

Abstract
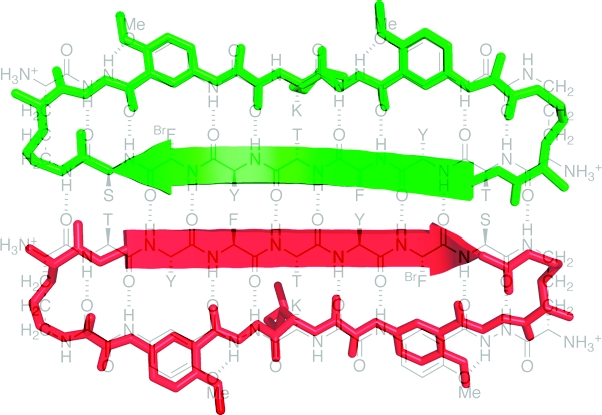
This paper describes the X-ray crystallographic structure of a designed cyclic β-sheet peptide that forms a well-defined hydrogen-bonded dimer that mimics β-sheet dimers formed by proteins. The 54-membered ring macrocyclic peptide (1a) contains molecular template and turn units that induce β-sheet structure in a heptapeptide strand that forms the dimerization interface. The X-ray crystallographic structure reveals the structures of the two “Hao” amino acids that help template the β-sheet structure and the two δ-linked ornithine turn units that link the Hao-containing template to the heptapeptide β-strand. The Hao amino acids adopt a conformation that resembles a tripeptide in a β-strand conformation, with one edge of the Hao unit presenting an alternating array of hydrogen-bond donor and acceptor groups in the same pattern as that of a tripeptide β-strand. The δ-linked ornithines adopt a conformation that resembles a hydrogen-bonded β-turn, in which the ornithine takes the place of the i+1 and i+2 residues. The dimers formed by macrocyclic β-sheet 1a resemble the dimers of many proteins, such as defensin HNP-3, the λ-Cro repressor, interleukin 8, and the ribonuclease H domain of HIV-1 reverse transcriptase. The dimers of 1a self-assemble in the solid state into a barrel-shaped trimer of dimers in which the three dimers are arranged in a triangular fashion. Molecular modeling in which one of the three dimers is removed and the remaining two dimers are aligned face-to-face provides a model of the dimers of dimers of closely related macrocyclic β-sheet peptides that were observed in solution.
Introduction
The formation of dimers through edge-to-edge interaction among β-sheets is a fundamental mode of protein interaction. Homodimers with β-sheet dimerization interfaces occur widely in the Protein Data Bank and are often formed from monomers that present an exposed β-strand. In these structures, the monomers pair symmetrically through antiparallel β-sheet formation; the topology of β-strands does not permit symmetrical pairing through parallel β-sheet formation in a protein with only one hydrogen-bonding edge of the β-strand exposed. Figure 1 illustrates the structures of four such dimers,1−4 which were selected from the Interchain β-Sheet (ICBS) Database.(5)
Figure 1.

Structures of β-sheet dimers: defensin HNP-3 (upper left, PDB ID 1DFN),(1) the λ-Cro repressor (upper right, PDB ID 1COP),(2) interleukin 8 (lower left, PDB ID 1IL8),(3) and the ribonuclease H domain of HIV-1 reverse transcriptase (lower right, PDB ID 1HRH).(4)
In contrast to the dimers of folded proteins, peptides and unfolded proteins often form aggregates consisting of infinite networks of β-sheets.6,7 Many of these aggregates consist of parallel, in-register β-sheets, which can form when both hydrogen-bonding edges of a peptide strand are exposed. Further interactions among the faces of the resulting β-sheets lead to layered structures in which tightly self-complementing “steric zippers” bind together the faces of the paired β-sheets.(6)
We recently reported macrocyclic β-sheet peptides that dimerize through antiparallel edge-to-edge interaction among β-sheets.(8) The macrocyclic peptides (1) consist of a 54-membered ring comprising a heptapeptide “upper” β-strand, two δ-linked ornithine turn units, and a “lower” β-strand mimic comprising two “Hao” amino acids that mimic a tripeptide β-strand and one additional amino acid.9−11 The Hao units help pre-organize the “upper” β-strand while blocking the “lower” edge and preventing the formation of an infinite network. Unless stabilized by intermolecular disulfide linkages between the R1 and R7 positions, the structures do not exist as isolated dimers in aqueous solution.(12) Instead, the dimers (1·1) self-assemble through hydrophobic face-to-face interactions to form tetramers, comprising dimers of dimers, or related higher-order assemblies.
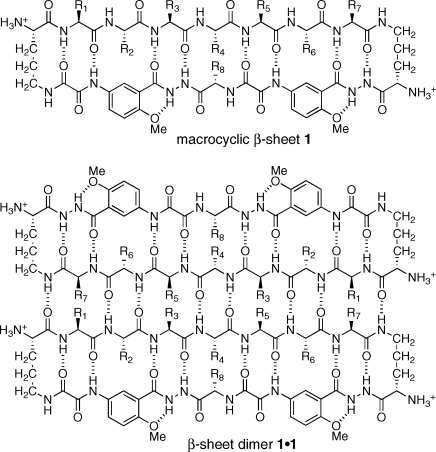
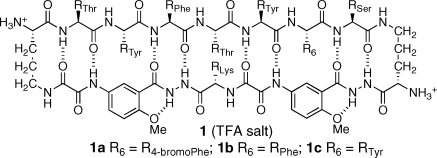
In this paper, we report the X-ray crystallographic structure of the β-sheet dimer of macrocyclic β-sheet peptide 1a. This compound is a close homologue of macrocyclic β-sheet peptides 1b and 1c, which we described in our original report.(8) In homologue 1a, the phenylalanine or tyrosine at the R6 position of 1b or 1c is replaced with 4-bromophenylalanine to facilitate X-ray diffraction studies. The sequence of the heptapeptide strand (R1−R7) of these macrocycles is loosely based upon that of the exposed β-sheet edge (TTFTYTT) of the redesigned protein G variant NuG2, which crystallizes as a β-sheet dimer.(13)
Results
Macrocycle 1a was prepared by synthesis of the corresponding linear peptide on chlorotrityl resin followed by cleavage of the protected linear peptide from the resin, cyclization of the linear peptide in solution, deprotection, and RP-HPLC purification.(8) Crystals were grown by the sitting drop vapor diffusion method from 30% aqueous isopropanol solution with HEPES buffer and MgCl2. X-ray diffraction data were collected with synchrotron radiation using the multi-wavelength anomalous dispersion (MAD) technique. The incorporation of 4-bromophenylalanine in 1a afforded the opportunity to solve the phase problem by performing a MAD experiment, commonly employed in protein crystallography. X-ray diffraction data were collected using three wavelengths for MAD phasing. A second crystal was used to collect a high-resolution data set. Refinement was performed to 1.35 Å resolution with the inclusion of 264 molecules of water and six molecules of isopropanol. Table 1 summarizes the data collection, phasing, and refinement statistics.
Table 1. Data Collection, Phasing, and Refinement Statistics for Macrocycle 1a.
| crystal used for refinement | crystal used for MAD phasing | |||
|---|---|---|---|---|
| Data Collection | ||||
| space group | P212121 | P212121 | ||
| cell dimensions | ||||
| a, b, c (Å) | 64.05, 64.19, 64.25 | 64.04, 64.01, 64.00 | ||
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | ||
| peak | inflection | remote | ||
| wavelength (Å) | 0.9785 | 0.9200 | 0.9205 | 0.8670 |
| resolution (Å) | 1.35 | 2.0 | 2.0 | 2.0 |
| Rmergea | 0.077 (0.453) | 0.067 (0.090) | 0.063 (0.089) | 0.056 (0.079) |
| I/σI | 19.2 (3.3) | 33.2 (27.8) | 34.2 (27.6) | 40.1 (37.7) |
| completeness (%)a | 95.1 (99.9) | 99.1 (94.5) | 99.1 (94.4) | 99.7 (99.6) |
| redundancya | 6.9 (5.9) | 14.5 (13.6) | 14.5(13.6) | 14.6 (14.6) |
| Refinement | ||||
| resolution (Å) | 1.35 | |||
| no. reflections | 53 252 | |||
| Rwork/Rfree | 0.150/0.157 | |||
| no. atoms | ||||
| macrocycle | 1537 | |||
| isopropanol | 24 | |||
| water | 264 | |||
| B-factors (Å2) | ||||
| macrocycle | 14.5 | |||
| ligand/ion | 25.8 | |||
| water | 32.7 | |||
| rms deviations | ||||
| bond lengths (Å) | 0.008 | |||
| bond angles (°) | 1.8 | |||
Highest resolution shell is shown in parentheses.
Figure 2 illustrates the X-ray crystallographic structure of the dimers of 1a. There are six crystallographically distinct dimers in the asymmetric unit. Structural deviation among the dimers is small, with <0.7 Å rms deviation among all pairwise comparisons of the dimer coordinates. The largest deviations throughout the structures involve conformations of the 4-bromophenylalanine (R7) and lysine (R8) side chains. Two conformations of the R2 tyrosine side chain are observed in one of the 12 monomer units, and two conformations of the R6 tyrosine side chain are observed in another of the 12 monomer units.(14) The four-stranded β-sheet comprising the dimer exhibits a gentle twist, in a fashion characteristic of protein β-sheets.(15)
Figure 2.

X-ray crystallographic structure of the dimer of 1a. Cartoon (A) and atomic (B) representations of a representative dimer; overlay of the six dimers in the asymmetric unit (C). Polar contacts corresponding to main-chain hydrogen bonds are shown as magenta dashes in panel B; polar contacts corresponding to side-chain hydrogen bonds are shown as yellow dashes in panel B.
Each dimer exhibits intra- and intermolecular hydrogen bonding characteristic of antiparallel β-sheets. A network of hydrogen bonds runs among the four peptide main chains comprising the dimer and is shown as magenta dashes in Figure 2B. The hydrogen bonds have N−O distances (2.8−3.0 Å) and geometries typical of β-sheets. Eight hydrogen bonds bridge the main chains of the dimer interface. All hydrogen-bond donors and acceptors of the “inner” two peptide main chains are satisfied, with the exception of the α-amino group of the ornithine residue, which forms hydrogen bonds with solvent water molecules. The “outer” two peptide main chains (those containing the Hao amino acids) form hydrogen bonds with the “inner” two peptide main chains. The outer edges of the Hao-containing peptide chains are solvent exposed. Ordered water molecules (not shown in Figure 2) form hydrogen bonds with the exposed backbone oxygens and nitrogens on these edges. Three hydrogen bonds are formed between the threonine and serine side chains at the dimer interface and are shown as yellow dashes in Figure 2B. Hydrogen-bond donors and acceptors on the threonine and serine side chains also interact with bulk solvent (which accounts for 59% of the crystal volume), as do those on the tyrosine and lysine side chains.
The six dimers in each asymmetric unit comprise two loosely packed trimers of dimers. The trimers of dimers are virtually identical in structure and have minimal contact within the lattice.(16) Figure 3 illustrates the structure of one of the two trimers. In each trimer, the three dimers are arranged in a triangular fashion. The R3 and R5 phenylalanine and tyrosine side chains point inward to create a loose hydrophobic core of the trimer. The R1 and R7 serine and threonine side chains also point inward and create hydrophilic caps to the core. The R2, R4, R6, and R8 tyrosine, threonine, 4-bromophenylalanine, and lysine side chains point outward. An unusual feature of this barrel-like structure is the large volume of the cavity at center of the barrel (1033 Å3).(17)
Figure 3.

X-ray crystallographic structure of the trimer of dimers of 1a. The asymmetric unit contains two trimers of dimers, which are virtually identical in structure.
The interface between each dimer within the trimer is stabilized by aromatic edge-to-face interactions of the R3 phenylalanine and R5 tyrosine side chains and van der Waals contacts with the Hao moieties. The sum of the area buried by both surfaces in this interface is 820 Å2, and the shape complementarity correlation Sc of the interface is 0.817.(18) The instability afforded by the creation of a void in the center of the barrel-like structure appears to be compensated by the amount of surface area buried by interfaces between the dimers and the high degree of surface complementarity.
Discussion
The controlled self-assembly of designed peptides, proteins, and peptide analogues has emerged as a major theme in the broadly defined areas of supramolecular chemistry and foldamers. The development of strategies that permit the creation of biomimetic and bio-inspired structures that fold and self-assemble like proteins permits the rational design of synthetic assemblies that function like the biomolecular assemblies in living organisms. In cases of rigid, conformationally constrained peptides for which the solid and solution-state structures are not expected to differ appreciably, X-ray crystallography provides a method of structure determination that is complementary to solution-state NMR spectroscopy.(19)
The X-ray crystallographic structure of macrocyclic β-sheet peptide 1a provides direct insight into the structures of the peptidomimetic δ-linked ornithine and Hao building blocks that our laboratory has developed and used extensively in the creation of compounds that mimic the structures and interactions of β-sheets.9,10,20 The δ-linked ornithine adopts a conformation that resembles a hydrogen-bonded β-turn, in which the ornithine takes the place of the i+1 and i+2 residues (Figure 4).10,21 The ornithine side chain adopts a well-defined conformation that resembles the chair conformer of a cyclohexane ring. In this conformation, the ornithine pro-S δ-hydrogen and α-hydrogen are in contact and occupy the positions that would be occupied by carbons in the cyclohexane ring.
Figure 4.

X-ray crystallographic structures of the δ-linked ornithine turn and Hao units of 1a. Hydrogen atoms have been added for clarity and were not determined crystallographically.
The Hao amino acid building block adopts a conformation that resembles a tripeptide in a β-strand conformation (Figure 4): the hydrazine and aromatic carboxamide groups occupy the position of the first amino acid of the tripeptide; the aromatic ring occupies the position of the second amino acid; and the oxalic and aromatic amino groups occupy the position of the third amino acid.(9) One edge of the Hao unit presents an alternating array of hydrogen-bond donor and acceptor groups in the same pattern as that of a tripeptide β-strand, while the other edge does not present this hydrogen-bonding pattern and is blocked by intramolecular hydrogen bonding and the aromatic ring. The blocking created by these features is reminiscent of the negative design that occurs in natural β-sheet proteins and prevents uncontrolled edge-to-edge interactions.(22)
The macrocyclic structure formed by 1a is similar to that of the θ-defensin peptides.(23) These cyclooctadecapeptides form hydrogen-bonded cyclic β-sheet structures that are reinforced by three disulfide bridges between the non-hydrogen-bonded residues of the macrocycles. Figure 5 illustrates the structures of the monomer subunit of macrocyclic β-sheet peptide 1a and the potent anti-HIV θ-defensin retrocyclin-2. In both structures, the main chain forms a 54-membered ring macrocycle. Interestingly, retrocyclin-2 forms trimers. The postulated barrel-like structure of these trimers is similar to the structure of the trimers of dimers observed for macrocyclic β-sheet peptide 1a (Figure 3).(23d)
Figure 5.

Comparison of the X-ray crystallographic structure of the monomer subunit of 1a (upper) to an NMR structure of the θ-defensin retrocyclin-2 (lower, PDB ID 2ATG). The NMR structure of retrocyclin-2 was determined in the presence of SDS micelles. Hydrogen atoms have been omitted from this structure for clarity.
Macrocycle 1a comprises 12 amino acids, in contrast to the 18 of the θ-defensin, because each Hao amino acid takes the place of three α-amino acids and each δ-linked ornithine takes the place of the two central amino acids of the β-turns. The Hao amino acids help reinforce a folded β-sheet structure in 1a, like the disulfide bridges help reinforce a β-sheet structure in the θ-defensins. Interactions between the electron-rich tyrosines at the R2 and R6 positions of 1a and the electron-deficient oxalic groups may contribute to this stabilization. The δ-linked ornithine units are particularly effective in forming β-hairpin structures and may also contribute to the stabilization.(10)
The dimers formed by macrocyclic β-sheet 1a resemble the dimers of many proteins, such as defensin HNP-3,(1) the λ-Cro repressor,(2) interleukin 8,(3) and the ribonuclease H domain of HIV-1 reverse transcriptase,(4) which are shown in Figure 1. The dimerization interfaces of all of these structures comprise a hydrogen-bonded antiparallel β-sheet in which each monomer contributes a β-strand. This fundamental form of biomolecular recognition has also been observed in other designed peptides and proteins.(24)
X-ray crystallography is excellent for observing β-sheet dimers, because the high concentration in the solid state and additional crystal packing forces help stabilize the dimers. In aqueous solution, β-sheet dimers require additional forces beyond hydrogen bonding to be stable. Hydrophobic interactions between molecules can reinforce the hydrogen bonds and provide an important mechanism for stabilizing the dimers. The additional hydrophobic buttressing of the helices in interleukin 8, for example, stabilizes the dimer in aqueous solution.(3) Self-assembly through hydrophobic face-to-face interactions stabilizes the dimers of artificial β-sheets 1 in aqueous solution.(8) Covalent bonds provided by disulfide linkages can also stabilize the dimers.12,24b
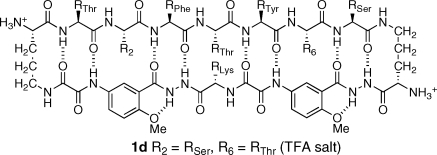
The X-ray crystallographic structure of macrocyclic β-sheet 1a provides deeper insights into the solution-phase structures of macrocycles 1.(8) Macrocycle 1d, for example is a double mutant of 1a (R2 = RSer, R6 = RThr) that forms dimers of dimers in aqueous solution. The association state of these tetramers was established through extensive analytical ultracentrifugation and pulsed field gradient (PFG) NMR diffusion studies. Macrocycles 1b and 1c were also determined to be dimers of dimers, albeit less rigorously, through PFG NMR diffusion studies alone. NMR studies also show that macrocycles 1a−c are largely folded in the monomeric state, while macrocycle 1d is largely unfolded. Interactions between the aromatic residues at the R2 and R6 positions and the two Hao amino acid units observed in the X-ray crystallographic structure of 1a (Figure 2) may explain the greater propensities of macrocycles 1a−c to fold.
We used the X-ray crystallographic structure of the trimer of dimers formed by 1a to create a molecular model of the dimer of dimers of 1d. To do this, we simply removed one dimer unit from the trimer of dimers (Figure 3) and moved the dimers together in a face-to-face fashion to form a β-sheet sandwich. We minimized the resulting structure using MacroModel v6.5 with the MMFF force field and GB/SA water solvation and mutated residues 2 and 6 to alanine and then to serine and threonine, reminimizing after each mutation. Figure 6 illustrates the structure of the resulting dimer of dimers.
Figure 6.

Model of the dimer of dimers of 1d. Top (A) and side (B) views. The model is based on the X-ray crystallographic structure of the trimer of dimers of 1a (Figure 3) and the known propensity of 1d to form a dimer of dimers in aqueous solution.
In the dimer of dimers of macrocyclic β-sheet peptide 1d modeled in Figure 6, much of the interdimer contact arises from the phenylalanine and tyrosine residues in the central peptide strands. The surface area buried by the dimer−dimer interface in this model is smaller than that buried by each dimer−dimer contact in the trimer of dimers of macrocyclic β-sheet peptide 1a shown in Figure 3 (460 vs 820 Å2), and the shape complementarity correlation Sc of the interface is lower (0.554 vs 0.817). The observed polymorphism of these macrocyclic β-sheet peptides likely reflects differences in the environments in which the samples were studied rather than their differences in sequence. The high (75 mM) concentration of 1a in the solid state may favor the trimer of dimers, while the lower concentrations (≤10 mM) at which 1b−d have been studied in solution may favor the dimer of dimers. The salts and isopropanol used for the crystallization may also favor the trimer of dimers. The observed polymorphism of macrocyclic β-sheet peptides 1 is reminiscent of the polymorphism of Alzheimer’s β-amyloid fibrils reported by Tycko and co-workers.(25)
Conclusion
The X-ray crystallographic structure of 1a provides with unparalleled confidence the structures of the dimers formed by macrocyclic β-sheets 1. The structure reveals in detail the peptidomimetic templates, the folded monomers, and the β-sheet dimers and corroborates the understanding of these units that had been developed through extensive solution-phase studies. This cross-validation goes both ways, validating solution-phase structures that had previously only been inferred while establishing that the crystalline state does not appreciably distort the structure of the macrocycle. X-ray crystallography also reveals a mode of higher-order self-assembly of the dimers that had not been anticipated from solution-phase studies of the closely related homologues. Both the anticipated dimers of dimers and the crystallographically observed trimer of dimers share in common the buttressing of the hydrogen-bonded dimers through additional hydrophobic interactions.
The artificial β-sheet dimers elucidated by this study are similar in structure to those formed by naturally occurring β-sheet proteins. We anticipate using insights provided from this study to better design molecules that interact through β-sheet formation to control β-sheet interactions between proteins and to inhibit the β-sheet interactions associated with peptide and protein aggregation.
Experimental Section
Sample Preparation
Macrocyclic β-sheet peptide 1a was synthesized and purified as described previously for macrocyclic β-sheet peptides 1b−d.(8) Crystals of 1a were grown with an Aldrich Basic Crystallography Kit by the sitting drop vapor diffusion method at 25 °C. Large cubic crystals (ca. 0.4 × 0.4 × 0.4 mm) grew from aqueous solution with 0.2 M MgCl2, 0.1 M HEPES pH 7.5 buffer, and 30% isopropanol in space group P212121 with cell dimensions a = 64.05 Å, b = 64.19 Å, c = 64.25 Å, α = β = γ = 90°, and 12 molecules of 1a in the asymmetric unit (AU). To avoid rapid evaporation of the mother liquor during crystal collection, drops were opened at 277 K. Crystals were soaked in well solution containing 30% glycerol as a cryoprotectant and flash-frozen at 100 K in liquid N2 prior to data collection.
Data Collection
X-ray diffraction data were collected using three wavelengths at the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 9-2 on a Mar325 CCD detector. A second crystal was used to collect a high-resolution data set at beamline 7-1 on an ADSC Quantum315 CCD detector. Data were collected at 100 K. Indexing, integration, and scaling were performed with the program HKL2000.(26)
Phasing, Model Building, and Refinement
All 12 bromine atoms in the AU were located using the program SOLVE.(27) The resulting MAD phases had a mean figure of merit of 0.68. Clear, continuous electron density was observed for all 12 cyclic peptides in the AU. The density was improved using one round of density modification and solvent flipping in CNS.(28) Model building was performed with the graphics program COOT.(29) Geometric restraints for the Hao amino acid were generated by the PRODRG server.(30) The model was refined to 1.35 Å using the maximum likelihood algorithm within Refmac5 in the CCP4 program suite.(31) Multiple rounds of model building and refinement followed. The addition of water and six well-defined isopropanol molecules improved the Rwork and Rfree to 0.150 and 0.157, respectively. All the amino acid residues in the structure lay within the favored β-sheet region of the Ramachandran plot.
Acknowledgments
Dedicated to the memory of Warren DeLano. J.S.N. and O.K. thank the National Institutes of Health for grant support (GM-49076). S.-C.T. and T.P.K. thank the Pew Foundation. D.E. and M.R.S. thank the HHMI and National Institutes of Health (AG-029430).
Supporting Information Available
Crystallographic information file for macrocyclic β-sheet peptide 1a (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.(32)
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hill C. P.; Yee J.; Selsted M. E.; Eisenberg D. Science 1991, 251, 1481–1485. [DOI] [PubMed] [Google Scholar]
- Matsuo H.; Shirakawa M.; Kyogoku Y. J. Mol. Biol. 1995, 254, 668–680. [DOI] [PubMed] [Google Scholar]
- a Clore G. M.; Appella E.; Yamada M.; Matsushima K.; Gronenborn A. M. Biochemistry 1990, 29, 1689–1696. [DOI] [PubMed] [Google Scholar]; b Baldwin E. T.; Weber I. T.; St Charles R.; Xuan J. C.; Appella E.; Yamada M.; Matsushima K.; Edwards B. F.; Clore G. M.; Gronenborn A. M.; Wlodawer A. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. F. II; Hostomska Z.; Hostomsky Z.; Jordan S. R.; Matthews D. A. Science 1991, 252, 88–95. [DOI] [PubMed] [Google Scholar]
- a http://www.igb.uci.edu/servers/icbs/.; b Dou Y.; Baisnée P.-F.; Pollastri G.; Pécout Y.; Nowick J.; Baldi P. Bioinformatics 2004, 20, 2767–2777. [DOI] [PubMed] [Google Scholar]
- a Nelson R.; Sawaya M. R.; Balbirnie M.; Madsen A. O.; Riekel C.; Grothe R.; Eisenberg D. Nature 2005, 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sawaya M. R.; Sambashivan S.; Nelson R.; Ivanova M. I.; Sievers S. A.; Apostol M. I.; Thompson M. J.; Balbirnie M.; Wiltzius J. J. W.; McFarlane H. T.; Madsen A. Ø; Riekel C.; Eisenberg D. Nature 2007, 447, 453–457. [DOI] [PubMed] [Google Scholar]; c Wiltzius J. J.; Sievers S. A.; Sawaya M. R.; Cascio D.; Popov D.; Riekel C.; Eisenberg D. Protein Sci. 2008, 17, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Ivanova M. I.; Sievers S. A.; Sawaya M. R.; Wall J. S.; Eisenberg D. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 18990–18995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Luhrs T.; Ritter C.; Adrian M.; Riek-Loher D.; Bohrmann B.; Dobeli H.; Schubert D.; Riek R. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Petkova A. T.; Yau W.-M.; Tycko R. Biochemistry 2006, 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Paravastu A. K.; Leapman R. D.; Yau W. M.; Tycko R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakshoor O.; Demeler B.; Nowick J. S. J. Am. Chem. Soc. 2007, 129, 5558–5569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowick J. S.; Chung D. M.; Maitra K.; Maitra S.; Stigers K. D.; Sun Y. J. Am. Chem. Soc. 2000, 122, 7654–7661. [Google Scholar]
- Nowick J. S.; Brower J. O. J. Am. Chem. Soc. 2003, 125, 876–877. [DOI] [PubMed] [Google Scholar]
- The name of unnatural amino acid “Hao” is a mnemonic for the three components that it comprises: hydrazine, an aminoaromatic group, and oxalic acid.
- Khakshoor O.; Nowick J. S. Org. Lett. 2009, 11, 3000–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nauli S.; Kuhlman B.; Baker D. Nat. Struct. Biol. 2001, 8, 602–605. [DOI] [PubMed] [Google Scholar]; b Nauli S.; Kuhlman B.; Le Trong I.; Stenkamp R. E.; Teller D.; Baker D. Protein Sci. 2002, 11, 2924–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ϕ and ψ angles of the R1−R8 amino acid residues lie entirely within the β-sheet region of the Ramachandran plot (−164° < ϕ < −110°, 120° < ψ < 160°). The conformations of the side chains of the R1−R8 amino acid residues are within 5° of the favored (canonical) rotamers. The alternate conformations of the tyrosine residues are both favored rotamers and differ by 120°.
- The twist between the two β-strands at the dimer interface (2.5°) is comparable to the twist between the β-strands in the dimer interfaces illustrated in Figure 1 (defensin HNP-3, 1.5°; λ-Cro repressor, 12°; interleukin-8, 8°; and the ribonuclease H domain of HIV-1 reverse transcriptase, 14°) and is smaller than the ca. 15° twist typically observed in globular proteins such as lysozyme and carboxypeptidase.
- Each trimer makes six relatively small lattice contacts to other trimers. The contacts are made at the corners of the trimer rather than the edges or faces and are roughly two-fold symmetric. Three of the contacts bury 543 Å2 surface area per interface. Each of these interfaces involves van der Waals contacts between a pair of bromophenyl rings and a pair of aromatic moieties from Hao. The remaining three contacts are even smaller, burying only 251 Å2 surface area per interface. Each of these interfaces involves van der Waals contacts between a pair of bromophenyl rings and a pair of hydrogen bonds between backbone amides of ornithine residues.
- The cavity volume was calculated with the program CASTp using a 1.4 Å probe:; Dundas J.; Ouyang Z.; Tseng J.; Binkowski A.; Turpaz Y.; Liang J. Nucleic Acids Res. 2006, 34, W116–W118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. C.; Colman P. M. J. Mol. Biol. 1993, 234, 946–950. [DOI] [PubMed] [Google Scholar]
- For examples, see:; a Karle I. L.; Awasthi S. K.; Balaram P. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 8189–8193. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gong B.; Yan Y.; Zeng H.; Skrzypczak-Jankunn E.; Kim Y. W.; Zhu J.; Ickes H. J. Am. Chem. Soc. 1999, 121, 5607–5608. [Google Scholar]; c Zhu J.; Parra R. D.; Zeng H.; Skrzypczak-Jankun E.; Zeng X. C.; Gong B. J. Am. Chem. Soc. 2000, 122, 4219–4220. [Google Scholar]; d Jiang H.; Léger J.-M.; Huc I. J. Am. Chem. Soc. 2003, 125, 3448–3449. [DOI] [PubMed] [Google Scholar]; e Dolain C.; Zhan C.; Léger J.-M.; Daniels L.; Huc I. J. Am. Chem. Soc. 2005, 127, 2400–2401. [DOI] [PubMed] [Google Scholar]; f Crisma M.; Saviano M.; Moretto A.; Broxterman Q. B.; Kaptein B.; Toniolo C. J. Am. Chem. Soc. 2007, 129, 15471–15473. [DOI] [PubMed] [Google Scholar]; g Liu J.; Wang D.; Zheng Q.; Lu M.; Arora P. S. J. Am. Chem. Soc. 2008, 130, 4334–4337. [DOI] [PubMed] [Google Scholar]; h Chatterjee S.; Vasudev P. G.; Raghothama S.; Ramakrishnan C.; Shamala N.; Balaram P. J. Am. Chem. Soc. 2009, 131, 5956–5965. [DOI] [PubMed] [Google Scholar]; i Sánchez-García D.; Kauffmann B.; Kawanami T.; Ihara H.; Takafuji M.; Delville M.-H.; Huc I. J. Am. Chem. Soc. 2009, 131, 8642–8648. [DOI] [PubMed] [Google Scholar]; j Raghavender U. S.; Aravinda S.; Shamala N.; Kantharaju; Rai R.; Balaram P. J. Am. Chem. Soc. 2009, 131, 15130–15132. [DOI] [PubMed] [Google Scholar]; k Raghavender U. S.; Kantharaju; Aravinda S.; Shamala N.; Balaram P. J. Am. Chem. Soc. 2010, 132, 1075–1086. [DOI] [PubMed] [Google Scholar]
- a Nowick J. S. Org. Biomol. Chem. 2006, 4, 3869–3885. [DOI] [PubMed] [Google Scholar]; b Nowick J. S. Acc. Chem. Res. 2008, 41, 1319–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The average dihedral angles of the δ-linked ornithine turn units are as follow: N−CO−Cα−Cβ,−69°; CO−Cα−Cβ−Cγ, 162°; Cα−Cβ−Cγ−Cδ,−64°; Cβ−Cγ−Cδ−Nε,−69°; and Cγ−Cδ−Nε−CO, 95°.
- Richardson J. S.; Richardson D. C. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tang Y.-Q.; Yuan J.; Ösapay G.; Ösapay K.; Tran D.; Miller C. J.; Quellette A. J.; Selsted M. E. Science 1999, 286, 498–502. [DOI] [PubMed] [Google Scholar]; b Trabi M.; Schirra H. J.; Craik D. J. Biochemistry 2001, 40, 4211–4221. [DOI] [PubMed] [Google Scholar]; c Tran D.; Tran P. A.; Tang Y.-Q.; Yuan J.; Cole T.; Selsted M. E. J. Biol. Chem. 2002, 277, 3079–3084. [DOI] [PubMed] [Google Scholar]; d Daly N. L.; Chen Y.-K.; Rosengren K. J.; Marx U. C.; Phillips M. L.; Waring A. J.; Wang W.; Lehrer R. I.; Craik D. J. Biochemistry 2007, 46, 9920–9928. [DOI] [PubMed] [Google Scholar]
- a Ilyina E.; Roongta V.; Mayo K. H. Biochemistry 1997, 36, 5245–5250. [DOI] [PubMed] [Google Scholar]; b Venkatraman J.; Gowda G. A. N.; Balaram P. J. Am. Chem. Soc. 2002, 124, 4987–4994. [DOI] [PubMed] [Google Scholar]
- a Petkova A. T.; Leapman R. D.; Guo Z.; Yau W. M.; Mattson M. P.; Tycko R. Science 2005, 307, 262–265. [DOI] [PubMed] [Google Scholar]; b Paravastu A. K.; Leapman R. D.; Yau W. M.; Tycko R. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Terwilliger T. J. Synchrotron Radiat. 2004, 11, 49–52. [DOI] [PubMed] [Google Scholar]
- Brünger A. T.; Adams P. D.; Clore G. M.; DeLano W. L.; Gros P.; Grosse-Kunstleve R. W.; Jiang J. S.; Kuszewski J.; Nilges M.; Pannu N. S.; Read R. J.; Rice L. M.; Simonson T.; Warren G. L. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Schüttelkopf A. W.; van Aalten D. M. F. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 50, 760−763. [Google Scholar]
- Crystallographic data are also available from the Cambridge Crystallographic Data Centre (CDCC ID code 782650) and the Research Collaboratory for Structural Biology Protein Data Bank (PDB ID code 3NI3).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.