

Abstract
The appearance over many days of Lac+ frameshift mutations in Escherichia coli strain FC40 incubated on lactose selection plates is a classic example of apparent “adaptive” mutation in an episomal gene. We show that endogenously overproduced carotenoids reduce adaptive mutation under selective conditions by a factor of around two. Carotenoids are known to scavenge singlet oxygen suggesting that the accumulation of oxidative base damage may be an integral part of the adaptive mutation phenomenon. If so, the lesion cannot be 7,8-dihydro-8-oxoguanine since adaptive mutation in FC40 is unaffected by mutM and mutY mutations. If active oxygen species such as singlet oxygen are involved in adaptive mutation then they should also induce frameshift mutations in FC40 under non-selective conditions. We show that such mutations can be induced under non-selective conditions by protoporphyrin photosensitisation and that this photodynamic induction is reduced by a factor of just over two when endogenous carotenoids are present. We argue that the involvement of oxidative damage would in no way be inconsistent with current understanding of the mechanism of adaptive mutation and the role of DNA polymerases.
Keywords: Adaptive mutation, Carotenoids, Singlet oxygen, Active oxygen species, Frameshift mutations
1. Introduction
In 1988 John Cairns and co-workers [1] described an experimental system in which bacteria, when plated under conditions where their growth was severely restricted by a single defective nutritional gene, mutated over several days to a phenotype that was able to grow. There are now many examples of this phenomenon. In some instances these late appearing colonies are actually slow growing mutants present in the culture at the time of plating. In many instances, however, the mutants can be shown to have arisen on the plate and it is characteristic that they do so at a rate far higher than would be expected from the amount of DNA replication that occurs (for review see [2]). Growth restriction is usually effected in such experiments by deprivation of a required amino acid or by supplying a sugar that cannot be metabolised. Because mutants at loci other than those that would allow growth have generally not been observed (but see later) the phenomenon has become loosely known as “adaptive” mutation, although other terms such as selection-induced mutation, stationary phase mutation, or starvation-associated mutation have also been employed.
The most widely studied model for adaptive mutation is Escherichia coli strain FC40 which contains an F′ bearing a lacI gene with a +1 frameshift mutation that is spectacularly mutable under selective conditions on lactose plates [3]. In contrast to other systems that have been studied, the mutations on the episome (almost all of which are single base deletions) arise only in strains that are proficient for RecA function, for genetic recombination, and for sexual conjugation (although actual conjugation is apparently not essential) [3–8]. In this system it has also been shown the specificity of “adaptive” mutation is not absolute and that mutations may also arise at genes not under selection [9,10].
It has generally been assumed that the mutations initially arise as misincorporations by one or other of the cell’s DNA polymerases. Studies with an allele of dnaE which gives rise to a DNA polymerase III with greater than normal fidelity showed that it also conferred a decrease in the rate of adaptive mutation, suggesting that Pol III was responsible for many of the initial mutation events [11,12]. The fact that adaptive mutation in FC40 is recA-dependent might, of course, indicate the involvement of polymerases under SOS control. Recent work has indicated that adaptive mutation in FC40 is indeed under SOS control [13]. Of the three polymerases under SOS control, Pol V (UmuD′,C) can be excluded as essential since bacteria unable to process UmuD to its active form show normal adaptive mutation [3] and a deletion of umuD,C also has little effect [13]. Pol II is also not essential since bacteria carrying a deletion of polB show an enhanced rate of adaptive mutation [14]. Indeed, this result suggests that when it is present Pol II competes with the mutation-generating polymerase(s) and is more accurate than them. There remains Pol IV, the product of the dinB gene, and recent work indicates an involvement of this polymerase in adaptive mutation in FC40 (H. Ohmori and P.L. Foster, unpublished observations).
Although polymerase infidelity seems likely to account for many of the errors, there are other possible sources. We have previously shown that in some other systems adaptive mutation appears to be due to oxidative DNA lesions (for review see [15]). Because there is evidence that exposure of both single- and double-stranded M13 DNA to singlet oxygen can lead to the generation of single base deletions when the DNA is transfected into cells [16,17] it was suggested [15] that DNA damage caused by oxidative species might also have a role in “adaptive” mutation in the FC40 system. The fact that deletion of polB, the gene for DNA polymerase II, confers a mutator phenotype for “adaptive” mutation is suggestive because DNA polymerase II has an important role in the repair of oxidative DNA damage [14]. Carotenoids are potent scavengers of active oxidative species, particularly singlet oxygen [18] (A. Eisenstark, personal communication). In an auxotrophic strain carrying a mutY mutation, where the target lesions are known to be oxidised bases, it was shown that expression of intracellular carotenoids reduced the rate of adaptive mutation by a factor of about two [19]. We have therefore, examined whether the rate of “adaptive” mutation in FC40 is altered when there is endogenous production of carotenoid scavengers.
2. Materials and methods
2.1. Bacterial strains and plasmids
The scavenger strain, FC29, and the strain undergoing “adaptive” mutation, FC40, have been described [3]. FC29 is rifampicin-sensitive and carries an F′ with a deletion allele of lacZ, FC40 has an F′ with a lacI–lacZ fusion with a +1 base-pair frameshift mutation (lacI33) in the lacI coding sequence [20]. Both strains carry a chromosomal deletion of the lac operon. Derivatives of FC40 used in this work are shown in Table 1. Absence of rpoS-dependent catalase activity was confirmed after 8 days incubation on L-agar plates by dropping hydrogen peroxide solution on to individual colonies (cf. [21]). Strains containing hemH1 (zbb-3055::Tn10) overexpress protoporphyrin IX. Plasmid pPL376 is a derivative of pBR322 containing six genes necessary for carotenoid biosynthesis originally cloned from Erwinia herbicola via cos-mid vector pHC79 [23]. The genes are upregulated by rpoS in stationary phase, and bacteria carrying the plasmid give bright yellow colonies after two days incubation. Plasmid pART53 is a derivative of pPL376 that does not express carotenoids and which has lost approximately a kilobase of DNA presumably including promoter sequences for the carotenoid genes. All manipulations used standard techniques [24].
Table 1.
Derivatives of E. coli FC40 used in this work
| Strain | Parent | Additional allele |
|---|---|---|
| CM1423 | FC40 | rpoS::Tn10 transduced from MT1rpoS [19] |
| CM1424 | FC40pPL376 | rpoS::Tn10 transduced from MT1rpoS [19] |
| CM1461 | FC40 | hemH1 (zbb-3055::Tn10) transduced from AW804 (B. Weiss) |
| CM1443 | FC40 | mutM::miniTn10 transduced from TT101 [22] |
| CM1429 | FC40 | mutY68::kanR transduced from CM1307 [22] |
2.2. Experimental protocols
For experiments under lactose selection conditions, bacteria were grown for 24 h at 37°C in glycerol salts medium plus ampicillin (100 μg/ml) in the case of strains containing plasmids. Scavenger bacteria (FC29) were centrifuged and resuspended in one tenth volume of phage buffer. An initial optical density reading on a 1/20 dilution gave an estimate of the number of bacteria and at least 109 were plated on lactose selection plates. Test bacteria (FC40 and its derivatives) were centrifuged and resuspended in the same volume of phage buffer. Following an optical density determination an estimated 108 bacteria were plated on lactose selection plates along with the scavenger bacteria. Viable counts were made of all bacterial suspensions so that the number of bacteria plated was known more exactly. Inoculated plates were incubated at 37°C and the number of Lac+ mutants was scored daily for 7 days. Mutant colonies appearing after day 2 were taken to have arisen on the plate. Scavenger bacteria were also plated alone to ensure that they remained non-revertible. In early experiments mutant colonies were picked off and checked for rifampicin resistance to ensure that they had arisen in the test strain and not the scavenger strain. To allow for small differences in the number of test bacteria plated, mutant counts were normalised per 108 bacteria plated, as shown by the viable counts.
In parallel with the mutation plates, 104 FC40 bacteria were plated along with at least 109 scavenger bacteria and incubated at 37°C. At intervals the bacteria were washed off plates with 10 ml phage buffer and the number of viable test bacteria determined on rifampicin plates on which the scavenger bacteria cannot grow. This enabled the accurate determination of the number of viable bacteria per plate over the course of the experiment without the problems caused by the presence of small undetected Lac+ colonies when higher FC40 plating densities were used.
For photodynamic induction of mutations in CM1461, bacteria were grown overnight in M9 salts medium containing glycerol (0.4% v/v) and thiamine (5 μg/ml w/v) and sub-cultured into 10 ml L-broth. Ampicillin was present in liquid cultures when bacteria carried pPL376. After 4–5 h, when the culture was entering stationary phase, the bacteria were centrifuged and resuspended in 10 ml phage buffer [25]. The suspension was transferred to a plastic petri dish and exposed at a distance of 11 cm from two Osram Liteguard 40 W warm light fluorescent tubes placed 10 cm apart. At intervals 0.2 ml samples were removed and plated on five lactose selection plates (composed of minimal salts [26] plus lactose (filter sterilised, 0.2% w/v) thiamine (5 μg/ml) and Difco Bacto agar (1.5% w/v)). After 2 days at 37°C in a darkened incubator most pre-existing mutants had formed visible colonies. At this stage plates were transferred to the bench and covered with foil to exclude light since it had been found that the rate of adaptive mutation was lower at this temperature. This was despite the eventual appearance of a visible lawn due to growth on contaminating nutrients in the absence of scavenger bacteria. Over the next 3 days a few slower-growing pre-existing mutants appeared together with most of the mutants induced by exposure to light. In parallel, aliquots of the irradiated suspension were diluted and plated on to L-agar plates to determine the associated lethality. Colonies were counted after 24 h. The difference between the nominal mutation frequencies per survivor of exposed versus unexposed samples was taken to be the induced mutation frequency.
Sensitivity to 254 nm UV light was determined by exposing bacteria suspended in buffer to a Philips 6 W TUV lamp and plating on L-agar.
3. Results
3.1. Effect of carotenoids on adaptive mutation
If active oxygen species are involved in mutation under selection conditions in FC40 then carotenoids should scavenge them and reduce the rate of adaptive mutation. Endogenous production of carotenoids was achieved by introducing plasmid pPL376 into FC40. Plasmid pPL376 is a derivative of pBR322 containing six genes from Erwinia herbicola necessary for carotenoid biosynthesis, under the control of the stationary phase transcription factor sigma [23]. E. coli strains in which these genes are expressed have bright yellow colonies [19]. We found that the rate of appearance of Lac+ mutants on selection plates was reduced in FC40pPL376 to less than half of that found with FC40 itself (Fig. 1a).
Fig. 1.
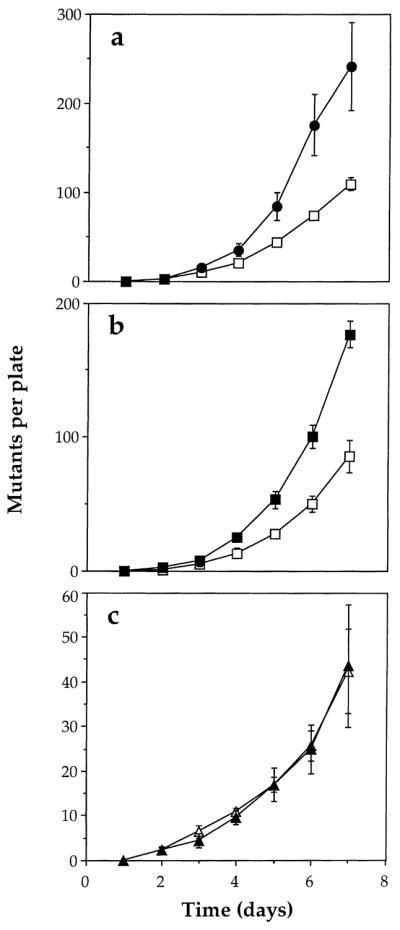
Effect of endogenous carotenoids on the number of Lac+ mutants arising as a function of time of incubation on selective agar. (a) FC40 (●) and FC40pPL376 (□); (b) FC40pPL376 (□) and FC40pART53 (■); (c) CM1423 (rpoS) (□) and CM1424 (rpoS pPL376) (▲). FC40pPL376 is the only strain expressing carotenoids. Means and standard errors of at least three experiments.
Because we had previously found that the survival of bacteria under conditions of amino acid starvation can be reduced if they carry a high copy number plasmid [27] it seemed possible that an effect of pPL376 on viability might explain the difference in Fig. 1a. In parallel with the mutation plates, 104 test bacteria were plated along with at least 109 scavenger bacteria and incubated at 37°C. At intervals the bacteria were washed off plates with 10 ml phage buffer and the number of viable test bacteria determined on rifampicin plates on which the scavenger bacteria cannot grow. This enabled the accurate determination of the number of viable bacteria per plate over the course of the experiment without the problems caused by the presence of small undetected Lac+ colonies when higher plating densities were used. No difference was seen between FC40 and FC40pPL376, with both strains the number of viable cells increased to a similar extent during the incubation despite the presence of an excess of scavenger bacteria. After 4 days the number of FC40 bacteria had increased by a factor of 3.24 ± 0.73, and the number of FC40pPL376 by 3.92 ± 1.5. The reduction in number of Lac+ mutants cannot therefore, be attributed to a reduced number of viable bacteria on the plate.
To demonstrate that the effect of the plasmid was due to the synthesis of carotenoids we generated plasmid pART53 by deleting around a kilobase of DNA presumably including promoter sequences. Strain FC40 carrying pART53 was not yellow and showed an adaptive mutation response similar to FC40 and clearly different from FC40pPL376 (Fig. 1b). The residual growth of both strains was similar; the viable count of FC40pPL376 had increased by a factor of 3.9 ± 0.7 and of FC40pART53 by a factor of 4.05 ± 0.69 by day 4.
A further control experiment using only pPL376 was possible because the carotenoid synthesising genes are under the control of the stationary phase sigma factor produced by the rpoS gene. Strains carrying rpoS mutations fail to induce a large number of genes involved in survival and cell turnover under starvation conditions (for review see [28]). An rpoS mutation was introduced into FC40 (to give CM1423) and into FC40pPL376 (to give CM1424). The latter strain was not yellow indicating that the carotenoid synthesising genes were not significantly expressed. As might be expected, both strains showed less residual growth on lactose plates (an increase in viable count of around two-fold over 6 days) and this was reflected in a reduced rate of adaptive mutation per plate compared to FC40. There was, however, no significant difference in the rate of adaptive mutation between the two strains (Fig. 1c), consistent with the effect of pPL376 in FC40 being attributable to synthesis of carotenoids and not to any other property of the plasmid.
The most obvious property of carotenoids to account for their effect is their ability to scavenge oxidative species. Nevertheless in principle some other unknown property affecting mutation expression might have been responsible for the lowering of the rate of adaptive mutation. Results from experiments which measure the mutation rate under non-selective conditions indicate that an effect on mutation expression is unlikely. Classical fluctuation experiments were carried out in which the bacteria were grown for 24 h in glycerol-thiamine-minimal medium and then plated on lactose plates for estimation of the number of lac+ mutants in each tube able to give rise to colonies in 2 days. Genes whose expression is controlled by rpoS become induced as the culture passes from the late logarithmic to the early stationary phase of growth. At the time of plating the carotenoid synthesising genes were induced as indicated by the yellow colour of the culture. In three experiments the mutation rates were estimated by the method of Lea and Coulson [29] to be 6.48 × 10−9 for FC40 and 6.41 × 10−9 for FC40pPL376. The presence of the carotenoids therefore, did not detectably influence either the expression of mutations under these non-selective conditions or the mutation rate during the last two or three cell cycles when carotenoids were present.
3.2. Induction of Lac+ mutants by oxidative damage
So far we have evidence consistent with a proportion of the mutations that emerge under lactose selection conditions being a consequence of oxidative damage. We next sought to establish whether lactose utilising mutants in FC40 could be induced by singlet oxygen, the species chiefly scavenged by carotenoids. Preliminary attempts to generate singlet oxygen photodynamically were unsuccessful since the concentrations of exogenous photosensitiser that were thought (on the basis of experiments with extracellular DNA and phages) to be needed to generate singlet oxygen were toxic to the cells. We therefore, engineered the endogenous production of the photosensitiser protoporphyrin by transducing in the hemH1 mutation [30] which specifies a defect in ferrochelatase (EC 4.99.1.1), the enzyme that inserts iron into protoporphyrin IX to make heme. The bacteria accumulate endogenous protoporphyrin and become hypersensitive to light due to the photodynamic generation of active oxygen species including singlet oxygen [31].
Demonstrating the induction of mutants in FC40 by any possible mutagen, including active oxygen species produced by photodynamic action, is not straightforward since FC40 derivatives can utilise contaminants in lactose selection plates and growth continues at a slow rate after plating. This has the advantage that there should be no impediment to the expression of any induced mutations but magnifies the propensity of this strain for continuously giving rise to Lac+ mutants when incubated on lactose selection plates (i.e. the phenomenon of “adaptive” mutation). Leaky growth is controlled in conventional “adaptive” mutation experiments by incorporating an excess of scavenger bacteria, but we feared that this might inhibit the expression of newly-induced mutations. We had, however, observed that the phenomenon of adaptive mutation is less marked at lower temperatures and we utilised this property to minimise the number of spontaneous (“adaptive”) mutants relative to any that might be induced. After exposing to light and allowing 2 days incubation in the dark at 37° for any induced mutations to become expressed, plates were placed on the bench (at around 25°) in the dark to allow newly expressed mutants to grow into visible colonies.
Fig. 2a shows a representative experiment with the protoporphyrin-containing strain CM1461 in which the number of mutants appearing on plates initially loaded with a similar number of bacteria and exposed to light for various periods of time is shown with increasing number of days incubation. At day 2 the mutants that are visible are largely those that were pre-existing in the culture at the time the experiment was begun. From day 3 onwards “adaptive” mutants began to appear, but there were always more mutants on plates loaded with bacteria that had been exposed to light and the difference was still increasing at day 5. While some slower-growing mutants induced by light almost certainly fail to appear in 5 days, beyond this time the number of spontaneous mutants becomes too great for reliable counting. Light was not mutagenic to the parental strain FC40 under the same conditions. In three experiments the data for exposed and unexposed bacteria were so close that they could not be clearly displayed graphically; data from a representative experiment are shown instead in tabular form (Table 2).
Fig. 2.

Appearance of mutants on lactose selection plates in CM1461, a hemH1 derivative of FC40 overexpressing protoporphyrin IX, following exposure to fluorescent light for 0, 10, 20, 30, or 40 min. The first 2 days were at 37°C, the subsequent 3 days on the bench (approximately 25°C). (a) Panel shows raw data from a representative experiment; (b) panel shows the number of mutants on plates of exposed bacteria minus the number on plates of unexposed bacteria.
Table 2.
Appearance of Lac+ mutants on lactose selection plates of FC40 following exposure to fluorescent lighta
| Exposure to light (minutes) | Viable bacteria plated | Mutants per plate |
|||
|---|---|---|---|---|---|
| Day 2 | Day 3 | Day 4 | Day 5 | ||
| 0 | 1.74 × 109 | 23.8 | 45.5 | 73.5 | 100 |
| 15 | 1.86 × 109 | 19.8 | 44.5 | 74.5 | 108 |
| 30 | 1.74 × 109 | 15.8 | 44.3 | 70.8 | 102 |
The first 2 days were at 37°, the subsequent 3 days on the bench (approximately 25°) (representative experiment).
In Fig. 2b the difference between the number of mutants on plates with light-exposed bacteria and the number on plates of unexposed bacteria is plotted for CM1461 as a function of time of incubation. We have taken the difference between the exposed and unexposed counts on day 5 as being the number of induced mutants, albeit an underestimate. Fig. 3 shows results from three experiments with CM1461 in which the data are shown as induced mutant frequencies per 108 viable bacteria plated as a function of time of illumination. The data clearly demonstrate that exposure to light induces Lac+ mutations in CM1461, the hemH1 derivative of FC40.
Fig. 3.
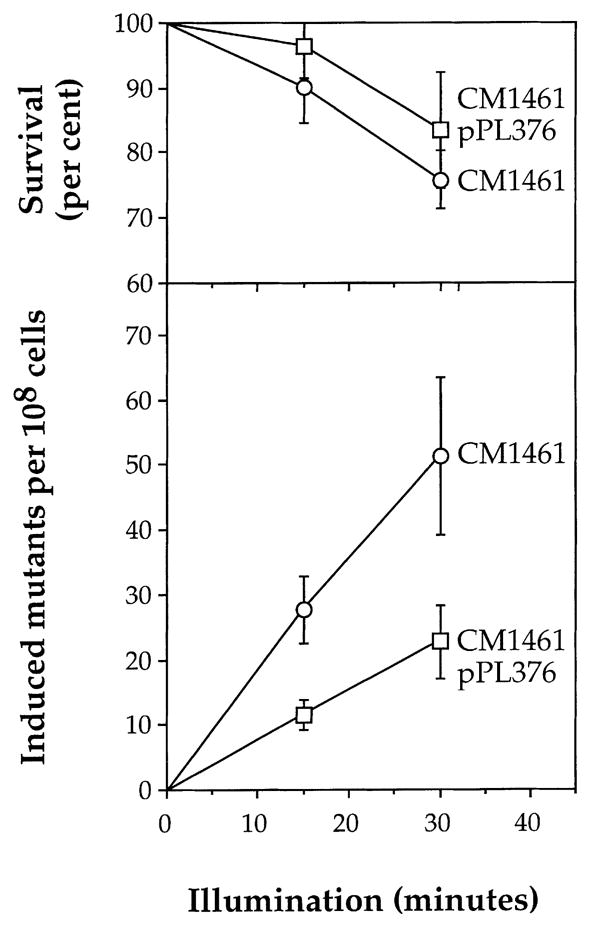
Survival and induction of Lac+ mutants in CM1461 (circles), and a derivative containing pPL376 (squares) as a function of time of exposure to fluorescent light. Mutation data adjusted to 108 viable cells plated. Each point is the mean and standard error of three experiments.
In other systems where damage caused by active oxygen species has been implicated, including that mediated by endogenous porphyrins, it has been found that intracellular carotenoids can reduce the effect [19,32,33]. Plasmid pPL376, conferring the ability to synthesise carotenoids was therefore, introduced into CM1461. It can be seen from Fig. 3 that exposure to light also increases the number of Lac+ mutants appearing in strain CM1461pPL376 but to a lesser extent than in CM1461; the effectiveness of light was reduced by more than a factor of two in the presence of carotenoids expressed by pPL376. The mean reduction calculated for all points was by 55.4 ± 7.4%.
To eliminate the trivial explanation that the carotenoids were physically shielding the DNA by absorbing incident light, we examined their effect with 254 nm UV light. At this wavelength absorption by carotenoids is comparable to that at around 400 nm, which is the active part of the spectrum for the photodynamic action of protoporphyrin IX. At 254 nm however, the phototoxicity is mediated not by active oxygen species but by photoproducts formed directly in the DNA. There was no protective effect of pPL376 carotenoids against the lethal action of 254 nm UV (Fig. 4). Thus, we can conclude that active oxygen species such as photodynamically produced singlet oxygen can indeed generate reversions to Lac+ in strain FC40 and that their effect can be blocked by endogenous carotenoids.
Fig. 4.

Survival of CM1461 and its carotenoid-containing derivative CM1464pPL376 following exposure to 254 nm UV light. Cell density approximately 2 × 109 per ml (representative experiment).
3.3. Effect of DNA repair mutations on adaptive mutation
In other adaptive mutation systems mutations have been attributed to the accumulation of 7,8-dihydro-8-oxoguanine (8-oxoG), a lesion produced in DNA by active oxygen species [22]. Derivatives of FC40 were constructed with mutations in the mutY and mutM genes whose products are involved in the response to 8-oxoG. These strains proved to be indistinguishable from FC40 in the rate at which Lac+ mutants appeared on selective plates (Fig. 5). There is thus, no evidence that 8-oxoG is involved in adaptive mutation in FC40.
Fig. 5.
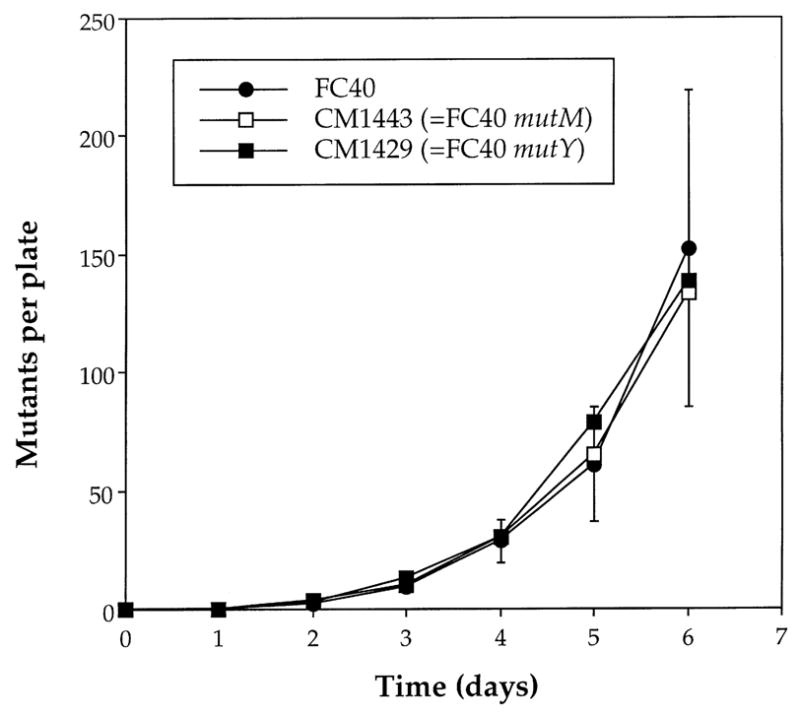
Adaptive mutation in FC40 and derivatives carrying mutations in mutM (CM1443) or mutY (CM1429). Means and standard errors of three experiments.
4. Discussion
The key observation in the present paper is that the rate of adaptive mutation in FC40 is reduced by a factor of around two when the bacteria contain endogenous carotenoids. What is the most likely explanation for this effect? There is now abundant evidence that one of the chief functions of carotenoid pigments in living things is to protect against active oxygen species (for review see [34]). The ability of carotenoids to quench singlet oxygen is well known [35–38] and appears to be responsible for their protective action in bacteria [28]. On the basis of currently available evidence, therefore, scavenging of singlet oxygen seems the most plausible explanation of the effect of carotenoids in the present experiments and the data are consistent with the hypothesis that oxidative species may be involved in the formation of adaptive mutations in FC40.
The effect of endogenous carotenoids in decreasing the rate of episomal adaptive mutation in FC40 is similar to that observed with chromosomal mutations from amino acid auxotrophy in other strains [19] where it appears that oxidative DNA damage accumulation within the bacteria is a major contributor to “adaptive” mutation [15,39]. Thus, strains lacking certain enzymes (the products of the mutY and mutM genes) that deal with the presence of the miscoding base 7,8-dihydro-8-oxoguanine (8-oxoG) in DNA are mutators under starvation conditions and overproduction of these enzymes leads to a reduction in the rate of “adaptive” mutation [22,40]. Derivatives of FC40 carrying mutations in mutY and mutM did not, however, show a mutator effect for adaptive mutation so that, in contrast to the chromosomal mutations to prototrophy that have been studied, the hypothetical lesion responsible for the episomal frameshift mutations in FC40 cannot be 8-oxoG or an 8-oxoG:adenine mismatched pair.
This is consistent with earlier work by van den Akker et al. who showed that when single-stranded M13 DNA was damaged by singlet oxygen and transfected into E. coli the predominant class of mutations generated in the lacZα gene comprised –G deletions and most of these were in two mutational hotspots [17]. These authors concluded that singlet oxygen gives rise to a guanine lesion probably without base-pairing ability (and therefore, not 8-oxoG) that can be bypassed by DNA polymerase at the cost of a one-base deletion. A very similar conclusion was reached by Wagner and Fuchs [41] working with double-stranded DNA exposed to methylene blue and light which generates oxidative species including singlet oxygen. They also showed that 8-oxoG site-specifically located in a plasmid was inefficient in inducing frameshifts. There is thus, an unidentified lesion produced by oxidative species including singlet oxygen that might also be responsible for the –G:C frameshift mutations that arise in FC40 under lactose selection and that are prevented by the presence of carotenoids. It might be a degradation product of guanine or even an abasic site. There is, for example, independent evidence from an in vitro system that abasic sites can cause DNA polymerase III to generate −1 frameshifts [42].
Singlet oxygen can be produced by enzymic reactions within the cell and by lipid peroxidation (for review see [43–47]). It may also be produced by dismutation of superoxide [48]. In contrast to oxidative free radicals, singlet oxygen probably reacts largely with guanine, and 8-oxoG is a major, though not the only product [43,49]. If lesions generated by singlet oxygen and other active oxidative species are involved in adaptive mutation, it follows that it should be possible to induce Lac+ revertants in FC40 by treatments that generate such species. This expectation was confirmed by the experiments shown in Figs. 3 and 4 where active oxygen species including singlet oxygen were produced from endogenous protoporphyrin IX during exposure to light.
The FC40 system has had a high profile in the adaptive mutation story, but hitherto mechanistic speculations have focused upon the requirements for conjugational competence and recombination ability. It must be emphasised that there is nothing in the present results that is inconsistent with these requirements or is in any way incompatible with current models (cf. [4,7,10,50–54]). At the heart of all these models is the generation of mutations during recombination-driven episomal DNA replication. In the present experiments significant cell multiplication occurred during the 7 days incubation and it may be presumed that episomal replication also occurred. There is also evidence for amplification of the episomal region containing the lac gene in some cells under these conditions [54,55]. From other systems there is indirect evidence that DNA turnover may be greater under starvation conditions than had been earlier deduced from studies with exogenous label [56]. The question at issue is whether the mutations that arise are solely those due to polymerase infidelity. The present data suggest that there may also be a component due to spontaneous oxidative lesions accumulating in the DNA during starvation which eventually encounter a replication fork where they may trigger a slippage step resulting in the omission of one base in the newly synthesised strand. It would not be surprising if the propensity of different polymerases to give rise to −1 frameshifts at sites of damage were related to the probability of them doing the same at undamaged sites. The local base sequence and the nature of the lesion may also influence whether such a slippage step is more or less likely to occur than stalling of the polymerase or insertion of an erroneous base.
One consequence of polymerase stalling under starvation conditions might be that the DNA would come apart in the region of the replication fork, effectively generating a double strand break. Although double strand DNA breaks could in principle also be formed directly by oxidative attack there is no evidence that they are a particularly common oxidative lesion. Nevertheless one could speculate that oxidative lesions could also give rise to the double strand DNA breaks that are an essential part of the models of Foster and Rosenberg and their collaborators (loc.cit.).
In summary, the evidence presented suggests that adaptive mutation in FC40 may be similar to stationary phase mutation in a number of other systems in that it includes a component due to DNA damage (largely oxidative in nature) (cf. [15]). This is consistent with the recently established involvement in adaptive mutation in FC40 of the DNA damage-inducible SOS system [13] and the SOS-controlled DNA polymerase IV (H. Ohmori and P.L. Foster, unpublished observations).
Acknowledgments
We thank Dr. B. Weiss for sending us strain AW804 containing hemH1 zbb-3055::Tn10 and Dr. A. Eisenstark for sending us plasmid pPL376.
References
- 1.Cairns J, Overbaugh J, Miller S. The origin of mutants. Nature. 1988;335:142–145. doi: 10.1038/335142a0. [DOI] [PubMed] [Google Scholar]
- 2.Foster PL. Adaptive mutation: the uses of adversity. Annu Rev Microbiol. 1993;47:467–504. doi: 10.1146/annurev.mi.47.100193.002343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cairns J, Foster PL. Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics. 1991;128:695–701. doi: 10.1093/genetics/128.4.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris RS, Longerich S, Rosenberg SM. Recombination in adaptive mutation. Science. 1994;264:258–260. doi: 10.1126/science.8146657. [DOI] [PubMed] [Google Scholar]
- 5.Foster PL, Trimarchi JM. Adaptive reversion of a episomal frameshift mutation in Escherichia coli requires conjugal functions but not actual conjugation. Proc Natl Acad Sci USA. 1995;92:5487–5490. doi: 10.1073/pnas.92.12.5487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Foster PL, Trimarchi JM. Conjugation is not required for adaptive reversion of an episomal frameshift mutation in Escherichia coli. J Bacteriol. 1995;177:6670–6671. doi: 10.1128/jb.177.22.6670-6671.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radicella JP, Park PU, Fox MS. Adaptive mutation in Escherichia coli: a role for conjugation. Science. 1995;268:418–420. doi: 10.1126/science.7716545. [DOI] [PubMed] [Google Scholar]
- 8.Galitski T, Roth JR. Evidence that F plasmid transfer replication underlies apparent adaptive mutation. Science. 1995;268:421–423. doi: 10.1126/science.7716546. [DOI] [PubMed] [Google Scholar]
- 9.Foster PL. Non-adaptive mutations occur on the F′ episome during adaptive mutation conditions in Escherichia coli. J Bacteriol. 1997;179:1550–1554. doi: 10.1128/jb.179.5.1550-1554.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Torkelson J, Harris RS, Lombardo MJ, Nagendran J, Thulin C, Rosenberg SM. Genome-wide hypermutation in a sub-population of stationary-phase cells underlies recombination-dependent adaptive mutation. EMBO J. 1997;16:3303–3311. doi: 10.1093/emboj/16.11.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Foster PL, Gudmundsson G, Trimarchi JM, Cai H, Goodman MF. Proofreading-defective DNA polymerase II increases adaptive mutation in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:7951–7955. doi: 10.1073/pnas.92.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harris RS, Bull HJ, Rosenberg SM. A direct role of DNA polymerase III in adaptive reversion of a frameshift mutation in Escherichia coli. Mutat Res. 1997;375:19–24. doi: 10.1016/s0027-5107(96)00244-8. [DOI] [PubMed] [Google Scholar]
- 13.McKenzie GJ, Harris RS, Lee PL, Rosenberg SM. The SOS response regulates adaptive mutation. Proc Natl Acad Sci USA. 2000 doi: 10.1073/pnas.120161797. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Escarceller M, Hicks J, Cudmundsson G, Trump G, Tonat D, Lovett S, Foster PL, McEntee K, Goodman MF. Involvement of Escherichia coli DNA polymerase II in response to oxidative damage and adaptive mutation. J Bacteriol. 1994;176:6221–6228. doi: 10.1128/jb.176.20.6221-6228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bridges BA. The role of DNA damage in stationary phase (‘adaptive’) mutation. Mutat Res. 1998;408:1–9. doi: 10.1016/s0921-8777(98)00008-1. [DOI] [PubMed] [Google Scholar]
- 16.Decuyper-Debergh D, Piette J, Van de Vorst A. Singlet oxygen-induced mutations in M13 lacZ phage DNA. EMBO J. 1987;6:3155–3161. doi: 10.1002/j.1460-2075.1987.tb02626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van den Akker E, Lutgerink JT, Lafleur MVM, Joenje H, Retel J. The formation of one –G deletions as a consequence of singlet-oxygen-induced DNA damage. Mutat Res. 1994;309:45–52. doi: 10.1016/0027-5107(94)90041-8. [DOI] [PubMed] [Google Scholar]
- 18.Tuveson RW, Sandmann G. Protection by cloned carotenoid genes expressed in Escherichia coli against phototoxic molecules activated by near-UV light. Meth Enzymol. 1993;214:323–330. doi: 10.1016/0076-6879(93)14075-t. [DOI] [PubMed] [Google Scholar]
- 19.Bridges BA, Timms AR. Effect of endogenous carotenoids and defective RpoS sigma factor on spontaneous mutation under starvation conditions in Escherichia coli: evidence for the possible involvement of singlet oxygen. Mutat Res. 1998;403:21–28. doi: 10.1016/s0027-5107(98)00013-x. [DOI] [PubMed] [Google Scholar]
- 20.Calos MP, Miller JH. Genetic and sequence analysis of frameshift mutations induced by ICR-191. J Mol Biol. 1981;153:39–66. doi: 10.1016/0022-2836(81)90525-8. [DOI] [PubMed] [Google Scholar]
- 21.Zambrano MM, Siegele DA, Almiron M, Jormo A, Kolter R. Microbial competition: Escherichia coli mutants that take over stationary phase cultures. Science. 1993;259:1757–1760. doi: 10.1126/science.7681219. [DOI] [PubMed] [Google Scholar]
- 22.Bridges BA, Sekiguchi M, Tajiri T. Effect of mutY and mutM/fpg-1 mutations on starvation-associated mutation in Escherichia coli: implications for the role of 7, 8-dihydro-8-oxoguanine. Mol Gen Genet. 1996;251:352–357. doi: 10.1007/BF02172526. [DOI] [PubMed] [Google Scholar]
- 23.Perry KL, Simonitch TA, Harrison-Lavoie KJ, Liu ST. Cloning and regulation of Erwinia herbicola pigment genes. J Bacteriol. 1986;168:607–612. doi: 10.1128/jb.168.2.607-612.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller JH. Experiments in Molecular Genetics. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1972. [Google Scholar]
- 25.Boyle JM, Symonds N. Radiation sensitive mutants of T4D.1. T4y: a new radiation sensitive mutant, effect of the mutation on radiation survival growth and recombination. Mutat Res. 1969;8:431–459. doi: 10.1016/0027-5107(69)90060-8. [DOI] [PubMed] [Google Scholar]
- 26.Davis BD, Mingioli ES. Mutants of Escherichia coli requiring methionine or vitamin B12. J Bacteriol. 1950;60:17–28. doi: 10.1128/jb.60.1.17-28.1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bridges BA. Starvation-associated mutation in Escherichia coli strains defective in transcription repair coupling factor. Mutat Res. 1995;329:49–56. doi: 10.1016/0027-5107(95)00016-c. [DOI] [PubMed] [Google Scholar]
- 28.Eisenstark A, Calcutt MJ, Becker-Hapak M, Ivanova A. Role of Escherichia coli rpoS and associated genes in defense against oxidative damage. Free Rad Biol Med. 1996;21:975–993. doi: 10.1016/s0891-5849(96)00154-2. [DOI] [PubMed] [Google Scholar]
- 29.Lea DE, Coulson CA. The distribution of numbers of mutants in bacterial populations. J Genetics. 1949;49:264–285. doi: 10.1007/BF02986080. [DOI] [PubMed] [Google Scholar]
- 30.Yang H, Inokuchi H, Adler J. Phototaxis away from blue light by an Escherichia coli mutant accumulating protoporphyrin IX. Proc Natl Acad Sci USA. 1995;92:7332–7336. doi: 10.1073/pnas.92.16.7332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guo G, Weiss B. Endonuclease V (nfi) mutant of Escherichia coli K-12. J Bacteriol. 1998;180:46–51. doi: 10.1128/jb.180.1.46-51.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burchard RP, Dworkin M. Light-induced lysis and carotenogenesis in Myxococus xanthus. J Bacteriol. 1966;91:535–545. doi: 10.1128/jb.91.2.535-545.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burchard RP, Gordon SA, Dworkin M. Action spectrum for the photolysis of Myxococcus xanthus. J Bacteriol. 1966;91:896–897. doi: 10.1128/jb.91.2.896-897.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Edge R, McGarvey DJ, Truscott TG. The carotenoids as anti-oxidants: a review. J Photochem Photobiol B: Biol. 1997;41:189–200. doi: 10.1016/s1011-1344(97)00092-4. [DOI] [PubMed] [Google Scholar]
- 35.Foote CS, Denny RW. Chemistry of singlet oxygen. VIII. Quenching by β-carotene. J Am Chem Soc. 1968;90:6233–6235. [Google Scholar]
- 36.Conn PF, Schalch W, Truscott TG. The singlet oxygen and carotenoid interaction. J Photochem Photobiol B: Biol. 1991;11:41–47. doi: 10.1016/1011-1344(91)80266-k. [DOI] [PubMed] [Google Scholar]
- 37.Olive PL. DNA precipitation assay: a rapid and simple method for detecting DNA damage in mammalian cells. Env Mol Mutagen. 1988;11:487–495. doi: 10.1002/em.2850110409. [DOI] [PubMed] [Google Scholar]
- 38.Tinkler JH, Bohm F, Schalch W, Truscott TG. Dietary carotenoids protect human cells from damage. J Photochem Photobiol B: Biol. 1994;26:283–285. doi: 10.1016/1011-1344(94)07049-0. [DOI] [PubMed] [Google Scholar]
- 39.Bridges BA. Mutation in resting cells: the role of endogenous DNA damage. In: Lindahl T, editor. Genetic Instability in Cancer. ICRF/Cold Spring Harbor Press; 1996. pp. 155–167. [PubMed] [Google Scholar]
- 40.Bridges BA. mutY ‘directs’ mutation? Nature. 1995;375:741–741. doi: 10.1038/375741a0. [DOI] [PubMed] [Google Scholar]
- 41.Wagner J, Fuchs RPP. Frameshift mutagenesis induced in Escherichia coli after in vitro treatment of double-stranded DNA with methylene blue plus white light: evidence for the involvement of lesion(s) other than 8-oxo-7, 8-dihydro-2′-deoxyguanosine. Chem Res Toxicol. 1997;10:568–574. doi: 10.1021/tx960169j. [DOI] [PubMed] [Google Scholar]
- 42.Reuven NB, Tomer G, Livneh Z. The mutagenesis proteins UmuD′ and UmuC prevent lethal frameshifts while increasing base substitution mutations. Mol Cell. 1998;2:191–199. doi: 10.1016/s1097-2765(00)80129-x. [DOI] [PubMed] [Google Scholar]
- 43.Epe B. Genotoxicity of singlet oxygen. Chem Biol Interactions. 1991;80:239–260. doi: 10.1016/0009-2797(91)90086-m. [DOI] [PubMed] [Google Scholar]
- 44.Joenje H, Lafleur VM, Retel J. Biological consequences of oxidative DNA damage. In: Vigo-Pelfrey C, editor. Membrane Lipid Oxidation. CRC Press; Boca Raton: 1991. pp. 87–113. [Google Scholar]
- 45.Piette J. Mutagenic and genotoxic properties of singlet oxygen. Photochem Photobiol. 1990;4:335–339. doi: 10.1016/1011-1344(90)85039-y. [DOI] [PubMed] [Google Scholar]
- 46.Kanofsky JR. Singlet oxygen production by biological systems. Chem Biol Interactions. 1989;70:1–28. doi: 10.1016/0009-2797(89)90059-8. [DOI] [PubMed] [Google Scholar]
- 47.Naqui A, Chance B, Cadenas E. Reactive oxygen intermediates in biochemistry. Annu Rev Biochem. 1986;55:137–166. doi: 10.1146/annurev.bi.55.070186.001033. [DOI] [PubMed] [Google Scholar]
- 48.Khan AU. Singlet molecular oxygen from superoxide anion and sensitised fluorescence of organic molecules. Science. 1970;168:476–477. doi: 10.1126/science.168.3930.476. [DOI] [PubMed] [Google Scholar]
- 49.Lutgerink JT, Van den Akker E, Smeets I, Pachen D, Van Dijk P, Aubry JM, Joenje H, Lafleur MVM, Retel J. Interaction of singlet oxygen with DNA and biological consequences. Mutat Res. 1992;275:377–386. doi: 10.1016/0921-8734(92)90040-v. [DOI] [PubMed] [Google Scholar]
- 50.Foster PL, Trimarchi JM. Adaptive reversion of a frameshift mutation in Escherichia coli by simple base deletions in homopolymeric runs. Science. 1994;265:407–409. doi: 10.1126/science.8023164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foster PL, Trimarchi JM, Maurer RA. Two enzymes, both of which process recombination intermediates, have opposite effects on adaptive mutation in Escherichia coli. Genetics. 1996;142:25–37. doi: 10.1093/genetics/142.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rosenberg SM, Harris RS, Longerich S, Galloway AM. Recombination-dependent mutation in non-dividing cells. Mutat Res. 1996;350:69–76. doi: 10.1016/0027-5107(95)00092-5. [DOI] [PubMed] [Google Scholar]
- 53.Foster PL. Adaptive mutation: has the unicorn landed? Genetics. 1998;148:1453–1459. doi: 10.1093/genetics/148.4.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Andersson DI, Slechta ES, Roth JR. Evidence that gene amplification underlies adaptive mutability of the bacterial lac operon. Science. 1998;282:1133–1135. doi: 10.1126/science.282.5391.1133. [DOI] [PubMed] [Google Scholar]
- 55.Foster PL. Population dynamics of a Lac+ strain of Escherichia coli during selection for lactose utilisation. Genetics. 1994;138:253–261. doi: 10.1093/genetics/138.2.253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bridges BA, Ereira S. DNA synthesis and viability of a mutT derivative of Escherichia coli WP2 under conditions of amino acid starvation and relation to stationary-phase (‘adaptive’) mutation. J Bacteriol. 1998;180:2906–2910. doi: 10.1128/jb.180.11.2906-2910.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]