

Abstract
Fragment hopping, a new fragment-based approach for de novo inhibitor design focusing on ligand diversity and isozyme selectivity, is described. The core of this approach is the derivation of the minimal pharmacophoric element for each pharmacophore. Sites for both ligand binding and isozyme selectivity are considered in deriving the minimal pharmacophoric elements. Five general-purpose libraries are established: the basic fragment library, the bioisostere library, the rules for metabolic stability, the toxicophore library, and the side chain library. These libraries are employed to generate focused fragment libraries to match the minimal pharmacophoric elements for each pharmacophore and then to link the fragment to the desired molecule. This method was successfully applied to neuronal nitric oxide synthase (nNOS), which is implicated in stroke and neurodegenerative diseases. Starting with the nitroarginine-containing dipeptide inhibitors we developed previously, a small organic molecule with a totally different chemical structure was designed, which showed nanomolar nNOS inhibitory potency and more than 1000-fold nNOS selectivity. The crystallographic analysis confirms that the small organic molecule with a constrained conformation can exactly mimic the mode of action of the dipeptide nNOS inhibitors. Therefore, a new peptidomimetic strategy, referred to as fragment hopping, which creates small organic molecules that mimic the biological function of peptides by a pharmacophore-driven strategy for fragment-based de novo design, has been established as a new type of fragment-based inhibitor design. As an open system, the newly established approach efficiently incorporates the concept of early “ADME/Tox” considerations and provides a basic platform for medicinal chemistry-driven efforts.
Introduction
Lead generation is a critical first step in the drug discovery process. Over the past decade, high-throughput screening (HTS) of corporate compound collections has emerged as the paradigm for hit or lead discovery. Despite the fact that this approach has often been successful, it has some inherent challenges and limitations. Typically, targets are interrogated with 106 to 107 discrete compounds in parallel, which fall far short of potential chemical diversity space, estimated to be upward of 1060 molecules containing up to 30 non-hydrogen atoms.1 For some target classes the HTS hit rate is low and results in few good chemical starting points for inhibitor optimization.2 The good hits identified from historical compound collections usually have moderate biological activity (Ki: 1−10 μM) but with relatively high molecular weights (the average molecular weight is 400 Da) and excessive lipophilicities,3 which are frequently not amenable for lead optimization to generate compounds with drug-like properties.4 Stimulated by the introduction of the “rule of five”,5 many research programs profile compound collections for lead identification with low average molecular weights. This trend leads to the generation of fragment-based screening approaches.6 Low molecular weight chemical fragments (usually 120–250 Da) are initially selected on the basis of their ability to bind to the target of interest or to inhibit/promote its biological function. Following the binding event, various affinity-based techniques7 have been proposed to accurately and efficiently identify the weak binding fragment (typically the binding affinities of fragments are in the 1 mM to 30 μM range), such as nuclear magnetic resonance-based screening8 (SAR by NMR9), mass spectroscopy-based identification10 (especially tethering11), X-ray crystallography-based approaches,12 or surface plasmon resonance spectroscopy-based screening.13 Alternatively, substrate activity screening highlights the roles of bioassay-based techniques in the identification of effective fragments.14 These fragments, which can be considered as the building blocks of a more complex lead structure, are then evolved or combined/merged into compounds. The exploration of the approaches to construct molecules from fragments leads to the generation of in situ fragment assembly techniques, such as click chemistry,15 dynamic combinatorial library design,16 and tethering with extenders.17
Fragment-based screening offers a number of attractive features compared to HTS. First, whereas compounds from HTS libraries are more restricted in their rotational degrees of freedom, and thus less able to adapt to a given target site, a high proportion of the atoms of a fragment hit are directly involved in the desired receptor–ligand interaction, which allows for optimal positioning within the receptor pocket. Therefore, a fragment is a more efficient binder (high binding energies per unit molecular mass). Second, a fragment-based strategy provides a combinatorial advantage. The number of fragments screened is in the range of only hundreds to a few thousands, but they can explore a larger chemical space than a preassembled large compound library. On the other hand, developing and maintaining a small set of fragments with simpler structures is easier than maintaining a massive HTS library. Third, when the binding of a fragment is identified, the subsequent structural optimization can benefit from extensive design and can lead to a higher success rate and greater flexibility for generating novel chemical entities. And last, starting with a low-molecular-mass fragment is likely to produce leads with rather small and simple structures, which allow for enhancement during the lead optimization process.
However, there are some internal problems and challenges in current fragment-based approaches. First, a fragment-based strategy can provide a combinatorial advantage relative to preassembled large chemical libraries, that is, a collection of 103 fragments can typically probe the chemical diversity space of 109 molecules, a tremendous increase relative to HTS; however, it is still a small fraction of the total diversity space.18 Second, because most fragments have low binding affinities as a result of limited interactions with the target, the identification of relevant fragments and determination of how to link them productively in three-dimensional space are still quite intractable problems in some cases,14,18,19 although many affinity-based assay techniques have been developed. Third, ligand specificity for its targets is a particularly important goal of drug discovery in the postgenomic era because a myriad of functional proteins have been characterized, and the enzymatic pockets within a target family/or superfamily, which execute the same/similar metabolic reactions and functions, are often quite similar. An important challenge in modern medicine is how to design compounds that can modulate a specific enzyme while leaving related isozymes unaffected. Known fragment-based approaches, however, are only able to identify and characterize fragment binding sites on the target protein (often called “hot spots”, that is, the regions of a protein surface that are major contributors to the ligand binding free energy19,20). In fact, many binding sites in the active site that are responsible for target specificity and/or selectivity are not included in these “hot spots”.
Utilizing the basic tenets of fragment-based inhibitor design in our earlier structure-based design of inhibitors of neuronal nitric oxide synthase (nNOS), this pharmacophore-driven approach was proposed that attempted to search for selective inhibitors of nNOS over the other two isozymes.21 Nitric oxide synthase (NOS, EC 1.14.13.39) is a multidomain enzyme consisting of an N-terminal catalytic oxygenase domain, a central linker region, and a C-terminal electron-supplying reductase domain that catalyzes the five-electron, two-step oxidation of L-arginine (L-Arg) to produce L-citrulline and nitric oxide (NO). NO is an important signaling molecule involved in a wide range of physiological functions as well as pathophysiological states mainly through the soluble guanylate cyclase/cGMP pathway.22 Three distinct NOS isoforms, neuronal (nNOS), endothelial (eNOS), and inducible (iNOS), have been identified in mammals; these isozymes have 50–60% sequence identity and share identical overall architecture.23 The N-terminal catalytic oxygenase domain binds heme (Fe-protoporphyrin IX), substrate L-Arg, and (6R)-5,6,7,8-tetrahydrobiopterin (H4B). The central linker region binds calmodulin (CaM), and the C-terminal reductase domain has the binding sites for flavin mononucleotide (FMN), flavin adenine dinucleotide (FAD), and nicotinamide adenine dinucleotide phosphate (NADPH). All three NOS isozymes are functional only as tight homodimers, and CaM binding to the central linker region mediates electron transfer from the reductase domain of one subunit trans to the oxygenase domain of the other subunit of the dimer.24 The NOS isozymes differ in cellular distribution, regulation, and activity. Under normal physiological conditions, both constitutively expressed isozymes (nNOS and eNOS) are regulated by intracellular Ca2+/CaM and generate trace amounts of NO as an intercellular messenger. The eNOS-derived NO is a vasodilator essential for vascular homeostasis and also inhibits platelet aggregation and leukocyte adhesion, while NO generated by nNOS participates in neurotransmission in both the central and peripheral nervous systems. iNOS binds CaM irreversibly and is instead regulated mainly by transcriptional control of enzyme expression in response to cytokines.
In line with the central biological role of NO, there are a number of pathological processes associated with its over- or underproduction.25 The impaired NO production by eNOS is associated with hypertension, atherosclerosis, and arterial thrombosis.26 Excess formation of NO from nNOS has been implicated in stroke and neurodegenerative diseases.27 iNOS-produced NO appears to be involved in a broad range of inflammatory pathologies, such as septic shock, rheumatoid arthritis, and multiple sclerosis.28 Owing to these many pathological conditions, in addition to the basic physiological functions related to NOS, indiscriminate inhibition of NOS would be detrimental.29 Therefore, selective inhibition of one isozyme over the others is essential. In particular, compounds that control the overproduction of NO by nNOS or iNOS, while leaving undisturbed the vasoprotective function of eNOS, are desired.30
The crystal structures of the dimeric oxygenase domain for all three NOS isoforms have been solved, which provide the possibility for structure-based inhibitor design.31 However, this has proven to be a challenging problem because the active sites of NOS isozymes are highly conserved. Sixteen out of eighteen residues within 6 Å of the substrate binding site are identical, and the side chain of one of these two dissimilar amino acids points out of the substrate-binding site.21 Previously, we synthesized and evaluated nitroarginine-containing dipeptide or peptidomimetic inhibitors and identified a family of compounds that had high potency and selectivity for inhibition of nNOS over eNOS and iNOS. The most potent nNOS inhibitors among these compounds were L-Nω-nitroarginine-2,4-L-diaminobutyramide (1),32 (4S)-N-[4-amino-5-(aminoethyl)aminopentyl]-N′-nitroguanidine (2),33,34 and L-Nω-nitroarginine-(4R)-amino-L-proline amide (3)35,36 (Figure 1). The selectivity of these dipeptide/peptidomimetic inhibitors for nNOS over eNOS and/or iNOS was investigated by crystallographic analysis37 and by GRID/CPCA in the earlier studies.38
Figure 1.

Chemical structures and NOS inhibitory activities of L-nitroarginine-containing dipeptide/peptidomimetic inhibitors.
In this paper the concept of minimal pharmacophoric elements is proposed, and focused (or targeted) fragment libraries that match the requirements of the minimal pharmacophoric elements are subsequently generated. On the basis of these focused fragment libraries a pharmacophore-driven strategy for fragment-based de novo design, we term fragment hopping, is proposed that utilizes the minute structural differences of the active sites of the three NOS isozymes and leads to the design of a class of non-peptide inhibitors that are highly selective for nNOS over the other two isozymes.
Results and Discussion
Fragment Hopping, a Pharmacophore-Driven Strategy for Fragment-Based de Novo Design
Figure 2 summarizes the pharmacophore-driven strategy for fragment-based inhibitor design. The first step of the strategy is to determine the pharmacophores of a specific drug target. If the target structure can be determined by X–ray crystallography or NMR spectroscopy, various experimental approaches can be used to determine the potential pharmacophores. The multiple solvent crystal structures method (MSCS)39 and various affinity-based biophysical techniques mentioned above are efficacious tools for understanding how small molecules bind to the active site of the enzymes. The energetic hot spots of enzymes for ligand binding can be unraveled in combination with alanine scanning.40 The computational methods for active site analysis are useful when the receptor structure is known, or, if unknown, the structure can be constructed by homology modeling.41 Two of the most popular and venerable algorithms are GRID,42 which calculates 3D energy maps around protein binding sites, thus highlighting favorable sites for small functional groups, and multiple copy simultaneous search43 (MCSS), which randomly places thousands of copies of small functional groups into the binding site, and the copies of small functional groups are subject to energy minimization. The copies with the lowest energies highlight hot spots of ligand binding. Many other computational methods, such as the knowledge-based equivalents of GRID (X-SITE44 and SuperStar45) and energy-based approaches (PocketFinder,46 Q-SiteFinder47), also can be used to explore sensitive and specific hot spots in the active site. Computational solvent mapping48 and binding site determination technology, based on grand canonical thermodynamics ensemble Monte Carlo simulations (Lotus),49 can be regarded as an important new breakthrough in this field. GRID/CPCA is an excellent tool for understanding the selectivity of inhibitors for a specific target over the other structure-related enzymes.50 If the structure of the receptor is unknown, the pharmacophore can be identified by structure–activity analysis of ligands or by various computational methods, such as Catalyst, DISCO, and GASP.51 Self-organizing maps (SOM) can be used as a ligand-based approach to predict compound selectivity.52
Figure 2.

Schematic flow diagram for fragment hopping, the pharmacophore-driven strategy for fragment-based de novo design.
Three-point or four-point pharmacophore models can be generated from the above analyses.53 However, the key point of the above pharmacophore investigation is to derive the minimal pharmacophoric elements for each pharmacophore, which means that a combinatorial application of different pharmacophore identification methods is required to provide as much information as possible. The minimal pharmacophoric element can be an atom, a cluster of atoms, a virtual graph, or vector(s). On the basis of the derived minimal pharmacophoric elements, the second step of this approach is to query two main general-purpose libraries: (1) A basic fragment library, which is constructed on the basis of the fragments extracted directly from known drugs and/or drug candidates. The fragments are either from well-known libraries, such as the MDL comprehensive medicinal chemistry (CMC) database,54 the World Drug Index (WDI),55 the Maccs Drug Data Report (MDDR),56 or from the literature.57 These are summarized in Supporting Information Figure 1. (2) A bioisostere library, which is constructed on the basis of known bioisosteric principles reported in the literature (Supporting Information Figure 2).58 The basic fragment library is searched first to find all of the possible fragments that are able to match the requirements of the minimal pharmacophoric elements for each pharmacophore. Then the bioisostere library is utilized to generate a focused fragment library with diverse structures. The generated focused fragment library is then interrogated with the rules for metabolic stability (see Supporting Information Figure 3)59 and a toxicophore library (see Supporting Information Figure 4)60 to provide a focused library for a specific pharmacophore. The focused library is then converted into a LUDI fragment library, and the LUDI program is used to search the optimal binding position for each fragment of each pharmacophore.61
The third step of this approach is to link these fragments. A constructed side chain library is used for this purpose, in which the synthetic accessibility is considered.55b,c,62 This library, shown in Supporting Information Figure 5, has been converted into a LUDI linking library. SciFinder Scholar 2006,63 in conjunction with the bioisostere library, also plays a key role in securing the synthetic accessibility of the formed chemical bond. The bioisostere library plays an assistant role in enhancing the binding capabilities and optimizing the chemical properties of the generated ligands. The generated ligand is interrogated again with the rules for metabolism stability and the toxicophore library.
The ligands generated by this iterative process are then docked into the active site using AutoDock3.0,64 scored with consensus scoring functions,65 and filtered with absorption, distribution, metabolism, excretion, and toxicity (ADME/Tox) considerations.5,66 If the ligands generated are not satisfactory, the molecule is reconstructed using the generated focused fragment libraries, the side chain library, and the bioisostere library (Figure 2).
Application of Fragment Hopping: The de Novo Design of Selective Neuronal Nitric Oxide Synthase Inhibitors
1. Active Site Analysis
The active site of NOS was investigated prior to the design of inhibitors by two different methods, GRID and MCSS. The active site of NOS has been divided into 4 pockets (S, M, C1, and C2) as described in the earlier study.38 The residue numbering for rat nNOS and bovine eNOS are used in the following discussion because these are the sources of the NOS X-ray crystal structure data. In the GRID analysis, hydrophobic interactions are calculated with the DRY probe (see Table 1 for a compilation of the probes used). The C3 and NM3 probes describe the steric interactions. The polar probes consist of N1, NH=, O, and O1. The COO− probe is negatively charged, while the probes N3+, NM3, N1+ are the positively charged single-atom probes. Since the natural ligand for all three isozymes of NOS is L-Arg, a polar and basic residue that needs a polar and acidic environment to stabilize it in the active site, more polar and positive probes were used in the active site analysis, including two multiatom probes Aramidine and Ami-dine. There are two significant molecular interaction fields (MIFs) in the active site of nNOS for steric effect probes C3 and NM3, as indicated in Figure 3A for the C3 probe. One is located in the S pocket, which is encompassed by residues P565, A566, V567, F584, S585, G586, W587, and the heme cofactor. The maximal interaction energy is −5.00 kcal/mol for C3 and −12.50 kcal/mol for NM3, respectively (Table 1). The second region is located in the M pocket, which is defined by D597 and the heme propionate of the pyrrole A ring. The corresponding maximal interaction energy is 4.00 kcal/mol for C3 and −10.00 kcal/mol for NM3. The MIFs for the polar hydrogen bond (H-bond) donor probes N1 and O1 are distributed almost everywhere in the active site. This is understandable because the substrate L-Arg is very polar and hydrogen donor-rich. Among these MIFs, there are two main regions for the N1 probe: One is in the S pocket determined by the backbone amide of W587 and the side chain carboxylate of E592. This is where the guanidine group of L-Arg is located. The second one is in the M pocket determined by the side chains of D597, R603, and Y588. The main MIF region for the O1 probe is determined by the M pocket enclosed by D495, Y562, R481, D597, R603, Q478, and Y588. The MIF of the polar H-bond acceptor probes NH= and O is much clearer compared to those of the H-bond donor probes. It is mainly located in the M pocket also determined by D495, Y562, R481, D597, R603, Q478, and Y588. The negatively charged probe COO− is only located in the S pocket, which is mainly driven by the presence of the heme Fe cation.
Table 1.
Summary of the Main Probe Interactions Observed in the Active Site of Rat nNOS. The Relevant Residues and Cofactors and the Maximal Interaction Energy Involved in a Particular Interaction Are Given
| probe | chemical group | maximal energy (kcal/mol) | main residues and cofactors | |
|---|---|---|---|---|
| C3 | methyl group | S | −5.00 | P565, A566, V567, F584, S585, G586, W587, heme |
| M | −4.00 | D597, heme propionate | ||
| NM3 | trimethylammonium cation | S | −12.50 | P565, A566, V567, F584, S585, G586, W587, heme |
| M | −10.00 | D597, heme propionate | ||
| N1 | neutral flat NH (e.g., amide) | S | −9.50 | W587, E592 |
| M | −7.30 | Y588, D597, R603 | ||
| O1 | alkyl hydroxyl OH group | M | −12.00 | Q478, R481, D495, Y562, Y588, D597, R603 |
| NH= | sp2 NH with lone pair | M | −12.00 | Q478, R481, D495, Y562, Y588, D597, R603 |
| O | sp2 carbonyl oxygen | M | −8.50 | Q478, R481, D495, Y562, Y588, D597, R603 |
| S | −7.00 | backbone of A566 and V567 | ||
| COO− | carboxylic acid anion | S | −17.00 | heme Fe cation |
| N3+ | sp3 amine NH3 cation | M | −13.50 | D597, Y588 |
| S | −11.40 | E592 | ||
| M | −11.00 | heme propionate | ||
| N1+ | sp3 amine NH cation | M | −11.00 | D597, Y588 |
| S | −9.50 | E592 | ||
| M | −8.50 | heme propionate | ||
| ARamidine | aromatic cationic amidine group | M | −20.00 | D597 |
| S | −12.00 | E592 | ||
| amidine | aliphatic cationic amidine group | M | −21.00 | D597 |
| S | −15.00 | E592 | ||
| DRY | hydrophobic probe | C1 | −1.85 | Y706, L337, M336, W306 (the other NOS monomer) |
Figure 3.

Results of GRID analysis of the substrate binding site of nNOS (PDB: 1p6i). The residues and cofactors (heme and H4B) are represented in an atom-type style. The S and M pockets are indicated. A: GRID contours of the C3 probe at an energy level of −3.50 kcal/mol. B: GRID contours of the N3+ probe at an energy level of −9.60 kcal/mol; “×” represents the position of the heme iron atom.
As shown in Figure 3B, there are three main MIFs for the positively charged single-atom probes N3+ and N1+ in the active site of nNOS. In the M pocket one region is determined by D597 and Y588 (the maximal interaction energy is −13.50 kcal/mol for N3+ and −11.00 kcal/mol for N1+). The second one is determined by the two heme propionates (the maximal interaction energy is −11.00 kcal/mol and −8.50 kcal/mol for N3+ and N1+, respectively). The last MIF is located in the S pocket, which is determined by E592, the same position where the guanidine group of substrate L-Arg is located in the active site. There are two main MIFs for the multiatom probes Aramidine and Amidine, determined by D597 and E592, respectively. The hydrophobic interaction is mainly determined by the hydrophobic residues in the C1 pocket (Table 1).
The nine aromatic, aliphatic, polar, or charged functional groups have been mapped into the active site of NOS by MCSS calculations (Table 2). Similar to what was obtained from the GRID analysis, the minima with the most favorable interaction energies for the apolar and bulky groups, such as benzene (minima no. 1–no. 3: −24.16 kcal/mol ~ −21.88 kcal/mol), cyclohexane (minima no. 1–no. 2: −9.80 kcal/mol ~ −9.65 kcal/mol), and _sobutene (minima no. 1–no. 2: −11.29 kcal/mol ~ −11.22 kcal/mol), are located in the hydrophobic cavity of the S pocket (Figure 4A). Among them, the heavy atom-only models of cyclohexane and isobutene are appropriate to explore steric effects in the active site. The region displaying the second most favorable interaction for these two functional groups is located in the M pocket (Figure 4A), which is defined by D597 and the heme propionate of the pyrrole A ring. (cyclohexane, minima no.3–no.4: −9.02 kcal/mol to −8.90 kcal/mol and isobutene, minima no. 3–no. 11: −7.33 kcal/mol to −6.24 kcal/mol). The polar functional groups (N-methylacetamide and methanol), containing H-bond donors, generated more MCSS minima than the H-bond acceptor (ether). The lowest energy minima of all three polar functional groups were located in the S pocket and bound to the heme iron atom. The minima of the H-bond donors displaying the second most favorable interaction energies formed H-bonds with the heme propionate groups (N-methylacetamide minimum no. 8 and methanol minimum no. 3 in Table 2), while the minimum of ether displaying the second most favorable interaction energies (minimum no. 2 in Table 2) formed H-bonds with Q478 and R603 of the M pocket. The minima with the most favorable interaction energies for the positively charged functional groups were located in the M pocket (Table 2). Minima no. 1–no. 3 of methylammonium (−121.90 kcal/mol to −120.62 kcal/mol) and minimum no. 2 of trimethylamine cation (−82.79 kcal/mol) bound to both heme propionates (Figure 4B). Minima no. 4 and no. 5 of methylammonium (−115.83 kcal/mol and −115.09 kcal/mol, respectively) bound to E592 and the heme propionate of the pyrrole A ring in the same way that the α-amino group of Nω-nitro-L-arginine (L-NNA) acts in its crystal structure complexed with NOS.38 Minimum no. 6 of methylammonium bound to D597 only. On the other hand, minimum no. 1 of the trimethylamine cation was located in a position close to E592 and D597. This position is where the α-amino group of compounds 1–3 is located in the crystal structure of nNOS. It is this site that is mainly responsible for the nNOS/eNOS selectivity of the nitroarginine-containing dipeptide/peptidomimetic inhibitors (Figure 4C).37,38 It is interesting that the MCSS interaction energies between the positively charged functional groups and E592 in the S pocket were much higher than those between the positively charged functional groups and D597 and/or the heme propionate. As indicated in Table 2 and Figure 4B, the minima bound to E592 are minimum no. 9 of methylammonium (−73.97 kcal/mol) and no. 22 of the trimethylammonium cation (−32.36 kcal/mol).
Table 2.
Minima Found by MCSS in the Active Site of Rat nNOS
| MCSS functional groups | initial no. of copies | no. of minima | range of Ubinda for minima (kcal/mol) |
selected minima |
||
|---|---|---|---|---|---|---|
| from | to | Ubind; pocket | residues/cofactors involved | |||
| benzene | 2500 | 27 | −1.75 | −24.16 | no. 1: −24.16; S | V567, F584, heme |
| no. 8: −12.99; M | D597, heme | |||||
| cyclohexane | 5000 | 98 | −0.09 | −9.80 | no. 1: −9.80; S | V567, F584, heme |
| no. 3: −9.02; M | D597, heme | |||||
| isobutane | 5000 | 22 | −5.75 | −11.29 | no. 1: −11.29; S | V567, F584, heme |
| no. 3: −7.33; M | D597, heme | |||||
| N-methylacetamide | 5000 | 85 | −2.44 | −54.77 | no. 8: −43.97; M | heme |
| no. 12: −42.15; M | heme, R603 | |||||
| no. 13: −41.91; M | Q478, E592 | |||||
| methanol | 5000 | 64 | −3.19 | −32.66 | no. 3: −30.65; M | heme |
| no. 5: −29.52; M | D597, Q478 | |||||
| no. 7: −25.26; S | E592 | |||||
| ether | 5000 | 27 | −3.21 | − 25.20 | no. 2: −19.91; M | Q478, R603 |
| acetate ion | 5000 | 42 | −5.93 | −79.51 | no. 1: −79.51; C1 | R481 |
| no. 2: −77.73; S | heme Fe | |||||
| no. 5: −42.93; M | Q478, R481 | |||||
| methylammonium | 5000 | 16 | −0.75 | −121.90 | no. 1: −121.90;M | heme |
| no. 4: −115.83; M | E592, heme | |||||
| no. 6: −85.32; M | D597 | |||||
| no. 9: −73.97; S | E592 | |||||
| trimethylamine ion | 5000 | 28 | −2.18 | −87.43 | no. 1: −87.43; M | D597, E592 |
| no. 2: −82.79; M | heme | |||||
| no. 22: −32.36; S | E592 | |||||
Ubind, the binding energy for a given functional group in each minimized replica obtained from the MCSS calculation, is defined as Ubind= Uprotein–group + Ugroup0, where Uprotein–group represents the nonbonded interactions between nNOS and the given functional group. Ugroup represents the internal energy of the functional group within the complex, and Ugroup0 represents the internal energy of the isolated functional group in vacuum.
Figure 4.

Representative MCSS-minimized positions of the functional groups in the active site of rat nNOS (PDB: 1p6i). Cofactors heme and H4B are shown in cyan and magenta, respectively. The S and M pockets are indicated. A. Benzene (red, minimum no. 1 is in the S pocket, while minimum no. 8 is in the M pocket). Cyclohexane (yellow, minimum no. 1 is in the S pocket, while minimum no. 3 is in the M pocket) and isobutene (green, minimum no. 1 is in the S pocket, while minimum no. 3 is in the M pocket). B. Methylammonium (red, 4 minima are shown in the M pocket. Minima no. 1 and no. 7: −79.74 kcal/mol bind to the heme propionate, minimum no. 4 binds to E592 and the heme propionate, and minimum no. 6 is close to D597. Minimum no. 9 is shown in the S pocket). Trimethylammonium cation (yellow, 2 minima are shown in the M pocket. Minimum no. 1 is close to D597 and E592, and minimum no. 2 binds to the heme propionate. Minimum no. 22 is shown in the S pocket). The labels of the minima denote their ranking. C. The relative positions of minima no. 4 and no. 6 of methylammonium, minimum no. 1 of trimethylammonium cation, and the α-amino group of compound 2 in Figure 2. The labels of the minima denote their ranking.
2. Minimal Pharmacophoric Elements and Lead Structure Design
Our earlier research found that a single-residue difference in the active site, rat nNOS D597 versus bovine eNOS N368, is mostly responsible for the favored selectivity of nNOS over eNOS; this high selectivity is determined by the α-amino groups of 1–3.37,38 The maximum electrostatic stabilization arising from residues D597 and E592 and the α-amino group of inhibitors 1–3 forces inhibitors to adopt a curled conformation. Such stabilization is rather weak in eNOS because N368 does not bear a negative charge. Inhibitors 1–3 in eNOS adopt an extended conformation, and the α-amino group is shifted away from the corresponding selective region defined in nNOS.37,38 The minimal pharmacophoric elements are proposed in Figure 5 on the basis of the above active site analyses and many structure–activity relationship studies conducted by us and others.30 An amidino group is positioned in the same place as the guanidino group of substrate L-Arg. This group is the minimal binding element to form a charge–charge interaction and also H-bonds with the carboxylate side chain of E592 and the backbone amide of W587. One sp3-hybridized nitrogen cation is placed in the selective region defined by D597 of nNOS and N368 of eNOS. The other three nitrogen atoms are placed close to the heme propionate to form a charge–charge interaction and H-bonds. In the S pocket steric and hydrophobic effects play important roles in ligand binding. The steric effect is prominent at the position close to D597 and the heme propionate, as indicated by the circles in Figure 5.
Figure 5.
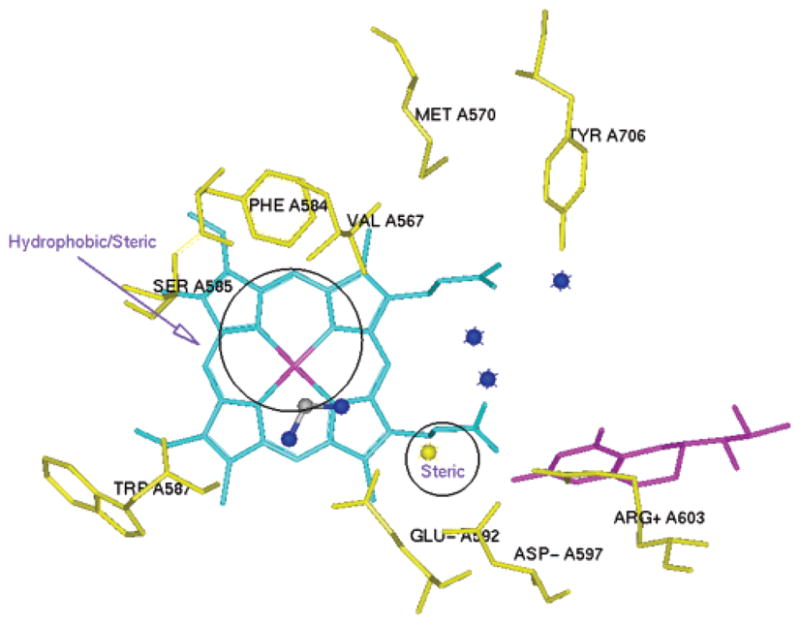
Minimal pharmacophoric elements for selective nNOS inhibitor design. An aminidino group is positioned close to E592. A yellow nitrogen atom is close to D597. The regions where hydrophobic and/or steric interactions play important roles are indicated by circles. Three blue nitrogen atoms are placed close to the heme propionate.
A focused fragment library was generated based on the minimal pharmacophoric elements for each pharmacophore. The fragments were then docked into the active site of nNOS where the corresponding pharmacophore is located. It is noteworthy that the amidino group and the nitrogen atoms are directional and require rather rigorous positioning for optimal ligand–receptor interactions. Thus, the fragments that are able to match their requirements are limited. However, the options for fragments for hydrophobic and steric interactions are rather broad when the basic fragment library and the bioisostere library are queried. That is, targeting hydrophobic and steric interactions would offer diverse fragments for each pharmacophore initially. A small subset of this focused fragment library is described here. To match the requirements of the amidino group and hydrophobic/steric effects, the 2-aminopyridine group was selected as a basic fragment. One advantage of the 2-aminopyridine fragment is that the pKa value of 2-amino-6-methylpyridine is 6.69.67 This fragment could act as a charge switch: in the small intestine the fragment could be in its neutral form, which is favorable for absorption; in the NOS active site, the local acidic environment could convert it into the positively charged form, which is favorable for binding. Starting with the nitrogen atom close to D597 in Figure 5, the pyrrolidino fragment was generated as a substitute for the α-amino group of 1–3. The pyrrolidino group is not only able to meet the charge–charge interaction requirement for nNOS selectivity but also to match the steric effect requirement for NOS binding. Another advantage of using the pyrrolidine ring is that the secondary amino group is more lipophilic and has less polar surface area (PSA) compared to the primary amino group of 1–3, which is better for in vivo inhibitor delivery.68 The ethylenediamine fragment was chosen to form a charge–charge interaction and H-bonds with the two heme propionate groups. After the linking of these fragments, compound 4 in Figure 6 emerged as the desired molecule. The mode of action of this molecule with the active site of nNOS was confirmed by AutoDock docking analysis. Some analogues (5–9) and their corresponding trans isomers (10–15) were designed to verify the derived pharmacophores and to provide preliminary structure–activity relationships.
Figure 6.

Chemical structures of the initially designed molecules.
3. Chemistry
Compounds 8 and 9 and their corresponding trans isomers 14 and 15 were synthesized by the route shown in Scheme 1a. The epoxidation of 1-benzyl-3-pyrroline using 3-chloroperoxybenzoic acid (m-CPBA) generated 17 in a 77% yield. Sulfuric acid was used to protonate the tertiary amine to avoid an N-oxidation side reaction. Boc-protected 2-amino-6-picoline (16) was treated with 2 equiv of n-butyllithium (n-BuLi) and allowed to react with epoxide 17 to form alcohol 18 in a 90% yield. The alcohol was then converted to ketone 19 by a standard Swern oxidation. The amine compounds were prepared by a reductive amination reaction. Cis isomer 20 and trans isomer 21 were separated by silica gel column chromatography; the more nonpolar fraction corresponded to the cis structure. The structures of the isomers were characterized by mass spectrometry, 1H NMR, 13C NMR, 13C NMR–DEPT, 1H–1H COSY, 1H–13C HMQC, and 1H–1H NOESY spectra (Supporting Information Figure 6). The NOE of the hydrogens communicating between carbon 6 and carbon 10 was only observed in the trans compound as indicated in Supporting Information Figure 7B. No NOE was observed for the cis isomer (Supporting Information Figure 7A). When sodium cyanoborohydride was used as the reducing reagent, 3 Å molecular sieves were added as a water trap, dry methanol was used as the solvent, and 1 equiv of acetic acid was added as a proton source. The ratio of the cis to trans isomers was generally 45:55. Deprotection of the Boc group of compounds 20 or 21 afforded final products 8, 9, 14, and 15. It is noteworthy that the 1H NMR and 13C NMR spectra of the cis and trans isomers of the final products are quite different from each other, as shown in Supporting Information Figure 8. One prominent difference is that the 13C chemical shift of the carbon atom attached to the pyridine ring (carbon 6 in Supporting Information Figure 7) is 29.5 ppm for the cis compound, but 34.1 ppm for the trans isomer, which can be used for structural characterization in future inhibitor optimizations.
Scheme 1.

Compounds 4–6 and their trans isomers 10–12 (Figure 6) were synthesized by the route in Scheme 1b. There was only one reaction different from that of Scheme 1a. Alcohol intermediate 23 was oxidized to ketone 24 by a Dess–Martin oxidation in a yield of 96%, while the Swern oxidation reaction was rather inefficient and only afforded a 54% yield. The two isomers (25 and 26) also can be separated cleanly by silica gel column chromatography after the reductive amination reaction. The more nonpolar isomers, which were believed to be the cis isomers (4 and 6) based on the above multidimensional NMR analyses, were cocrystallized with rat nNOS. The X-ray crystallographic analysis described below confirms the cis stereochemistry for 4 and 6.
Compound 7 and its trans isomer 13 were prepared according to the synthetic route in Scheme 2. 3-Cyclopenten-1-ol was converted to its N-Boc amine derivative 27 by a Mitsunobu reaction and a hydrolysis reaction.69 The epoxidation of 27 using 3-chloroperoxybenzoic acid (m-CPBA) generated 28 and its trans isomer 28a, which were separated by silica gel column chromatography. The structural characterization of 28 and its trans isomer has been elucidated by Barrett et al.70 The ratio of the cis to trans isomers was 75:25. Three equivalents of n-BuLi were essential to successfully generate alcohol intermediate 29 in a 50% yield. Several methods were attempted for the conversion of 29 to 30. A combination of tetrapropylammonium perruthenate (TPAP)/4-methylmorpholine N-oxide (NMO) oxidation in 10% acetonitrile in dichloromethane successfully generated 30 in a 67% yield,71 while neither the Swern oxidation nor Dess–Martin oxidation was effective. Reductive amination provided amines 31 and 32, which can be separated cleanly by silica gel column chromatography; the ratio of the cis to trans isomer was 60:40.
Scheme 2.

4. NOS Inhibition and Structure–Activity Relationships
The racemic mixtures of 4 and 6 were used for the inhibitor complex crystal structure determination with the rat nNOS heme domain. Crystallographic analysis of new NOS inhibitors generated from the proposed fragment hopping method is described in detail elsewhere.72 To briefly summarize, the structure of nNOS complexed with 4 (PDB accession 3B3N) was solved to 1.98 Å resolution with R/Rfree = 0.23/0.27, and the complex with 6 (PDB accession 3B3M) was solved to 1.95 Å with R/Rfree = 0.20/0.23). Only one of the two cis enantiomers, the (3′S,4′S)-isomer, was bound to the active site. The enantiomer binding preference for 4 and 6 in the crystal structures is the same as that predicted by the above pharmacophore-driven strategy for fragment-based de novo design (fragment hopping). Figure 7 is the superimposition of the binding conformation (blue) and the predicted bioactive conformation (yellow) for 4 or 6 in the active site of nNOS. The 2-aminopyridino group of 4 and 6 binds to residue E592 in the active site. The nitrogen atom of the pyrrolidine ring is located in the selective region defined by residues nNOS D597/eNOS N368. The nitrogen atom next to the pyrrolidine ring forms a H-bond with one of the heme propionate groups. There is some difference concerning the location of the terminal amino group of 4. As indicated in Figure 7A, in the crystal structure it forms H-bonds and charge–charge interactions with the heme propionate of the pyrrole D ring and one structural water, while in the predicted bioactive conformation it is located in the middle of two heme propionates to form H-bonds and charge–charge interactions with both heme propionates. The location of the terminal hydroxyl group of 6 in the crystal structure is also different from that of the predicted bioactive conformation. The bioactive conformation was predicted to form a H-bond directly with the heme propionate of the pyrrole A ring. However, the binding conformation from the crystal structure in complex with nNOS exhibits a H-bonding interaction with the heme propionate through the medium of a structural water molecule (Figure 7B). Since the interaction of the terminal hydroxyl group of 6 with the enzyme is rather weak, the root-mean-square (rms) deviation of the side chain heavy atoms of 6 between the actual binding conformation and the predicted bioactive conformation is larger than that for 4.
Figure 7.

Superimposition of the binding conformation (blue) and predicted bioactive conformation (yellow) of 4 (A) and 6 (B) in the active site of rat nNOS. The heme (orange), H4B (violet), and structural water (green) involved in the binding of 4 and 6 to nNOS are shown. The distances of some important H-bonds between the residues, structural water, cofactors, and inhibitors are given in angstroms (Å).
Table 3 shows the results of the NOS enzyme assays. The nNOS inhibitory activity (Ki) for 4 is 388 nM with high selectivity for nNOS over eNOS (1100 fold). Compound 4 is a racemic mixture; theoretically only the active (3′S,4′S)-enantiomer binds to the active site, which means that the single enantiomer should generate enhanced inhibitory activity. The selectivity of 4 between nNOS and iNOS is 150-fold. These selectivities are comparable to those attained with the nitroarginine dipeptide amide analogues made previously.32–36 A comparison of the NOS assay results between 4 and 10 reveals that the cis isomer is more potent and more selective than the corresponding trans isomer, as was predicted by fragment hopping. This indicates that the nitrogen atom attached to the pyrrolidine ring is a pharmacophore for nNOS inhibition and also is important for nNOS selectivity because it binds to the heme propionate and maintains the nitrogen atom of the pyrrolidine ring in the selective region defined by nNOS D597/eNOS N368.37,38 A comparison of the results for compounds 4, 5, and 6 indicates that the terminal amino group in the side chain of 4 also is an important pharmacophore for nNOS binding. The nNOS inhibitory activity for 5 is better than that for 6, which means that the phenyl group probably is located in a hydrophobic pocket in the active site of nNOS defined by M336 and L337. The residue corresponding to L336 in murine iNOS is N115. The nNOS/iNOS selectivity for 5 is better than that for 6, which further confirms that the phenyl group of 5 binds to the above hydrophobic pocket. The NOS inhibitory activities of the cis compounds, 8 and 9, and their trans isomers, 14 and 15, are very weak, which suggests that the benzyl group is too large to be accommodated by the M pocket. Derivative 7 is one atom longer than 4, but its nNOS inhibitory activity is 4-fold lower than that of 4. The bond length between the exocyclic primary amino nitrogen atom of 7 and the attached cyclopentane carbon atom (1.49 Å) is not long enough to allow the exocyclic amino group to form a direct H-bond interaction between the bridging water molecule and D597 (Figure 7). The selectivity between nNOS and eNOS also is decreased in 7, despite the exocyclic primary amino group of 7 being closer to nNOS D597 compared to the pyrrolidine nitrogen of 4. This suggests that maximal nNOS/eNOS selectivity requires that not only must a positively charged functional group be placed into the selective region defined by nNOS D597/eNOS N368, but it also has to be placed in an appropriate position to form strong interactions with the enzyme. It is interesting that the nNOS potency and the nNOS/eNOS selectivity of 13 are similar to those of its cis isomer 7, which means that the nitrogen atom of the ethylene diamine fragment which is attached to the five-membered ring is less important in the cyclopentane derivatives, compared to the pyrrolidine-type lead structure, although the overall trend of structure–activity/selectivity relationship between the cis and trans isomers is still the same.
Table 3.
NOS Enzyme Assay Results for the Lead Compounds
| compd |
Ki (μM) |
selectivity |
|||
|---|---|---|---|---|---|
| nNOS | eNOS | iNOS | n/e | n/i | |
| (±)-4 | 0.388 | 434.5 | 58.4 | 1114 | 150 |
| (±)-5 | 3.17 | 88.5 | 166.2 | 28 | 53 |
| (±)-6 | 9.44 | 366.6 | 143.1 | 39 | 15 |
| (±)-7 | 1.59 | 403.5 | 51.9 | 254 | 33 |
| (±)-8 | 47.94 | 121.6 | 609.4 | 3 | 13 |
| (±)-9 | 305.0 | 849.2 | 2523 | 3 | 8 |
| (±)-10 | 8.76 | 866.2 | 77.4 | 99 | 9 |
| (±)-11 | 13.28 | 85.2 | 143.8 | 6 | 11 |
| (±)-12 | 28.54 | 683.7 | 233.8 | 24 | 8 |
| (±)-13 | 2.34 | 242.3 | 71.5 | 104 | 31 |
| (±)-14 | 13.98 | 44.7 | 60.1 | 3 | 4 |
| (±)-15 | 190.3 | 677.7 | 1750 | 4 | 9 |
Designed lead compound 4 forms interactions with selective residue D597 via two structural water molecules. A similar interaction was observed in the crystal structure of nNOS in complex with the nitroarginine-containing dipeptide or peptidomimetic inhibitors 1–3.37 Figure 8A shows the binding mode of 2 in the crystal structure of rat nNOS. A superimposition of the binding conformations of 4 and 2 indicates that they have similar binding modes with nNOS (Figure 8B). The NOS inhibition and selectivity profile of 4 (Table 3) are very similar to those of compounds 1–3 (Figure 1). This suggests that the small organic molecule 4 derived from fragment hopping mimics the mode of action of the peptide or peptidomimetic inhibitors with the enzyme. The new peptidomimetic inhibitor was generated by the approach we proposed, and the prediction about the selective region in the active site of NOS being defined by nNOS D597/eNOS N368 was verified by inhibitor design. Furthermore, the flexibility of dipeptide inhibitors in Figure 1 was constrained in 4 by the introduction of a pyrrolidine ring. Peptide or peptidomimetic inhibitors are often very potent and exhibit high specificity for their targets. However, poor oral bioavailability, metabolic instability, and difficulty in passage through biomembranes are serious disadvantages. The methodology presented here offers a way to design small biologically active peptidomimetic molecules.
Figure 8.

A. Binding conformation of 2 in complex with rat nNOS. B. Superimposition of the binding conformations of 4 (blue) and 2 (yellow) in rat nNOS. Heme (orange), H4B (violet), and the structural water molecules (green) involved in the binding of the inhibitors to nNOS are shown. The distances of some important H-bonds between the residues, structural water, cofactors, and inhibitors are given in angstroms (Å).
5. Comparison of Fragment Hopping with Known Experimental and Computational Approaches
The fragments used in conventional fragment-based approaches are usually in the molecular weight range of 120–250 Da, containing 8–18 non-hydrogen atoms.6b This molecular size is essential because of the detection limit of biophysics-guided fragment screening;7 however, the essential structural requirement for a specific binding pocket is much smaller than these fragments in most cases. To elucidate the minimal pharmacophoric elements for each binding pocket of a druggable target and to derive various fragments with different chemotypes, a much wider chemical space can be explored. On the other hand, with the use of fragment hopping, key fragments can be identified and linked more efficiently in three-dimensional space. The selective region in the active site responsible for ligand selectivity is rather delicate in many cases. As discussed above, the chemical structure of compound 7 is very similar to that of 4. The primary amino group attached to the cyclopentane ring is also located in the selective region defined by nNOS D597/eNOS N368, but its selectivity is much lower than that of 4. As noted in our previous study,38 L-NNA also contains an α-amino group as in the case of compounds in Figure 1, yet L-NNA exhibits no selectivity between nNOS and eNOS. This is because the carboxylate group of L-NNA is shielding the α-amino group from the influence of the selective residues nNOS D597/eNOS N368. It is difficult for fragment-based approaches to differentiate this kind of small difference. Although conventional fragment-based approaches are able to identify and characterize those fragments located in the “hot spot” of the active site,73 the fragments that are responsible for isozyme selectivity are generally not located in the “hot spots”. As indicated in Figure 9, thirteen MCSS energy minima were obtained when the pyrrolidinium group was subjected to a MCSS calculation. These thirteen MCSS minima can be clustered into five groups. Group I: minima no. 1–no. 4 (−132.65 kcal/mol to −129.74 kcal/mol) are in the middle of the two heme propionates. Group II: minima no. 5 (−129.30 kcal/mol) and no. 6 (−127.60 kcal/mol) are located between E592 and the heme propionate of the pyrrole A ring. Group III: minima no. 7 (−111.82 kcal/mol) and no. 8 (−107.15 kcal/mol) form charge–charge interactions only with the heme propionate of the pyrrole A ring. Group IV: minima no. 9 (−104.75 kcal/mol) and no. 10 (−101.80 kcal/mol) form charge–charge interactions only with the heme propionate of the pyrrole D ring. Group V: minima no. 11–no. 13 (−99.25 kcal/mol to −97.86 kcal/mol) bind to D597 directly. None of the MCSS-minimized pyrrolidinium cations are placed in the position of the pyrrolidine ring found in the crystal structure of nNOS in complex with 4 (Figure 9). Fragment hopping determines the minimal pharmacophoric elements for each pharmacophore that are important for ligand selectivity. Then fragments are generated to match the requirement of minimal pharmacophoric elements based on the basic fragment and bioisostere libraries. After the focused fragment library is generated for each pharmacophore, conventional fragment-based approaches, such as NMR-based and/or X-crystallography-based fragment screening techniques, click chemistry, and dynamic combinatorial chemistry, can be utilized to investigate the binding mode of the above-generated fragments within the active site of the enzyme. The interaction of the generated fragments with the selective regions of the active site can be analyzed further by tethering or tethering with an extender. Therefore, fragment hopping as a pharmacophore-driven strategy is an open system that can incorporate other techniques and provide a more efficient pathway to generate more potent and more selective inhibitors. For example, compound 4 obtained in the present study is one of the most selective inhibitors of nNOS with nanomolar inhibitory potency.
Figure 9.

Representative MCSS-minimized positions of the pyrrolidinium group in the active site of rat nNOS in complex with 4. Cofactors heme and H4B are shown in cyan and magenta, respectively. The S pocket and the M pocket are noted. The label of the minima denotes their ranking. Hydrogen atoms attached to the nitrogen atom of the pyrrolidine ring are shown to indicate the spatial orientation of the pyrrolidinium cation.
In some cases, fragments identified by classical fragment-based methods do not have the same binding site as the linked molecule. Babaoglu and Shoichet parsed a typical, moderately potent (Ki = 1 μM) β-lactamase inhibitor into its component fragments. Crystallographic analysis of less complex fragments bound to the enzyme revealed that they do not recapitulate the binding mode of the original inhibitor.74 Instead, the compounds probe an entirely new region of the active site. That means that less complex molecules (that is, fragments with fewer points of interaction) should be able to bind in multiple ways to a given receptor binding site.75 Fragment hopping proposed here determines the positioning of the potential pharmacophores and then places diverse fragments to match each minimal pharmacophoric element. After linking, the molecule shows the same spatial orientation as the pharmacophores and is able to exhibit the maximal desirable biological activities. To date there have been about 38 computation-based de novo design programs reported.76,77 In conventional computation-based de novo design strategies, the output structures obtained from the computer program can be problematic with regard to synthetic accessibility76 and binding affinity prediction.78 Analyses of conventional computation-based de novo design techniques indicate that it is rare to generate novel lead structures with nanomolar activity initially.76 Recently de novo design methods, such as graph framework-based inhibitor design approaches, (e.g., scaffold hopping79) and privileged substructure-based design,80 have been proposed. The starting point for these types of methods is the selection of a template structure. Then isofunctional but structurally dissimilar substructures (scaffolds) are hopped into the different parts of the template structure. These methods can decrease the risks of molecular construction or synthetic accessibility, increase the hit rate for lead generation, and offer certain structural diversity. However, the skeleton of the newly designed molecules is confined to the basic architecture of the template structure, which usually comes from a known drug or drug candidate. Moreover, mimicking the different parts of the template structure with scaffolds often does not optimize the interaction between the ligand and the receptor to the maximal extent because the scaffold is quite large, and sometimes rigidity of the template structure does not allow an optimal match between ligand and receptor. To overcome the problem arising from binding affinity predictions in de novo design, the concept of minimal pharmacophoric elements proposed here can map an important interaction pattern between a ligand and a receptor based on a priori knowledge and experience. As noted in Figure 2, experiment-based methods, computation-based methods, receptor-based methods, or ligand-based methods can find their roles in deriving an accurate pharmacophore model. The approach proposed here is an open system that provides a basic platform for medicinal chemistry-driven efforts. To solve the problem arising from synthetic accessibility, common chemical bonds in drugs or drug candidates are considered preferentially in this approach (Supporting Information Figure 5).55b,c,62 The bioisostere principles and SciFinder search engine are two effective tools to design synthetically feasible molecules.
Conclusion
To the best of our knowledge this is the first time that the concept of minimal pharmacophore elements has been proposed, and this concept has been combined with fragment-based inhibitor design to generate inhibitors with novel structures. It is also the first published example of the de novo design of potent and selective nNOS inhibitors. These small organic molecules designed from the proposed approach mimic the biological function of the earlier nitroarginine-containing peptide inhibitors. Thus, a new peptidomimetic strategy, referred to as fragment hopping, which creates small organic molecules that mimic the biological function of peptides, has been established as a new type of fragment-based inhibitor design.
This new de novo design methodology is an open system; it merges new discovery and development of related research fields and provides a window for future modification. The final decision regarding inhibitor design in this approach is made by the medicinal chemist on the basis of his/her requirements and an understanding of a specific research project. A minimal pharmacophoric element is the core concept of the proposed approach. It can be derived by receptor-based active site analyses or ligand-based structure–activity relationship studies. In our approach, the bioisoterism concept proved to be a research tool of utmost importance in the generation of a focused fragment library or for the linking of fragments to form effective and accessible molecules with strong structural diversity. As an open system, the approach established here also efficiently incorporates early “ADME/Tox” considerations. The functional groups that potentially influence metabolic stability and toxicophores are taken into account during the generation of the focused fragment library and in the linking of fragments.
Using the established approaches, an inhibitor of nNOS, compound 4 in Figure 6, was discovered with nanomolar inhibitory potency and high selectivity for nNOS over eNOS and iNOS. The structure–activity relationship analyses of the derivatives are consistent with the model of minimal pharmacophoric elements. The crystal structure of rat nNOS in complex with 4 confirms that the binding conformation of 4 is the same as the predicted docking conformation. Compound 4, as a small organic molecule with a constrained conformation, can exactly mimic the mode of action of the dipeptide nNOS inhibitors. Therefore, the proposed pharmacophore-driven strategy for fragment-based de novo design provides a new peptidomimetic strategy. The molecular size of obtained compound 4 is rather small (17 non-hydrogen atoms), indicating that the lead structure designed by this newly established approach has high ligand efficiency,81 which provides a good starting point for further inhibitor optimization.
Supplementary Material
Acknowledgments
The authors are grateful for financial support from the National Institutes of Health, GM 49725 to R.B.S., GM57353 to T.L.P., and GM52419 to Dr. Bettie Sue Masters, in whose lab P.M. and L.J.R. work, and Grant No. AQ1192 from The Robert A. Welch Foundation to B.S.M. P.M. is also supported by Grant KAN200200651 from Academy of Science, Czech Republic.
Footnotes
Supporting Information Available: Complete refs 13, 42b; full experimental details; a basic fragment library, which is constructed on the basis of the fragments extracted directly from known drugs and/or drug candidates; a bioisostere library, which is constructed on the basis of known bioisosteric principles reported in the literature; rules for metabolic stability; a toxicophore library to provide a focused library for a specific pharmacophore; a constructed side chain library to link fragments, which is converted into a LUDI linking library; 1H NMR, 13C NMR, 13C NMR–DEPT, 1H–1H COSY, 1H–13C HMQC, and 1H–1H NOESY spectra; the PDB registration numbers, data collection, and refinement statistics of nNOS in complex with 4 and 6. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Bohacek RS, McMartin C, Guida WC. Med Res Rev. 1996;16:3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Posner BA. Curr Opin Drug Discovery Dev. 2005;8:487–494. [PubMed] [Google Scholar]
- 3.Teague SJ, Davis AM, Leeson PD, Oprea T. Angew Chem, Int Ed. 1999;38:3743–3748. doi: 10.1002/(SICI)1521-3773(19991216)38:24<3743::AID-ANIE3743>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 4.Oprea TI, Davis AM, Teague SJ, Leeson PD. J Chem Inf Comput Sci. 2001;41:1308–1315. doi: 10.1021/ci010366a. [DOI] [PubMed] [Google Scholar]
- 5.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Adv Drug Delivery Rev. 1997;23:3–25. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- 6.(a) Erlanson DA, McDowell RS, O’Brien T. J Med Chem. 2004;47:3463–3482. doi: 10.1021/jm040031v. [DOI] [PubMed] [Google Scholar]; (b) Rees DC, Congreve M, Murray CW, Carr R. Nat Rev Drug Discovery. 2004;3:660–672. doi: 10.1038/nrd1467. [DOI] [PubMed] [Google Scholar]
- 7.(a) Makara GM, Athanasopoulos J. Curr Opin Biotech. 2005;16:666–673. doi: 10.1016/j.copbio.2005.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Comess KM, Schurdak ME. Curr Opin Drug Discovery Dev. 2004;7:411–416. [PubMed] [Google Scholar]
- 8.Lepre CA, Moore JM, Peng JW. Chem Rev. 2004;104:3641–3675. doi: 10.1021/cr030409h. [DOI] [PubMed] [Google Scholar]
- 9.Shuker SB, Hajduk PJ, Meadows RP, Fesik SW. Science. 1996;274:1531–1534. doi: 10.1126/science.274.5292.1531. [DOI] [PubMed] [Google Scholar]
- 10.(a) Swayze EE, Jefferson EA, Sannes-Lowery KA, Blyn LB, Risen LM, Arakawa S, Osgood SA, Hofstadler SA, Griffey RH. J Med Chem. 2002;45:3816–3819. doi: 10.1021/jm0255466. [DOI] [PubMed] [Google Scholar]; (b) Erlanson DA, Wells JA, Braisted AC. Annu Rev Biophys Biomol Struct. 2004;33:199–223. doi: 10.1146/annurev.biophys.33.110502.140409. [DOI] [PubMed] [Google Scholar]
- 11.Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, Wells JA. Proc Natl Acad Sci USA. 2000;97:9367–9372. doi: 10.1073/pnas.97.17.9367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Hartshorn MJ, Murray CW, Cleasby A, Frederickson M, Tickle IJ, Jhoti H. J Med Chem. 2005;48:403–413. doi: 10.1021/jm0495778. [DOI] [PubMed] [Google Scholar]; (b) Nienaber VL, Richardson PL, Klighofer V, Bouska JJ, Giranda VL, Greer J. Nat Biotechnol. 2000;18:1105–1108. doi: 10.1038/80319. [DOI] [PubMed] [Google Scholar]
- 13.Dickopf S, Frank M, Junker HD, Maier S, Metz G, Ottleben H, Rau H, Schellhaas N, Schmidt K, Sekul R, Vanier C, Vetter D, Czech J, Lorenz M, Matter H, Schudok M, Schreuder H, Will DW, Nestler HP. Anal Biochem. 2004;335:50–57. doi: 10.1016/j.ab.2004.08.033. [DOI] [PubMed] [Google Scholar]
- 14.Wood WJ, Patterson AW, Tsuruoka H, Jain RK, Ellman JA. J Am Chem Soc. 2005;127:15521–15527. doi: 10.1021/ja0547230. [DOI] [PubMed] [Google Scholar]
- 15.(a) Lewis WG, Green LG, Grynszpan F, Radic Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (b) Whiting M, Muldoon J, Lin YC, Silverman SM, Lindstrom W, Olson AJ, Kolb HC, Finn MG, Sharpless KB, Elder JH, Fokin VV. Angew Chem, Int Ed. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- 16.Ramstrom O, Lehn JM. Nat Rev Drug Discovery. 2002;1:26–36. doi: 10.1038/nrd704. [DOI] [PubMed] [Google Scholar]
- 17.Erlanson DA, Lam JW, Wiesmann C, Luong TN, Simmons RL, DeLano WL, Choong IC, Burdett MT, Flanagan WM, Lee D, Gordon EM, O’Brien T. Nat Biotechnol. 2003;21:308–314. doi: 10.1038/nbt786. [DOI] [PubMed] [Google Scholar]
- 18.Erlanson DA, Hansen SK. Curr Opin Chem Biol. 2004;8:399–406. doi: 10.1016/j.cbpa.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 19.Murray CW, Verdonk ML. J Comput–Aided Mol Des. 2002;16:741–753. doi: 10.1023/a:1022446720849. [DOI] [PubMed] [Google Scholar]
- 20.(a) Vajda S, Guarnieri F. Curr Opin Drug Discovery Dev. 2006;9:354–362. [PubMed] [Google Scholar]; (b) Verdonk ML, Hartshorn MJ. Curr Opin Drug Discovery Dev. 2004;7:404–410. [PubMed] [Google Scholar]
- 21.Ji H, Tan S, Igarashi J, Li H, Derrick M, Martásek P, Roman LJ, Vásquez-Vivar J, Poulos TL, Silverman RB. doi: 10.1002/ana.21555. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alderton WK, Cooper CE, Knowles RG. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hobbs AJ, Higgs A, Moncada S. Annu Rev Pharmacol Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- 24.Siddhanta U, Presta A, Fan B, Wolan D, Rousseau DL, Stuehr DJ. J Biol Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 25.Vallance P, Leiper J. Nat Rev Drug Discovery. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- 26.Forstermann U, Munzel T. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 27.Reif DW, McCarthy DJ, Cregan E, Macdonald JE. Free Radic Biol Med. 2000;28:1470–1477. doi: 10.1016/s0891-5849(00)00250-1. [DOI] [PubMed] [Google Scholar]
- 28.Tinker AC, Wallace AV. Curr Top Med Chem. 2006;6:77–92. doi: 10.2174/156802606775270297. [DOI] [PubMed] [Google Scholar]
- 29.Babu BR, Griffith OW. Curr Opin Chem Biol. 1998;2:491–500. doi: 10.1016/s1367-5931(98)80125-7. [DOI] [PubMed] [Google Scholar]
- 30.(a) Salerno L, Sorrenti V, Di Giacomo C, Romeo G, Siracusa MA. Curr Pharm Des. 2002;8:177–200. doi: 10.2174/1381612023396375. [DOI] [PubMed] [Google Scholar]; (b) Erdal EP, Litzinger EA, Seo J, Zhu Y, Ji H, Silverman RB. Curr Top Med Chem. 2005;5:603–624. doi: 10.2174/1568026054679317. [DOI] [PubMed] [Google Scholar]; (c) Tafi A, Angeli L, Venturini G, Travagli M, Corelli F, Botta M. Curr Med Chem. 2006;13:1929–1946. doi: 10.2174/092986706777585031. [DOI] [PubMed] [Google Scholar]
- 31.(a) Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]; (b) Raman CS, Li H, Martásek P, Kral V, Masters BS, Poulos TL. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]; (c) Fischmann TO, Hruza A, Niu XD, Fossetta JD, Lunn CA, Dolphin E, Prongay AJ, Reichert P, Lundell DJ, Narula SK, Weber PC. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]; (d) Li H, Shimizu H, Flinspach M, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 32.Huang H, Martásek P, Roman LJ, Masters BS, Silverman RB. J Med Chem. 1999;42:3147–3153. doi: 10.1021/jm990111c. [DOI] [PubMed] [Google Scholar]
- 33.Hah JM, Roman LJ, Martásek P, Silverman RB. J Med Chem. 2001;44:2667–2670. doi: 10.1021/jm0101491. [DOI] [PubMed] [Google Scholar]
- 34.Hah JM, Martásek P, Roman LJ, Silverman RB. J Med Chem. 2003;46:1661–1669. doi: 10.1021/jm0202932. [DOI] [PubMed] [Google Scholar]
- 35.Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. J Med Chem. 2004;47:703–710. doi: 10.1021/jm030297m. [DOI] [PubMed] [Google Scholar]
- 36.Ji H, Gómez-Vidal JA, Martásek P, Roman LJ, Silverman RB. J Med Chem. 2006;49:6254–6263. doi: 10.1021/jm0604124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Flinspach ML, Li H, Jamal J, Yang W, Huang H, Hah JM, Gómez-Vidal JA, Litzinger EA, Silverman RB, Poulos TL. Nat Struct Mol Biol. 2004;11:54–59. doi: 10.1038/nsmb704. [DOI] [PubMed] [Google Scholar]
- 38.Ji H, Li H, Flinspach M, Poulos TL, Silverman RB. J Med Chem. 2003;46:5700–5711. doi: 10.1021/jm030301u. [DOI] [PubMed] [Google Scholar]
- 39.(a) Mattos C, Ringe D. Nat Biotechnol. 1996;14:595–599. doi: 10.1038/nbt0596-595. [DOI] [PubMed] [Google Scholar]; (b) Mattos C, Bellamacina CR, Peisach E, Pereira A, Vitkup D, Petsko GA, Ringe D. J Mol Biol. 2006;357:1471–1482. doi: 10.1016/j.jmb.2006.01.039. [DOI] [PubMed] [Google Scholar]
- 40.DeLano WL. Curr Opin Struct Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- 41.Ji H, Zhang W, Zhang M, Kudo M, Aoyama Y, Yoshida Y, Sheng C, Song Y, Yang S, Zhou Y, Lu J, Zhu J. J Med Chem. 2003;46:474–485. doi: 10.1021/jm020362c. [DOI] [PubMed] [Google Scholar]
- 42.(a) Goodford PJ. J Med Chem. 1985;28:849–857. doi: 10.1021/jm00145a002. [DOI] [PubMed] [Google Scholar]; (b) von Itzstein M, Wu WY, Kok GB, Pegg MS, Dyason JC, Jin B, Van Phan T, Smythe ML, White HF, Oliver SW, Colman PM, Varghese JN, Ryan DM, Woods JM, Bethell RC, Hotham VJ, Cameron JM, Penn CR. Nature. 1993;363:418–423. doi: 10.1038/363418a0. [DOI] [PubMed] [Google Scholar]
- 43.(a) Miranker A, Karplus M. Proteins. 1991;11:29–34. doi: 10.1002/prot.340110104. [DOI] [PubMed] [Google Scholar]; (b) Zoete V, Meuwly M, Karplus M. Proteins. 2005;61:79–93. doi: 10.1002/prot.20528. [DOI] [PubMed] [Google Scholar]
- 44.Laskowski RA, Thornton JM, Humblet C, Singh J. J Mol Biol. 1996;259:175–201. doi: 10.1006/jmbi.1996.0311. [DOI] [PubMed] [Google Scholar]
- 45.Boer DR, Kroon J, Cole JC, Smith B, Verdonk ML. J Mol Biol. 2001;312:275–287. doi: 10.1006/jmbi.2001.4901. [DOI] [PubMed] [Google Scholar]
- 46.An J, Totrov M, Abagyan R. Mol Cell Proteomics. 2005;4:752–761. doi: 10.1074/mcp.M400159-MCP200. [DOI] [PubMed] [Google Scholar]
- 47.Laurie AT, Jackson RM. Bioinformatics. 2005;21:1908–1916. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 48.(a) Dennis S, Kortvelyesi T, Vajda S. Proc Natl Acad Sci USA. 2002;99:4290–4295. doi: 10.1073/pnas.062398499. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Silberstein M, Dennis S, Brown L, Kortvelyesi T, Clodfelter K, Vajda S. J Mol Biol. 2003;332:1095–1113. doi: 10.1016/j.jmb.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 49.(a) Vajda S, Guarnieri F. Curr Opin Drug Discovery Dev. 2006;9:354–362. [PubMed] [Google Scholar]; (b) Moore WR., Jr Curr Opin Drug Discovery Dev. 2005;8:355–364. [PubMed] [Google Scholar]; (c) Clark M, Guarnieri F, Shkurko I, Wiseman J. J Chem Inf Model. 2006;46:231–242. doi: 10.1021/ci050268f. [DOI] [PubMed] [Google Scholar]
- 50.Kastenholz MA, Pastor M, Cruciani G, Haaksma EEJ, Fox T. J Med Chem. 2000;43:3033–3044. doi: 10.1021/jm000934y. [DOI] [PubMed] [Google Scholar]
- 51.Patel Y, Gillet VJ, Bravi G, Leach AR. J Comput-Aided Mol Des. 2002;16:653–681. doi: 10.1023/a:1021954728347. [DOI] [PubMed] [Google Scholar]
- 52.Noeske T, Sasse BC, Stark H, Parsons CG, Weil T, Schneider G. Chem Med Chem. 2006;1:1066–1068. doi: 10.1002/cmdc.200600147. [DOI] [PubMed] [Google Scholar]
- 53.Mason JS, Good AC, Martin EJ. Curr Pharm Des. 2001;7:567–597. doi: 10.2174/1381612013397843. [DOI] [PubMed] [Google Scholar]
- 54.(a) Fejzo J, Lepre CA, Peng JW, Bemis GW, Ajay, Murcko MA, Moore JM. Chem Biol. 1999;6:755–769. doi: 10.1016/s1074-5521(00)80022-8. [DOI] [PubMed] [Google Scholar]; (b) Bemis GW, Murcko MA. J Med Chem. 1996;39:2887–2893. doi: 10.1021/jm9602928. [DOI] [PubMed] [Google Scholar]
- 55.(a) Hajduk PJ, Bures M, Praestgaard J, Fesik SW. J Med Chem. 2000;43:3443–3447. doi: 10.1021/jm000164q. [DOI] [PubMed] [Google Scholar]; (b) Ertl P. J Chem Inf Comput Sci. 2003;43:374–380. doi: 10.1021/ci0255782. [DOI] [PubMed] [Google Scholar]; (c) Lewell XQ, Judd DB, Watson SP, Hann MM. J Chem Inf Comput Sci. 1998;38:511–522. doi: 10.1021/ci970429i. [DOI] [PubMed] [Google Scholar]
- 56.(a) Vieth M, Siegel MG, Higgs RE, Watson IA, Robertson DH, Savin KA, Durst GL, Hipskind PA. J Med Chem. 2004;47:224–232. doi: 10.1021/jm030267j. [DOI] [PubMed] [Google Scholar]; (b) Ertl P, Jelfs S, Muhlbacher J, Schuffenhauer A, Selzer P. J Med Chem. 2006;49:4568–4573. doi: 10.1021/jm060217p. [DOI] [PubMed] [Google Scholar]; (c) Boda K, Johnson AP. J Med Chem. 2006;49:5869–5879. doi: 10.1021/jm050054p. [DOI] [PubMed] [Google Scholar]; (d) Sheridan RP. J Chem Inf Comput Sci. 2002;42:103–108. doi: 10.1021/ci0100806. [DOI] [PubMed] [Google Scholar]
- 57.(a) Boehm HJ, Boehringer M, Bur D, Gmuender H, Huber W, Klaus W, Kostrewa D, Kuehne H, Luebbers T, Meunier-Keller N, Mueller F. J Med Chem. 2000;43:2664–2674. doi: 10.1021/jm000017s. [DOI] [PubMed] [Google Scholar]; (b) Muller G. Drug Discovery Today. 2003;8:681–691. doi: 10.1016/s1359-6446(03)02781-8. [DOI] [PubMed] [Google Scholar]; (c) Carr R, Jhoti H. Drug Discovery Today. 2002;7:522–527. doi: 10.1016/s1359-6446(02)02245-6. [DOI] [PubMed] [Google Scholar]; (d) Lewell XQ, Jones AC, Bruce CL, Harper G, Jones MM, McLay IM, Bradshaw J. J Med Chem. 2003;46:3257–3274. doi: 10.1021/jm0300429. [DOI] [PubMed] [Google Scholar]
- 58.(a) Patani GA, LaVoie EJ. Chem Rev. 1996;96:3147–3176. doi: 10.1021/cr950066q. [DOI] [PubMed] [Google Scholar]; (b) Lima LM, Barreiro EJ. Curr Med Chem. 2005;12:23–49. doi: 10.2174/0929867053363540. [DOI] [PubMed] [Google Scholar]; (c) Thornber CW. Chem Soc Rev. 1979;8:563–580. [Google Scholar]; (d) Wermuth CG. Molecular variations based on isosteric replacements. In: Wermuth CG, editor. The practice of medicinal chemistry. 2. Chapter 13. Academic Press; San Diego: 2003. pp. 189–214. [Google Scholar]; (e) Abraham DJ. Analog design. In: Cannon JG, editor. Burger’s medicinal chemistry and drug discovery, vol. 1 drug discovery. 6. Chapter 16. John Wiley and Sons, Inc; New York: 2003. pp. 687–714. [Google Scholar]
- 59.(a) van de Waterbeemd H, Smith DA, Beaumont K, Walker DK. J Med Chem. 2001;44:1313–1333. doi: 10.1021/jm000407e. [DOI] [PubMed] [Google Scholar]; (b) Nassar AEF, Kamel AM, Clarimont C. Drug Discovery Today. 2004;9:1020–1028. doi: 10.1016/S1359-6446(04)03280-5. [DOI] [PubMed] [Google Scholar]; (c) Rishton GM. Drug Discovery Today. 1997;2:382–384. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]; (d) Rishton GM. Drug Discov Today. 2003;8:86–96. doi: 10.1016/s1359644602025722. [DOI] [PubMed] [Google Scholar]
- 60.(a) Kazius J, McGuire R, Bursi R. J Med Chem. 2005;48:312–320. doi: 10.1021/jm040835a. [DOI] [PubMed] [Google Scholar]; (b) Nassar AEF, Kamel AM, Clarimont C. Drug Discovery Today. 2004;9:1055–1064. doi: 10.1016/S1359-6446(04)03297-0. [DOI] [PubMed] [Google Scholar]; (c) Williams DP, Naisbitt DJ. Curr Opin Drug Discovery Dev. 2002;5:104–115. [PubMed] [Google Scholar]; (d) Llorens O, Perez JJ, Villar HO. J Med Chem. 2001;44:2793–2804. doi: 10.1021/jm0004594. [DOI] [PubMed] [Google Scholar]
- 61.(a) Böhm HJ. J Comput-Aided Mol Des. 1992;6:61–78. doi: 10.1007/BF00124387. [DOI] [PubMed] [Google Scholar]; (b) Böhm HJ, Banner DW, Weber L. J Comput-Aided Mol Des. 1999;13:51–56. doi: 10.1023/a:1008040531766. [DOI] [PubMed] [Google Scholar]
- 62.Bemis GW, Murcko MA. J Med Chem. 1999;42:5095–5099. doi: 10.1021/jm9903996. [DOI] [PubMed] [Google Scholar]
- 63.Wagner AB. J Chem Inf Model. 2006;46:767–774. doi: 10.1021/ci050481b. [DOI] [PubMed] [Google Scholar]
- 64.Morris GM, Goodsell DS, Halliday RS, Huey R, Hart WE, Belew RK, Olson AJ. J Comput Chem. 1998;19:1639–1662. [Google Scholar]
- 65.Charifson PS, Corkery JJ, Murcko MA, Walters WP. J Med Chem. 1999;42:5100–5109. doi: 10.1021/jm990352k. [DOI] [PubMed] [Google Scholar]
- 66.(a) Norinder U, Haeberlein M. Adv Drug Delivery Rev. 2002;54:291–313. doi: 10.1016/s0169-409x(02)00005-4. [DOI] [PubMed] [Google Scholar]; (b) Cruciani G, Carosati E, De Boeck B, Ethirajulu K, Mackie C, Howe T, Vianello R. J Med Chem. 2005;48:6970–6979. doi: 10.1021/jm050529c. [DOI] [PubMed] [Google Scholar]
- 67.Paudler WW, Blewitt HL. J Org Chem. 1966;31:1295–1298. [Google Scholar]
- 68.Ertl P, Rohde B, Selzer P. J Med Chem. 2000;43:3714–3717. doi: 10.1021/jm000942e. [DOI] [PubMed] [Google Scholar]
- 69.Berrée F, Michelot G, Le Corre M. Tetrahedron Lett. 1998;39:8275–8276. [Google Scholar]
- 70.Barret S, O’Brien P, Steffens HC, Towers TD, Voith M. Tetrahedron. 2000;56:9633–9640. [Google Scholar]
- 71.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639–666. [Google Scholar]
- 72.Igarashi J, Li H, Jamal J, Ji H, Fang J, Silverman RB, Poulos TL. To be submitted. [Google Scholar]
- 73.Böhm M, Klebe G. J Med Chem. 2002;45:1585–1597. doi: 10.1021/jm011039x. [DOI] [PubMed] [Google Scholar]
- 74.Babaoglu K, Shoichet BK. Nat Chem Biol. 2006;2:720–723. doi: 10.1038/nchembio831. [DOI] [PubMed] [Google Scholar]
- 75.Hajduk PJ. Nat Chem Biol. 2006;2:658–659. doi: 10.1038/nchembio1206-658. [DOI] [PubMed] [Google Scholar]
- 76.(a) Schneider G, Fechner U. Nat Rev Drug Discovery. 2005;4:649–663. doi: 10.1038/nrd1799. [DOI] [PubMed] [Google Scholar]; (b) Honma T. Med Res Rev. 2003;23:606–632. doi: 10.1002/med.10046. [DOI] [PubMed] [Google Scholar]
- 77.(a) Böhm HJ. Curr Opin Biotechnol. 1996;7:433–436. doi: 10.1016/s0958-1669(96)80120-0. [DOI] [PubMed] [Google Scholar]; (b) Bohacek RS, McMartin C. Curr Opin Chem Biol. 1997;1:157–161. doi: 10.1016/s1367-5931(97)80004-x. [DOI] [PubMed] [Google Scholar]
- 78.(a) Leach AR, Shoichet BK, Peishoff CE. J Med Chem. 2006;49:5851–5855. doi: 10.1021/jm060999m. [DOI] [PubMed] [Google Scholar]; (b) Jorgensen WL. Science. 2004;303:1813–1818. doi: 10.1126/science.1096361. [DOI] [PubMed] [Google Scholar]; (c) Shoichet BK. Nature. 2004;432:862–865. doi: 10.1038/nature03197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.(a) Schneider G, Neidhart W, Giller T, Schmid G. Angew Chem, Int Ed. 1999;38:2894–2896. [PubMed] [Google Scholar]; (b) Barker EJ, Buttar D, Cosgrove DA, Gardiner EJ, Kitts P, Willett P, Gillet VJ. J Chem Inf Model. 2006;46:503–511. doi: 10.1021/ci050347r. [DOI] [PubMed] [Google Scholar]
- 80.Schnur DM, Hermsmeier MA, Tebben AJ. J Med Chem. 2006;49:2000–2009. doi: 10.1021/jm0502900. [DOI] [PubMed] [Google Scholar]
- 81.Hopkins AL, Groom CR, Alex A. Drug Discovery Today. 2004;9:430–431. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.