATP-binding fluorophore, demonstrated punctate, granular staining, suggesting that ATP can be localized in vesicles in MC3T3-E1 osteoblasts; further experimentation, such as density gradient fractionaion, however, is required to unambiguously demonstrate vesicular ATP localization.
Similar to a previous report in epithelial cells (26), we show that FSS-induced ATP release from osteoblastic cells is the result of Ca2+-dependent vesicular binding to the membrane. Three pharmacologic agents, which cause disruption of the Golgi apparatus (26), prevent vesicle formation from the Golgi apparatus (26,40) prevent vesicle fusion to the plasma membrane by interfering with vesicle-associated NSF proteins (26,40) all significantly decreased FSS-induced ATP release compared to untreated controls. Interestingly, none of these agents completely blocked either basal or FSS-induced ATP release. This observation would suggest that either each of these agents does not totally block vesicular fusion to the membrane or that a secondary pathway for ATP release exists.
While numerous studies have shown that ATP binding to P2 receptors results in an increase in [Ca2+]i that is dependent on both extracellular Ca2+ entry and intracellular Ca2+release, few have examined the role of [Ca2+]i in ATP release. Osteoblasts and osteocytes respond to fluid shear with a rapid increase in intracellular Ca2+ (3) that is essential for shear-induced changes in actin cytoskeletal organization and gene expression (10). While this [Ca2+]i response has been shown to be dependent on both extracellular Ca2+ entry and intracellular Ca2+ release, we have shown that only IP3-mediated Ca2+ release is required for the subsequent changes in cell architecture and protein production (10). However, others have reported that Ca2+ entry via ion channels is important to shear-induced secretion of prostaglandins (11), TGFβ (13) and nitric oxide (15) from bone cells, leading us to postulate that Ca2+ entry mediates signal amplification while intracellular Ca2+ release results in changes in gene expression in osteoblasts. Katsuragi, et al., (41) have shown that inhibition of phospholipase C / IP3-mediated intracellular Ca2+ release blocks angiotensin II stimulated ATP release in smooth muscle cells. However, in this study we found no consistent effect of inhibition of phospholipase C with U73122 on either basal or shear-induced ATP release. This lack of consistency may be due to the subsequent inhibition by U73122 of protein kinase C, which mediates a number of cellular responses, including phosphorylation of Ca2+ channels.
Calcium entry through membrane ion channels could also mediate ATP release in response to shear. This postulate is strengthened by the observation that when extracellular Ca2+ is removed, ATP release in response to mechanical load is attenuated (26). Osteoblasts express a number of ion channels capable of conducting Ca2+ (14) but, to date, only two have been shown to play a role in the [Ca2+]i response to shear: the mechanosensitive, cation-selective channel (MSCC) (3,33) and the dihydropyridine- and voltage-sensitive, L-type Ca2+ channel (VSCC) (33). Activation of the MSCC in response to loading has been associated with prostaglandin, TGFβ and NO release (11,13,15). Activation of the L-type VSCC has been linked to cellular responses of osteoblasts or osteocytes to shear (15) and hormonal stimulation (33). We have recently demonstrated that inhibition of this channel with either nifedipine or verapamil significantly reduces bone formation in mechanically loaded rat tibiae and ulnae, in vivo (16), indicating the importance of this channel in mechanotransduction in bone.
How L-type VSCC's are activated by mechanical perturbation is unclear. We have postulated that shear induced activation of the MSCC results in a membrane depolarization that, in turn, activates the VSCC current. However, the data reported here fails to support this premise. Inhibition of the MSCC with 10 μM GdCl3 did not significantly block ATP release, although a reduced ATP release level in response to shear was observed. Thus, either FSS depolarizes the membrane through a separate mechanism to activate the L-VSCC, or this stimulus can directly activate the L-VSCC.
One mechanism through which the L-VSCC could be activated directly is by the autocrine/paracrine action of ATP. ATP released from the cell can bind to P2 receptors that modulate a number of second messenger pathways, including [Ca2+]i. There are two subtypes of P2 receptors: ionotropic (P2X) receptors that allow entry of ions through receptor-mediated channels, and metabotropic (P2Y) receptors that induce G-protein-mediated intracellular Ca2+ release (42). While the P2Y receptor has been linked with IP3-mediated intracellular Ca2+ release, P2X receptors have been shown to activate both K+ channel and Ca2+ channels, including the L-type VSCC (43,44). A number of isoforms for each of these subtypes have been described, based on the nucleotide binding selectivity of the receptor and inhibition. Osteoblasts exhibit many of these isoforms for both P2X and P2Y (17), and ATP binding to these receptors has also been associated with fast, gapjunction-independent Ca2+ waves (45) and potentiation of the [Ca2+]i response to PTH in osteoblasts (46). Here, we demonstrate that MC3T3-E1 osteoblasts respond to a defined fluid shear with release of ATP within one minute of shear onset. Since the [Ca2+]i response to shear exhibits a time lag from onset to intracellular response of 30-60 sec. (3,33), it is possible that ATP initiates this event to enhance its own release.
Prostaglandin synthesis and release have been shown to occur rapidly in osteoblasts in response to shear (7,8,11) is essential for the anabolic response of bone to mechanical loading (36). PGE2 release from osteoblasts and osteocytes has been shown to be released in two stages. Upon application of shear, a brief burst of PGE2 is observed, peaking at 5–10 min. then returning to levels near baseline (7,11). This is followed at 45-60 min by a large, continuous increase in release that corresponds to an increase in Cox-2 production (7,8). The importance of Cox-2 function in bone formation was demonstrated when the Cox-2 specific inhibitor, NS398, was given to rats prior to mechanical loading: the presence of NS398 completely abolished tibial bone formation in response to four-point bending compared to loaded controls (36). While these results implicate Cox-2 in bone formation in response to exogenous load, it should be noted, however, that prostaglandin synthesis and release does not directly correlate with bone formation, as prostaglandins can similarly promote bone resorption (47). The addition of exogenous ATP to static MC3T3-E1 cells induced the release of PGE2, and the hydrolysis of ATP released in response to shear blocked flow-induced increase in PGE2 release, suggesting that FSS-induced ATP secretion induces PGE2 release through activation of a P2 receptor. Because Reich et al. (8,48) demonstrated that flow-induced PGE2 release was mediated through a Gq protein, we hypothesize that a metabotropic P2Y receptor is involved in this response.
While the measured amount of secreted ATP in response to shear was on the order of 100nM (i.e., 1000-fold less than the amount required to induce PGE2 release in static conditions), it should be noted that the data presented in Figures 1-4 represents ATP found in solution. This does not accurately demonstrate the local ATP concentration at the cell surface immediately upon its release from the cytosol, or the local concentration at P2 receptors. Further, while their expression has not yet been demonstrated in osteoblasts, other ATP-releasing cells, such as chondrocytes and endothelial cells, express membrane-bound NTP-degrading enzymes (ecto-5′-nucleotidases and exonucleotidases) that regulate the extracellular availability of ATP.
In summary, we show that osteoblasts exhibit a basal release of ATP in static cells and respond to a well-defined fluid shear regimen with a significant increase in ATP release. This release is mediated by Ca2+-dependent vesicular fusion to the membrane, although only the shear-induced release is sensitive to Ca2+. We further demonstrate that Ca2+ entry through the L-type VSCC, but not Ca2+ release from intracellular stores, is important to this response. PGE2 release in response to shear appears to be mediated by secreted ATP, suggesting that purinergic signaling may be an important component of the mechanotransduction response of bone.
Figure 6.
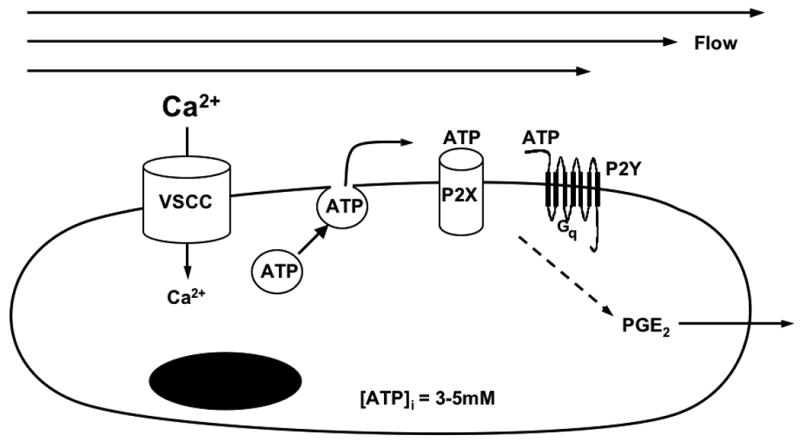
Proposed model for data presented herein. Fluid shear stress activates the L-type VSCC calcium channel through some unknown mechanism. Calcium influx through the L-type VSCC mobilizes vesicular stores of ATP to the plasmalemma, where vesicular fusion releases ATP into the extrcellular environment. ATP then diffuses to P2 receptors to function in an autocrine or paracrine manner, activating either ionotropic P2X or metabotropic P2Y receptors to increase PGE2 release.
Acknowledgments
The National Institutes of Health NIAMS Grants P01 AR45218 and R01 AR43222 (RLD), NIA Grant 13087-09 (HJD), and the National Aeronautics and Space Administration Predoctoral Fellowship Grant NGT5-50366 (DCG) supported this work. We are grateful to Dr. Suzanne M. Norvell for assistance with prostaglandin assays and to Dr. Robert M. Bigsby for his assistance with the luciferin/luciferase assay.
Supported by: NIH grants: NIAMS P01 AR45218 (RLD), NIDDK DK58246 (RLD), NIAG 13087 (HJD) and NASA Predoctoral Fellowship Grant 01-GSRP-028 (DCG).
Contributor Information
Damian C. Genetos, Email: dcg13@psu.edu.
Derik J. Geist, Email: dgeist@iupui.edu.
Liu Dawei, Email: daweliu@iupui.edu.
Henry J. Donahue, Email: hdonahue@psu.edu.
References
- 1.Bikle DD, Sakata T, Halloran BP. The impact of skeletal unloading on bone formation. Gravit Space Biol Bull. 2003;16(2):45–54. [PubMed] [Google Scholar]
- 2.Burr DB, Robling AG, Turner CH. Effects of biomechanical stress on bones in animals. Bone. 2002;30(5):781–6. doi: 10.1016/s8756-3282(02)00707-x. [DOI] [PubMed] [Google Scholar]
- 3.Hung CT, Allen FD, Pollack SR, Brighton CT. Intracellular Ca2+ stores and extracellular Ca2+ are required in the real-time Ca2+ response of bone cells experiencing fluid flow. J Biomechanics. 1996;29(11):1411–17. doi: 10.1016/0021-9290(96)84536-2. [DOI] [PubMed] [Google Scholar]
- 4.You J, Reilly GC, Zhen X, Yellowley CE, Chen Q, Donahue HJ, Jacobs CR. Osteopontin gene regulation by oscillatory fluid flow via intracellular calcium mobilization and activation of mitogen-activated protein kinase in MC3T3-E1 osteoblasts. J Biol Chem. 2001;276(16):13365–71. doi: 10.1074/jbc.M009846200. [DOI] [PubMed] [Google Scholar]
- 5.Wadhwa S, Godwin SL, Peterson DR, Epstein MA, Raisz LG, Pilbeam CC. Fluid flow induction of cyclo-oxygenase 2 gene expression in osteoblasts is dependent on an extracellular signal-regulated kinase signaling pathway. J Bone Miner Res. 2002;17(2):266–74. doi: 10.1359/jbmr.2002.17.2.266. [DOI] [PubMed] [Google Scholar]
- 6.Pavalko FM, Chen NX, Turner CH, Burr DB, Atkinson S, Hsieh YF, Qiu J, Duncan RL. Fluid shear-induced mechanical signaling in MC3T3-E1 osteoblasts requires cytoskeletal-integrin interactions. Am J Physiol. 1998;275(6 pt 1):C1591–1601. [PubMed] [Google Scholar]
- 7.Klein-Nulend J, Burger EH, Semeins CM, Raisz LG, Pilbeam CC. Pulsating fluid flow stimulates prostaglandin release and inducible G/H synthase mRNA expression in primary mouse bone cells. J Bone Miner Res. 1997;12(1):45–51. doi: 10.1359/jbmr.1997.12.1.45. [DOI] [PubMed] [Google Scholar]
- 8.Reich KM, Frangos JA. Effect of flow on prostaglandin E2 and inositol triphosphate levels in osteoblasts. Am J Physiol. 1991;261(3 pt 1):C428–32. doi: 10.1152/ajpcell.1991.261.3.C428. [DOI] [PubMed] [Google Scholar]
- 9.Owan I, Burr DB, Turner CH, Qiu J, Tu Y, Onyia JE, Duncan RL. Mechanotransduction in bone: osteoblasts are more responsive to fluid forces than mechanical strain. Am J Physiol. 1997;273(3 p2 1):C810–815. doi: 10.1152/ajpcell.1997.273.3.C810. [DOI] [PubMed] [Google Scholar]
- 10.Chen NX, Ryder KD, Pavalko FM, Turner CH, Burr DB, Qiu J, Duncan RL. Calcium regulates fluid shear-induced cytoskeletal reorganization and gene expression in osteoblasts. Am J Physiol. 2000;278(5):C989–997. doi: 10.1152/ajpcell.2000.278.5.C989. [DOI] [PubMed] [Google Scholar]
- 11.Ajubi NE, Klein-Nulend J, Alblas MJ, Burger EH, Nijweide PJ. Signal transduction pathways involved in shear stress-induced prostaglandin E2 production by cultured chicken osteocytes. Am J Physiol. 1999;276(1 pt 1):E171–8. doi: 10.1152/ajpendo.1999.276.1.E171. [DOI] [PubMed] [Google Scholar]
- 12.McAllister TN, Frangos JA. Steady and transient fluid shear stress stimulate NO release in osteoblasts through distinct biochemical pathways. J Bone Miner Res. 1999;14(6):930–6. doi: 10.1359/jbmr.1999.14.6.930. [DOI] [PubMed] [Google Scholar]
- 13.Sakai K, Mohtai M, Iwamoto Y. Fluid shear stress increases transforming growth factor beta 1 expression in human osteoblast-like cells: modulation by cation channel blockades. Calcif Tiss Intl. 1998;63(6):515–20. doi: 10.1007/s002239900567. [DOI] [PubMed] [Google Scholar]
- 14.Duncan RL, Akanbi KA, Farach-Carson MC. Calcium signals and calcium channels in osteoblastic cells. Semin Nephrology. 1998;18(2):178–190. [PubMed] [Google Scholar]
- 15.Rawlinson SC, Pitsillides AA, Lanyon LE. Involvement of different ion channels in osteoblasts' and osteocytes' early response to mechanical strain. Bone. 1996;19(6):609–14. doi: 10.1016/s8756-3282(96)00260-8. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Duncan RL, Burr DB, Turner CH. L-type calcium channels mediate mechanically induced bone formation in vivo. J Bone Miner Res. 2002;17(10):1795–800. doi: 10.1359/jbmr.2002.17.10.1795. [DOI] [PubMed] [Google Scholar]
- 17.Bowler WB, Dixon CJ, Halleux C, Maier R, Bilbe G, Fraser WD, Gallagher JA, Hipskind RA. Signaling in human osteoblasts by extracellular nucleotides. Their weak induction of the c-fos proto-oncogene via Ca2+ mobilization is strongly potentiated by a parathyroid hormone/cAMP-dependent protein kinase pathway independently of mitogen-activated protein kinase. J Biol Chem. 1999;274(20):14315–24. doi: 10.1074/jbc.274.20.14315. [DOI] [PubMed] [Google Scholar]
- 18.Jorgensen NR, Geist ST, Civitelli R, Steinberg TH. ATP- and gap junction-dependent intercellular calcium signaling in osteoblastic cells. J Cell Biol. 1997;139(2):497–506. doi: 10.1083/jcb.139.2.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nakamura E, Uezono Y, Narusawa K, Shibuya I, Oishi Y, Tanaka M, Yanagihara N, Nakamura T, Izumi F. ATP activates DNA synthesis by acting on P2X receptors in human osteoblast-like MG-63 cells. Am J Physiol Cell Physiol. 2000;279(2):C510–9. doi: 10.1152/ajpcell.2000.279.2.C510. [DOI] [PubMed] [Google Scholar]
- 20.Shimegi S. ATP and adenosine act as a mitogen for osteoblast-like cells (MC3T3-E1) Calcif Tissue Int. 1996;58(2):109–13. doi: 10.1007/BF02529732. [DOI] [PubMed] [Google Scholar]
- 21.Zimmermann H. Extracellular metabolism of ATP and other nucleotides. Naunyn Schmiedebergs Arch Pharmacol. 2000;362(4-5):299–309. doi: 10.1007/s002100000309. [DOI] [PubMed] [Google Scholar]
- 22.Bentolila V, Boyce TM, Fyhrie DP, Drumb R, Skerry TM, Schaffler MB. Intracortical remodeling in adult rat long bones after fatigue loading. Bone. 1998;23(3):275–81. doi: 10.1016/s8756-3282(98)00104-5. [DOI] [PubMed] [Google Scholar]
- 23.Braunstein GM, Roman RM, Clancy JP, Kudlow BA, Taylor AL, Shylonsky VG, Jovov B, Peter K, Jilling T, Ismailov II, Benos DJ, Schwiebert LM, Fitz JG, Schwiebert EM. Cystic fibrosis transmembrane conductance regulator facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume regulation. J Biol Chem. 2001;276(9):6621–30. doi: 10.1074/jbc.M005893200. [DOI] [PubMed] [Google Scholar]
- 24.Cotrina ML, Lin JH, Alves-Rodrigues A, Liu S, Li J, Azmi-Ghadimi H, Kang J, Naus CC, Nedergaard M. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci U S A. 1998;95(26):15735–40. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Romanello M, Pani B, Bicego M, D'Andrea P. Mechanically induced ATP release from human osteoblastic cells. Biochem Biophys Res Commun. 2001;289(5):1275–81. doi: 10.1006/bbrc.2001.6124. [DOI] [PubMed] [Google Scholar]
- 26.Knight GE, Bodin P, De Groat WC, Burnstock G. ATP is released from guinea pig ureter epithelium on distension. Am J Physiol Renal Physiol. 2002;282(2):F281–8. doi: 10.1152/ajprenal.00293.2000. [DOI] [PubMed] [Google Scholar]
- 27.Bodin P, Burnstock G. Increased release of ATP from endothelial cells during acute inflammation. Inflamm Res. 1998;47(8):351–4. doi: 10.1007/s000110050341. [DOI] [PubMed] [Google Scholar]
- 28.Frangos JA, McIntire LV, Eskin SG. Shear stress induced stimulation of mammalian cell metabolism. Biotech and Bioengineering. 1988;32:1053–60. doi: 10.1002/bit.260320812. [DOI] [PubMed] [Google Scholar]
- 29.Saunders MM, You J, Trosko JE, Yamasaki H, Li Z, Donahue HJ, Jacobs CR. Gap junctions and fluid flow response in MC3T3-E1 cells. Am J Physiol Cell Physiol. 2001;281(6):C1917–25. doi: 10.1152/ajpcell.2001.281.6.C1917. [DOI] [PubMed] [Google Scholar]
- 30.Yellowley CE, Li Z, Jacobs CR, Donahue HJ. Functional gap junctions between osteocytic and osteoblastic cells. J Bone Miner Res. 2000;15(2):209–217. doi: 10.1359/jbmr.2000.15.2.209. [DOI] [PubMed] [Google Scholar]
- 31.Schwiebert EM. Extracellular ATP-mediated propagation of Ca(2+) waves. Focus on “mechanical strain-induced Ca(2+) waves are propagated via ATP release and purinergic receptor activation”. Am J Physiol Cell Physiol. 2000;279(2):C281–3. doi: 10.1152/ajpcell.2000.279.2.C281. [DOI] [PubMed] [Google Scholar]
- 32.Ostrom RS, Gregorian C, Insel PA. Cellular release and response to ATP as key determinants of the set-point of signal transduction pathways. J Bio Chem. 2000;275(16):11735–39. doi: 10.1074/jbc.275.16.11735. [DOI] [PubMed] [Google Scholar]
- 33.Ryder KD, Duncan RL. Parathyroid hormone enhances fluid shear-induced [Ca2+]i signaling in osteoblastic cells through activation of mechanosensitive and voltage- sensitive Ca2+ channels. J Bone Miner Res. 2001;16(2):240–8. doi: 10.1359/jbmr.2001.16.2.240. [DOI] [PubMed] [Google Scholar]
- 34.Romanello M, D'Andrea P. Dual mechanism of intercellular communication in HOBIT osteoblastic cells: a role for gap-junctional hemichannels. J Bone Miner Res. 2001;16(8):1465–76. doi: 10.1359/jbmr.2001.16.8.1465. [DOI] [PubMed] [Google Scholar]
- 35.Davidson JS, Baumgarten IM, Harley EH. Reversible inhibition of intercellular junctional communication by glycyrrhetinic acid. Biochem Biophys Res Commun. 1986;134(1):29–36. doi: 10.1016/0006-291x(86)90522-x. [DOI] [PubMed] [Google Scholar]
- 36.Forwood MR. Inducible cyclo-oxygenase (COX-2) mediates the induction of bone formation by mechanical loading in vivo. J Bone Miner Res. 1996;11(11):1688–93. doi: 10.1002/jbmr.5650111112. [DOI] [PubMed] [Google Scholar]
- 37.Rotoli BM, Bussolati O, Dall' Asta V, Hoffmann EK, Cabrini G, Gazzola GC. CFTR expression in C127 cells is associated with enhanced cell shrinkage and ATP extrusion in Cl(-)-free medium. Biochem Biophys Res Commun. 1996;227(3):755–61. doi: 10.1006/bbrc.1996.1581. [DOI] [PubMed] [Google Scholar]
- 38.Barg S, Renstrom E, Berggren PO, Bertorello A, Bokvist K, Braun M, Eliasson L, Holmes WE, Kohler M, Rorsman P, Thevenod F. The stimulatory action of tolbutamide on Ca2+-dependent exocytosis in pancreatic beta cells is mediated by a 65-kDa mdr-like P-glycoprotein. Proc Natl Acad Sci U S A. 1999;96(10):5539–44. doi: 10.1073/pnas.96.10.5539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Graff RD, Lazarowski ER, Banes AJ, Lee GM. ATP release by mechanically loaded porcine chondrons in pellet culture. Arthritis Rheum. 2000;43(7):1571–9. doi: 10.1002/1529-0131(200007)43:7<1571::AID-ANR22>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 40.Bodin P, Burnstock G. Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J Cardiovasc Pharmacol. 2001;38(6):900–8. doi: 10.1097/00005344-200112000-00012. [DOI] [PubMed] [Google Scholar]
- 41.Katsuragi T, Sato C, Guangyuan L, Honda K. Inositol(1,4,5)trisphosphate signal triggers a receptor-mediated ATP release. Biochem Biophys Res Commun. 2002;293(2):686–90. doi: 10.1016/S0006-291X(02)00272-3. [DOI] [PubMed] [Google Scholar]
- 42.Di Viriglio F, Chiozzi P, Ferrari D, Falzoni S, Sanz JM, Morellu A, Torboli M, Bolognesi G, Baricordi OR. Nucleotide receptors: An emerging family of regulatory molecules in blood cells. Blood. 2001;97(3):587–600. doi: 10.1182/blood.v97.3.587. [DOI] [PubMed] [Google Scholar]
- 43.Hur EM, Park TJ, Kim KT. Coupling of L-type voltage-sensitive calcium channels to P2X2 purinoceptors in PC-12 cells. Am J Physiol. 2001;280(5):C1121–9. doi: 10.1152/ajpcell.2001.280.5.C1121. [DOI] [PubMed] [Google Scholar]
- 44.Inscho EW, Cook AK. P2 receptor-mediated afferent arteriolar vasoconstriction during calcium blockade. Am J Physiol Renal Physiol. 2002;282(2):F245–55. doi: 10.1152/ajprenal.0038.2001. [DOI] [PubMed] [Google Scholar]
- 45.Jorgensen NR, Henriksen Z, Brot C, Eriksen EF, Sorensen OH, Civitelli R, Steinberg TH. Human osteoblastic cells propagate intercellular calcium signals by two different mechanisms. J Bone Miner Res. 2000;15(6):1024–32. doi: 10.1359/jbmr.2000.15.6.1024. [DOI] [PubMed] [Google Scholar]
- 46.Kaplan AD, Reimer WJ, Feldman RD, Dixon SJ. Extracellular nucleotides potentiate the cytosolic Ca2+, but not cyclic adenosine 3′, 5′-monophosphate response to parathyroid hormone in rat osteoblastic cells. Endocrinology. 1995;136(4):1674–85. doi: 10.1210/endo.136.4.7895678. [DOI] [PubMed] [Google Scholar]
- 47.Suzawa T, Miyaura C, Inada M, Maruyama T, Sugimoto T, Ushikubi F, Ichikawa A, Narumiya S, Suda M. The role of prostalgandin E receptor subtypes (EP1, EP2, EP3, and EP4) in bone resorption: an analysis using specific agonists for the resorptive EPs. Endocrinology. 2000;141(4):1554–9. doi: 10.1210/endo.141.4.7405. [DOI] [PubMed] [Google Scholar]
- 48.Reich KM, Frangos JA. Protein kinase C mediates flow-induced prostaglandin E2 production in osteoblasts. Calcif Tissue Int. 1993;52(1):62–6. doi: 10.1007/BF00675628. [DOI] [PubMed] [Google Scholar]