

Abstract
AB5 toxins are important virulence factors for several major bacterial pathogens, including Bordetella pertussis, Vibrio cholerae, Shigella dysenteriae and at least two distinct pathotypes of Escherichia coli. The AB5 toxins are so termed because they comprise a catalytic A-subunit, which is responsible for disruption of essential host functions, and a pentameric B-subunit that binds to specific glycan receptors on the target cell surface. The molecular mechanisms by which these AB5 toxins cause disease have been largely unraveled, including recent insights into a novel AB5 toxin family, subtilase cytotoxin (SubAB). Furthermore, AB5 toxins have become a valuable tool for studying fundamental cellular functions, and are now being investigated for potential applications in the clinical treatment of human diseases.
Getting from A to B of AB5 toxins
AB5 toxins are an important weapon in the armory of virulence factors deployed by major bacterial pathogens, which collectively kill over a million people each year. Bordetella pertussis, which produces pertussis toxin (Ptx), is the aetiological agent of whooping cough, an acute respiratory infection that continues to cause high global morbidity and mortality in spite of the availability of effective vaccines. Cholera toxin (Ctx) produced by Vibrio cholerae and the closely related heat labile enterotoxins (LT) produced by enterotoxigenic Escherichia coli (ETEC) are principally responsible for the copious watery diarrhoea that is the hallmark of gastrointestinal infection with the respective organism. Cholera is endemic in Asia, but causes epidemics in nearly all parts of the globe 1. Indeed, it continues to represent a major threat to global human health, with the 2008 outbreak in Zimbabwe resulting in the deaths of over 4000 people. ETEC infection is highly prevalent in developing countries, where it is a major killer of young children. Visitors to regions where ETEC disease is endemic are also at high risk, hence the term “travellers’ diarrhoea”. ETEC diarrhoea is also a major problem for the livestock industry 2. Shiga toxin (Stx) is produced by Shigella dysenteriae and Shiga toxigenic E. coli (STEC), which also cause serious gastrointestinal disease in humans, ranging from diarrhoea to haemorrhagic colitis and life-threatening systemic sequelae such as the haemolytic uraemic syndrome (HUS). These manifestations are largely attributable to Stx-mediated damage to microvascular endothelial cells in the gut, kidneys and brain 3, 4. Outbreaks of STEC disease including HUS are common in developed countries (40 in the United States in 1999) 5; HUS has a 5-10% mortality rate for children and 35% for adults 5 and STEC infections are estimated to cause 500 deaths each year in the United States 2. The high prevalence and severity of all these bacterial infections emphasizes the importance of gaining a full understanding of the mode of action of the AB5 toxins that are central to disease pathogenesis.
Over the past two decades, approximately 30 crystal structures of AB5 toxins have been determined, including the complete holotoxins from the various pathogenic bacteria as well as the apo and holo forms of the individual A-subunit and B-subunit components 6. These major breakthroughs provided significant structural insights into the biological function and catalytic activity of the holotoxins. Based on sequence homology and the specific A-subunit catalytic activity, the AB5 toxins have been classified into four families (Figure 1). The B-subunit forms a ring-shaped pentamer that is responsible for the binding to the host cell surface, whereas the catalytic A-subunit disrupts the host’s cellular machinery. Despite sharing a similar structural architecture, the various AB5 family members can differ in their host cell surface receptor specificity, catalytic activity and intracellular trafficking. Here we review the various structural characteristics shared by the AB5 toxins and their implications in terms of toxicity, cellular targeting, and their use as cell biology reagents and potential therapeutics.
Figure 1. Crystal structures of members of the four recognised AB5 toxin families.
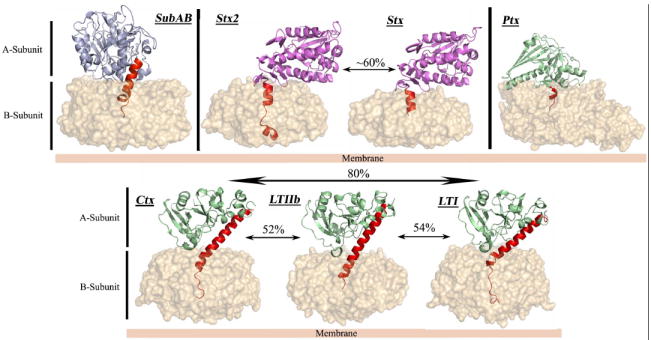
The B-subunit is represented as molecular surface. The A-subunits of SubAB, Ctx and LT, Stx and Ptx are shown in cartoon representation and coloured according to the respective catalytic activity (Light blue for subtilase activity, light green for ADP-ribosylase activity and purple for RNA N-glycosidase activity). The common structural element (Helix A2) is coloured in red, and the level of sequence identity of the A-subunit inside a family is also indicated.
The A-subunit: catalytic domain
The A-subunit of the bacterial AB5 toxins is a single polypeptide composed of two domains (A1 and A2) that are also linked together via a disulfide bond. The A1 domain comprises the catalytic domain responsible for the toxicity to the host cell. The A2 domain consists of an α-helix that penetrates into the central pore of the pentameric B-subunit, thereby non-covalently anchoring the A- and B-subunits together to create the holotoxin (Figure 1). This unusual arrangement is a common structural feature of all AB5 toxins, as first revealed by the crystal structure of LT from E. coli (for review see 6) . To date, the A-subunit crystal structures of seven members of the various AB5 toxin families have been determined either in their apo, holo or mutant forms (Figure 1) 7.
The AB5 toxins are subdivided into families according to A-subunit sequence homology and catalytic activity. The cholera toxin family (which includes Ctx and LT), and the Ptx family, trigger the ADP-ribosylation of the Gsα and Giα proteins in the cytosol, disrupting the respective G-protein signal transduction pathways. This results in an increase in intracellular cAMP levels and disregulation of ion transport mechanisms 6. The LT toxins have been serologically distinguished into two groups, termed type I and type II, the latter being further divided into LT-IIa and LT-IIb 8. The A-subunits of Ctx, LT-I and LT-II share approximately 55-80 % amino acid sequence identity 6, whereas the sequence identity between the catalytic subunit of Ptx (referred to as the S1 subunit) and the A-subunits of Ctx or LT is much lower (15-20%).
The Stx family members have RNA N-glycosidase activity, and inhibit eukaryotic protein synthesis by cleaving a specific adenine base from 28S rRNA, thereby causing cell death. STEC produce two major Stx classes (Stx1 and Stx2). Stx1 is essentially identical to classical Stx produced by S. dysenteriae (a single amino acid difference), and is about 60% identical at the amino acid level to Stx2, which also exists as several subtypes 9. Accordingly the three-dimensional structures of Stx and Stx2 revealed few structural differences 10. Interestingly however, people infected with Stx2-producing STEC strains are more likely to develop HUS than if they were infected by Stx1-producing STEC; this is most likely due to increased cytotoxicity of Stx2 for microvascular endothelial cells 3, 11.
The recent discovery of an additional STEC AB5 toxin, whose A-subunit shares homology with subtilase-like serine proteases and differs significantly from other AB5 toxins, led to the establishment of a fourth AB5 family 12, 13 (Figure 1). This subtilase cytotoxin (SubAB) was discovered in an O113:H21 STEC strain, which was responsible for an outbreak of HUS in South Australia in 1998, and has since been detected in numerous other STEC serotypes 3, 14, 15. SubAB is extremely cytotoxic in vitro for a range of cell types, and it is more toxic than Stx for Vero cells (kidney epithelial cells). It is also lethal for mice when injected intraperitoneally, producing pathological features that strongly resemble HUS in humans 12, 16. Subtilase family proteases are characterised by a conserved catalytic triad (comprising distinct Asp, His, and Ser motifs); substitution of the critical catalytic Ser in SubA with Ala abolished toxicity, indicating that the SubA subunit acts as a protease 12, 16. Proteomic and functional analysis subsequently revealed that BiP (also termed GRP78) is the specific molecular target for SubA 13. Unlike most subtilases, structural studies revealed that SubA possessed an unusually deep active site cleft (Figure 2), providing a basis for its exquisite substrate specificity for BiP 13, a highly conserved endoplasmic reticulum (ER) chaperone essential for survival of eukaryotic cells. BiP also maintains the permeability barrier of the ER membrane and plays a crucial role in the unfolded protein response (UPR) as the ER stress-signalling master regulator 17. Thus, cleavage of BiP has fatal consequences for the cell. Collectively, these recent findings for SubA reveal a novel mechanism in which bacterial toxins can induce cell death, via the specific proteolytic abrogation of chaperone function.
Figure 2. Molecular surface representations of a broad specificity protease and the SubAB A-subunit.
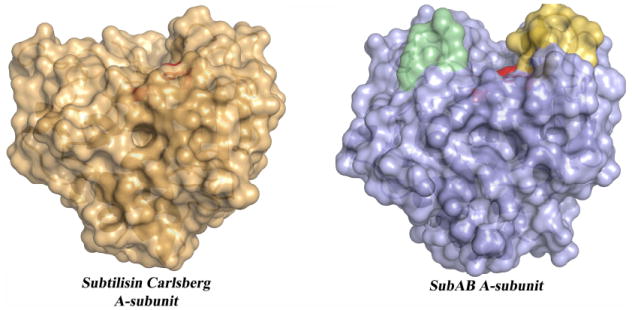
The position of the catalytic triad (His-Asp-Ser) in both X-ray structures is coloured in red. The Subtilisin Carlsberg (Pdb code: 1C3L) is a typical broad specificity protease with a shallow active site. The loops composed of the amino acid residues 81-88 and 234-239 forming a deeper binding cleft in SubAB are coloured in yellow and pale green, respectively.
In order to reach their molecular targets in the cytosol, the Ptx, Stx, Ctx and LT toxins recognize their cognate cell surface receptor via the pentameric B-subunit; this recognition event triggers endocytosis, followed by retrograde transport via the Golgi to the ER (Figure 3) 18. In the ER, protein disulfide isomerase (PDI) unfolds the A-subunit (A1) 19, which is then retro-translocated into the cytosol via the Sec61 channel 20, or via the derlin-1–Hrd1 complex 21-23. It then undergoes refolding and finally causes cellular toxicity. In Ctx and LT, the very end of the A2 domain contains an ER-targeting motif (KDEL or RDEL). However, this motif is clearly not essential, as it is not present in Stx. Furthermore, CtxB and LT-B undergo retrograde transport efficiently in the absence of their respective A subunits 24. SubAB also lacks a KDEL motif, and furthermore, the localization of its molecular target in the ER lumen implies that SubAB does not require retro-translocation into the cytosol to induce toxicity. Within the target cell, SubAB follows a retrograde pathway similar to that previously observed for Stx and Ctx. However, unlike Stx, Ctx and LT, which engage both clathrin-dependent and lipid raft pathways, trafficking of SubAB in Vero cells was shown to be entirely clathrin-dependent 25. The route through the Golgi is also distinct, with SubAB exploiting a novel p115–golgin-84-independent, COG–Rab6–COPI-dependent mechanism, and unlike Stx, retrograde transport is not dependent on the endosomal sorting nexins, SNX1 and SNX2 26.
Figure 3. [SC1]Schematic representation of the glycan surface recognition of the various AB5 toxins and their respective trafficking pathways into the cell.

The toxins’ A-subunits are shown as a pentagon and each subunit is coloured according to the specific catalytic activity (blue for protease activity, green and magenta for ADP-ribosyltransferase and N-glycosidase activities, respectively). The toxins bind to the cell-surface via their respective glycan receptors, where they are internalized to the early endosomal (EE) compartment. The names of these glycan receptors are indicated and inserted into schematic rings located on the surface of the cell and coloured according to their nature (grey and pale green for glycoproteins and glycolipids, respectively). The toxins are then trafficked to the Golgi then onto the endoplasmic reticulum (ER) where the A-subunit of the Stx, Ctx, LT and Ptx are separated from the B-pentamer. They are then exported out of the ER, where they are able to attack their respective molecular targets (the G-protein (brown triangle), the 28S ribosomal RNA, and the ribosome (teal[SC2]). In the case of SubAB, it is unknown whether SubA is separated from SubB; however the purified SubA and SubAB can cleave BiP (grey) in vitro 31.
The B5 receptor-binding domain: Same fold, different specificity
The B-subunits of the AB5 toxins recognise glycan receptors displayed on the cell surface. Receptor specificity is critical for the pathogenic process, as it determines host susceptibility, tissue tropism, and the nature and spectrum of the resultant pathology. In the past two decades, an increasing number of X-ray structures of AB5 toxin B-subunits, determined in their apo form as well as in complex with their respective A-subunits and various ligands or inhibitors, have been solved. This has provided crucial structural insights into the receptor specificity and mode of binding, which has informed inhibitor design 6, 27, 28. Despite low sequence identity, the B-subunits of the Cholera (Ctx, LTI and LTII), Shiga, and SubAB toxin families share the common oligosaccharide/oligonucleotide binding fold (OB) consisting of a five-stranded β-barrel flanked by an α-helix that assembles to form a homo-pentameric ring-shaped scaffold (Figure 4). By contrast, the B subunit of Ptx is a hetero-pentamer comprising four different monomers (S2, S3, two copies of S4, and S5). Despite sharing a common fold, the cell surface receptor specificity of the AB5 toxins varies from one family to the other and also within the same family. In the cholera toxin family, the Ctx and LT-I B pentamers specifically recognise the oligosaccharide moiety of the ganglioside GM1 29, whereas the LT-IIa and LT-IIb B subunits show affinity for gangliosides GD1b and GD1a, respectively 30. Stx/Stx1 and most variants of Stx2 bind the oligosaccharide component of the glycosphingolipid Gb3, which is expressed on the surface of the microvascular endothelium as well as kidney tubular epithelium, whereas a variant toxin, Stx2e, binds preferentially to Gb4 3. Glycan array (http://www.functionalglycomics.org/static/consortium/consortium.shtml) and subsequent functional studies have revealed that the recently discovered SubAB has unusual binding specificity for glycans terminating in α-3-linked N-glycolylneuraminic acid (Neu5Gc) 31. This sialic acid is not synthesised by humans (Box 1), but it is incorporated into the cell surface via dietary intake, with the most common source of Neu5Gc being dairy products and red meat. Ironically, these foods are the most common sources of STEC contamination, and so dietary choices can increase both the susceptibility of an individual to the toxin as well as the risk of infection with the pathogen that produces it 31.
Figure 4. The pentameric B-subunit of the AB5 toxin.
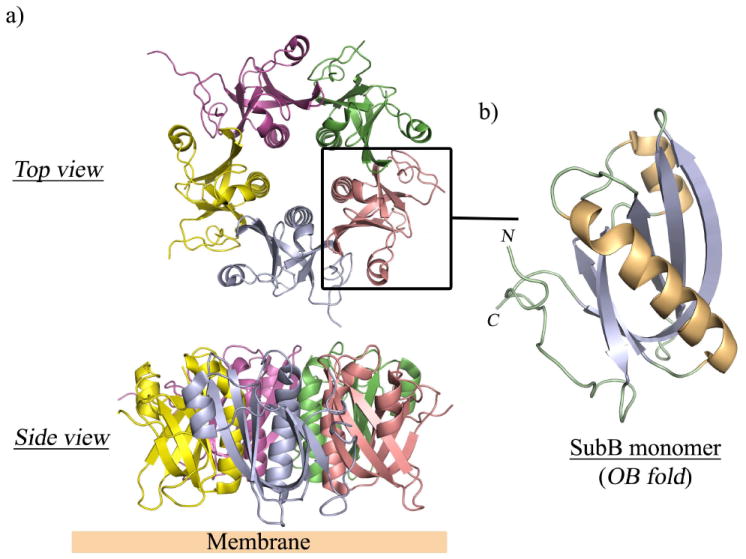
a) Overall architecture of the pentameric B-subunit of SubAB shown in two different orientations. Each monomer is coloured differently. b) Close up view of a monomer forming the common oligosaccharide/oligonucleotide (OB) fold. The monomer is coloured according to the secondary structural elements (helices, gold; β-strands, purple).
Box 1. Why have humans lost the ability to synthesise Neu5Gc?
Neu5Gc and Neu5Ac are sialic acids (Sia) that differ by a single hydroxyl group. They are attached to the termini of cell surface glycans of all vertebrates and are essential for development 67. Moreover, they are major targets of many pathogens 68. In humans the major Sia is Neu5Ac due to disruption of the gene (CMAH) encoding CMP-Neu5Ac hydroxylase, which converts Neu5Ac to Neu5Gc. This mutation occurred approximately 2 million years ago, after separation of the hominid lineage from the great apes 69. By contrast, Neu5Gc is the most abundant Sia in other mammals, including the great apes 70. Loss of Neu5Gc-containing glycans in humans was probably due to infection by pathogens, in particular malaria (Plasmodium falciparum), one of the most devastating human pathogens 71. A recent study analyzing different strains of P. reichenowi, which infects chimpanzees and gorillas, suggested that it gave rise to all strains of P. falciparum 72. In P. richenowi, the well-characterized red-blood cell binding protein EBA-175, which is required for infection, prefers Neu5Gc whereas P. falciparum EBA-175 prefers Neu5Ac 71. This switch in Sia preference was due to humans losing the ability to synthesise Neu5Gc, perhaps as a means to provide relief from infection.
Not only do the AB5 toxin B subunits show a broad variety in terms of receptor specificity, but the location and the number of binding sites for these glycans can also differ markedly (Figure 5). Interestingly, SubAB, LT and Ctx contain five receptor-binding sites (one per monomer), whereas Stx contains up to fifteen binding pockets (three per monomer) presenting various affinities 32. The receptor-binding site of the cholera toxin family members Ctx and LT is conserved and buried in a deep pocket formed by the L45, L3α and N-terminal loops located at the bottom of the B-subunit 33. The receptor GM1 makes extensive interactions with the B-subunit resulting in high affinity for the ganglioside 34. CtxB binds exclusively to GM1 whereas the LT B-subunit can also recognise N-acetyllactosamine-terminating glycolipids and glycoproteins 35 and certain blood group ABO antigens 36. Furthermore, the crystal structures of the B-subunit of a hybrid between CtxB and LT-IB in complex with a blood group A antigen analogue reveals a novel binding specificity and a binding site distinct from the GM1 site 37. More recently, the X-ray structure of the LT-I B-subunit showed that LT-I could bind the blood group A antigen ligand in the same shallow binding groove halfway down the subunit 38. The study provided important structural insight for the blood group antigen recognition by the enterotoxin and supported the idea that specificity of LT or even Ctx could play a role in the blood group dependence observed in ETEC infections and cholera.
Figure 5. Molecular surface representations of the B-subunits of Ctx, Stx1, SubAB, Ptx and LT-I with bound glycans.
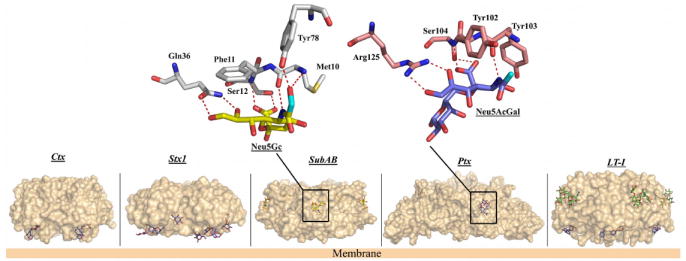
The Gb3 analogue (PK-MCO) is shown bound to Stx1. The antagonist BMSC-0011 (in blue) is shown bound to LT-I and Ctx. The second binding site of LT-I is also shown with the blood group A antigen analogue (in green) bound in a similar position as in SubAB, whereas Neu5Gc and Neu5Acα-3Gal are shown bound to SubAB and Ptx, respectively. A close-up view of the interactions for SubAB–Neu5Gc and Ptx–Neu5Acα2-3Gal is also represented. For clarity, only the amino acid residues involved in direct hydrogen bonds (dashed red lines) with the two ligands are shown. The position of the extra hydroxyl in Neu5Gc and the equivalent position in Neu5Acα2-3Gal are highlighted in cyan.
In the SubAB toxin, the non-human Neu5Gc also binds in a shallow pocket located halfway down the side of the B-subunit and formed by the β1 and β4 strands. The structure of SubB resembles the S2, S3 and S5 subunits of Ptx, with the S2 and S3 subunits also forming a similar binding pocket that binds sialylated glycoproteins (Figure 5) 31. Interestingly, the architecture of the active site of SubB provided a basis for understanding its unique specificity for Neu5Gc, and mutation of these key active-site residues abrogated binding and cytotoxicity 31. Collectively, these findings for SubAB provide a novel paradigm wherein a bacterial toxin’s receptor can be generated by metabolic incorporation of an exogenous factor derived from food.
AB5 toxins as innovative cellular tools
The highly specific actions of the AB5 toxin catalytic subunits have been exploited both to investigate the roles of the targeted processes in cellular physiology, and to specifically manipulate the associated signaling pathways. Most notably, the specific ADP-ribosylation of Gs and Gi by Ctx/LT and Ptx, respectively, has had a major impact on our understanding of the roles of G-protein-coupled receptor signaling in diverse physiological processes 39, 40. Experiments have typically involved treating cells with exogenous holotoxin in vitro, but in recent years, the role of Gi/o signaling in vivo has been examined using transgenic mice expressing the Ptx catalytic S1 subunit 41. The B-subunit pentamers of the AB5 toxins, particularly StxB and CtxB, have also proved to be highly valuable tools for detailed characterization of receptor-mediated endocytic pathways in eukaryotic cells (for review see 24).
The immunomodulatory and mucosal adjuvant properties of Ctx/LT are also well-established (reviewed in 42, 43). The Ctx/LT holotoxins elicit strong mucosal responses to co-administered antigens, through a combination of increasing the permeability of the mucosal epithelium, upregulating co-stimulatory molecule expression and enhancing presentation on antigen presenting cells, promoting isotype differentiation in B cells, and exerting an array of effects on T cell proliferation and cytokine production 42. Both A- and B-subunits contribute to these properties, and the precise ganglioside binding specificity of the respective B-subunits has been associated with differences in T helper type 1 (Th1) versus T helper type 2 (Th2) responses elicited by the two holotoxins 44. The isolated Ctx/LT B subunits have much reduced adjuvant activity, unless directly conjugated to the heterologous antigen 42. CtxB has also been used to promote mucosal tolerance to autoantigens or allergens 42. For example, patients with Behcet’s disease experienced significantly reduced numbers of uveitis relapses following oral administration of CtxB conjugated to an autoantigenic peptide derived from the human 60 kDa heat shock protein 45. More recent experiments demonstrated that administration of CtxB to the airways of allergic and naïve mice can suppress airway allergen-triggered eosinophilia, Th2 cytokine synthesis and bronchial hyperreactivity. These effects were both preventative and curative, and were mediated by stimulation of antigen-specific IgA production by B cells 46.
StxB also has potent immunomodulatory properties and has been used to deliver exogenous antigens to the major histocompatibility complex (MHC) Class I pathway of antigen presenting cells (which express the StxB receptor Gb3), thereby eliciting a strong cell mediated immune response (reviewed by 47). This property is mediated by the capacity of StxB to direct retrograde trafficking of conjugated antigens from endosomes to the ER compartment, enabling loading of the processed antigen onto nascent Class I MHC molecules prior to display on the cell surface. Thus, StxB has great potential as a non-living, non-toxic vaccine vector, which has already been demonstrated to impart anti-tumour protective immunity in mice 47. Its capacity to elicit cell-mediated immunity also points to potential for immunoprophylaxis and therapy of infectious diseases.
The exquisite specificity of the recently discovered SubAB toxin for BiP, and its capacity abolish BiP function in vitro and in vivo, also make it a powerful cell biological tool, particularly for investigating the roles of BiP and ER stress in a range of cellular processes. As BiP is essential for the survival of eukaryotic cells, Bip-/- cell lines and animal models are not available. Moreover, RNA knock-down approaches do no completely suppress BiP expression in transfected cells. However, treatment with SubAB results in rapid degradation of nearly all cellular BiP in a wide variety of cell lines, effectively mimicking a knock-out phenotype. The toxin has already been used to examine the role of BiP and/or ER stress in ER-associated degradation in HeLa cells 48, and regulation of gap junction function in mesangial cells 49. SubAB has also been employed to demonstrate the critical role of BiP in assembly and egress of cytomegalovirus from infected cells 50 and in production and processing of dengue virus proteins 51. Both viruses upregulate BiP during infection, and SubAB treatment significantly reduces infectious virus release. Like Ctx/LT, SubAB might also have immunomodulatory activities, affecting T-cell activation and inflammatory responses of a variety of cell types 52, 53. The toxin also preferentially inhibits secretion of immunoglobulins by activated murine B lymphocytes, leaving cytokine secretion relatively unscathed. SubAB preferentially cleaved newly synthesized BiP in these cells, and the C-terminal BiP fragment remained tightly bound to nascent immunoglobulin light chains, trapping them in the ER compartment 54. SubAB treatment also caused transient Akt phosphorylation and nuclear factor-kappaB (NF-κB) activation in rat renal tubular epithelials cells, effects which were mediated via the activating transcription factor 6 (ATF6) branch of the UPR 55. Interestingly however, at sub-cytotoxic concentrations, SubAB has anti-inflammatory properties and can inhibit lipopolysaccharide (LPS)-mediated NF-κB activation in a murine macrophage cell line, as well as protect mice from LPS-induced endotoxic lethality and experimental arthritis 56.
AB5 toxins as novel therapeutics
In addition to their potential use as immunomodulatory agents, AB5 toxins could be used as therapeutic agents against a range of diseases. In particular, their specific catalytic functions, their cell binding specificities, and their intracellular trafficking properties can be exploited in the treatment of cancer. Many tumours exhibit aberrent glycosylation patterns, enabling them to be targeted by AB5 toxin B-subunits, which exhibit high glycan binding specificity. StxB, for example, binds with high specificity to Gb3 (also referred to as CD77), surface expression of which is enhanced in a broad range of cancer cells, including colon, pancreatic, ovarian, breast and testicular cancers, as well as in myelomas, lymphomas, astrocytomas and meningiomas 47, 57-59. Thus, StxB has the capacity to target tumours in vivo preferentially over normal tissues, and then mediate uptake of holotoxin and intracellular delivery of the toxic A-subunit, which then inhibits cellular protein synthesis and kills the cancer cell. Indeed several studies have demonstrated inhibition by Stx holotoxin of tumour growth in vivo 47, 60, 61. Nevertheless, Gb3 is also expressed in a number of normal cells/tissues, particularly the kidneys, microvasculature endothelial cells, and CD77-positive dendritic cells and B lymphocytes, necessitating careful dose optimization. Notwithstanding the therapeutic potential of the Stx holotoxin, StxB has the capacity to deliver other toxic payloads or drugs, as well as molecular imaging reagents 24, 62, 63.
SubAB also has potential as an anti-cancer therapeutic. Many tumours up-regulate its unique substrate BiP in response to the ER stress induced by their own rapid growth. Such up-regulation is a crucial anti-apoptotic mechanism for the tumour and it is also linked to metastatic potential and resistance to chemotherapy 64. Thus, if SubA can be specifically targeted to these tumours, it has the potential to inactivate a cellular process that is crucial for survival of the cancer cell. Fortuitously, the glycan specificity of SubB might enable such targeting. SubB binds preferentially to glycans terminating in α2-3-linked Neu5Gc, a sugar that humans cannot synthesise. Humans can assimilate Neu5Gc from dietary sources, but in cancer patients, it is preferentially incorporated into the tumours 65. As in the case of Stx, careful dosing would be required if SubAB holotoxin were to be deployed, as there is the potential for toxicity to normal tissues expressing traces of Neu5Gc taken up from the diet, or through weaker, but nevertheless potentially significant, interactions with more ubiquitously distributed α-3-linked Neu5Ac glycans. Alternatively, SubA alone could be delivered via a more tumour-specific targeting molecule, such as epidermal growth factor (EGF), as its receptor (EGFR) is expressed at high levels in many tumours, including melanomas and gliomas, as well as breast, prostate, and ovarian cancers. Indeed, an EGF–SubA fusion protein was recently constructed, and it was internalized efficiently by tumour cells in an EGFR-dependent fashion. Moreover, treatment with EGF–SubA killed EGFR-positive rat glioma and human breast and prostate cancer cells at picomolar concentrations 66. Cell lines expressing moderate to high levels of EGFR, which is associated with invasiveness and metastatic potential, were the most susceptible. EGF–SubA acted synergistically with drugs, such as thapsigargin, that induce ER stress, enabling effective deployment at concentrations well below the cytotoxicity threshold for each component. EGF–SubA is also efficacious in vivo, significantly inhibiting tumour growth in mouse xenograft models of human breast and prostate cancer 66.
Concluding remarks
AB5 toxins are key virulence factors of several major bacterial pathogens that cause worldwide morbidity and mortality. Early studies on AB5 toxins classified the toxins into three families; however, the recent discovery of the SubAB toxin resulted in the establishment of a fourth member of this family. Unique among other AB5 toxins, the A-subunit of this toxin is a subtilase-like serine protease that specifically cleaves BiP, and unexpectedly, SubB binds a glycan that humans cannot synthesise, but which is incorporated from our diet. This novel SubAB toxin has raised several exciting questions including: i) Are there other AB5 toxins that possess different catalytic A-subunits? ii) How did SubA evolve its highly specific activity? iii) How does the OB fold of the B-pentamer evolve different binding sites and glycan specificities?
Through investigating how these toxins cause disease they have become essential reagents in a biochemist’s toolbox for investigating the process of cell trafficking and immune cell function. The ability of AB5 toxins to bind specific glycans has enabled experiments to directly assess the physiological role of the various glycans (eg GM1 that is recognized by Ctx). The ability to evolve the glycan specificity of the B-pentamer could also have potential benefit in the use of AB5 toxins in a clinical setting. The continued investigation into the AB5 family should lead to greater understanding of cellular trafficking and the specific physiological role of defined glycoconjugates, which will ultimately lead to increased use as potential therapies for a range of human diseases.
Glossary
- BiP
(Binding immunoglobulin protein): also called Grp78 (Glucose regulated protein); BiP is an essential ER resident chaperone that plays a major role in the assistance of newly synthesised proteins to acquire correct folding and quaternary structure.
- ETEC and STEC
Enterotoxigenic and Shiga Toxigenic E. coli strains are common E. coli infections. ETEC produces the heat-labile (LT) and is the most common cause of travellers’ diarrhea. The Shiga-Toxin secreting E. coli STEC also causes gastrointestinal disease that can lead to the life threatening syndrome HUS (renal failure)
- HUS
(haemolytic uraemic syndrome): a disease characterised by haemolytic anemia and acute renal failure. This syndrome occurs predominantly in children and is preceded by diarrhoea caused by the shiga-toxin producing E. coli. The toxin preferentially targets the kidneys by inhibiting protein synthesis leading to cell necrosis or apoptosis. The most common route of infection in children is via undercooked meat containing bacteria
- Neu5Gc
N-glycolylneuraminic acid is a terminating sialic acid glycan not synthesised by humans as they do not possess the gene necessary for its synthesis. Neu5Gc is abundant in dairy products and red meats, which ironically are the foods the most commonly contaminated by STEC
- SubAB
abbreviated name for the recently established 4th member of AB5 toxin family. It was named ‘subtilase cytotoxin’ (SubAB) because the A-subunit of the toxin shares sequence homology with a subtilase-like serine protease of Bacillus anthracis and exhibits protease activity towards the ER chaperone BiP, a protein playing a key role for cell survival
- UPR
(Unfolded Protein Response): a cellular stress response and is critical for the maintenance of cellular function in the ER. Under stress conditions, proteins in the ER can misfold or aggregate. To prevent protein aggregation and/or misfolding, the ER activates the UPR system, thus enabling an increase in the capacity for protein folding
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sack DA, et al. Cholera. Lancet. 2004;363:223–233. doi: 10.1016/s0140-6736(03)15328-7. [DOI] [PubMed] [Google Scholar]
- 2.Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paton JC, Paton AW. Pathogenesis and Diagnosis of Shiga Toxin-Producing Escherichia coli Infections. Clinical Microbiology Reviews. 1998;11:450–479. doi: 10.1128/cmr.11.3.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tarr PI, et al. Shiga-toxin-producing Escherichia coli and haemolytic uraemic syndrome. Lancet. 2005;365:1073–1086. doi: 10.1016/S0140-6736(05)71144-2. [DOI] [PubMed] [Google Scholar]
- 5.Griffin P. Shiga toxin-producing Escherichia coli infections in the United States. 4th International Symposium and Workshop on Shiga Toxin-producing Escherichia coli Infections. 2000:21. [Google Scholar]
- 6.Fan E, et al. AB(5) toxins: structures and inhibitor design. Curr Opin Struct Biol. 2000;10:680–686. doi: 10.1016/s0959-440x(00)00152-4. [DOI] [PubMed] [Google Scholar]
- 7.O’Neal CJ, et al. Crystal structures of an intrinsically active cholera toxin mutant yield insight into the toxin activation mechanism. Biochemistry. 2004;43:3772–3782. doi: 10.1021/bi0360152. [DOI] [PubMed] [Google Scholar]
- 8.van den Akker F, et al. Crystal structure of a new heat-labile enterotoxin, LT-IIb. Structure. 1996;4:665–678. doi: 10.1016/s0969-2126(96)00073-1. [DOI] [PubMed] [Google Scholar]
- 9.Paton AW, Paton JC. Detection and characterization of Shiga toxigenic Escherichia coli by using multiplex PCR assays for stx1, stx2, eaeA, enterohemorrhagic E. coli hlyA, rfbO111, and rfbO157. J Clin Microbiol. 1998;36:598–602. doi: 10.1128/jcm.36.2.598-602.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fraser ME, et al. Structure of shiga toxin type 2 (Stx2) from Escherichia coli O157:H7. J Biol Chem. 2004;279:27511–27517. doi: 10.1074/jbc.M401939200. [DOI] [PubMed] [Google Scholar]
- 11.Siegler RL, et al. Response to Shiga toxin 1 and 2 in a baboon model of hemolytic uremic syndrome. Pediatr Nephrol. 2003;18:92–96. doi: 10.1007/s00467-002-1035-7. [DOI] [PubMed] [Google Scholar]
- 12.Paton AW, et al. A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. J Exp Med. 2004;200:35–46. doi: 10.1084/jem.20040392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paton AW, et al. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature. 2006;443:548–552. doi: 10.1038/nature05124. [DOI] [PubMed] [Google Scholar]
- 14.Cergole-Novella MC, et al. Distribution of virulence profiles related to new toxins and putative adhesins in Shiga toxin-producing Escherichia coli isolated from diverse sources in Brazil. FEMS Microbiol Lett. 2007;274:329–334. doi: 10.1111/j.1574-6968.2007.00856.x. [DOI] [PubMed] [Google Scholar]
- 15.Khaitan A, et al. The operon encoding SubAB, a novel cytotoxin, is present in shiga toxin-producing Escherichia coli isolates from the United States. J Clin Microbiol. 2007;45:1374–1375. doi: 10.1128/JCM.00076-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, et al. Pathologic changes in mice induced by subtilase cytotoxin, a potent new Escherichia coli AB5 toxin that targets the endoplasmic reticulum. J Infect Dis. 2007;196:1093–1101. doi: 10.1086/521364. [DOI] [PubMed] [Google Scholar]
- 17.Hendershot LM. The ER function BiP is a master regulator of ER function. Mt Sinai J Med. 2004;71:289–297. [PubMed] [Google Scholar]
- 18.Plaut RD, Carbonetti NH. Retrograde transport of pertussis toxin in the mammalian cell. Cell Microbiol. 2008;10:1130–1139. doi: 10.1111/j.1462-5822.2007.01115.x. [DOI] [PubMed] [Google Scholar]
- 19.Tsai B, et al. Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell. 2001;104:937–948. doi: 10.1016/s0092-8674(01)00289-6. [DOI] [PubMed] [Google Scholar]
- 20.Matlack KE, et al. Protein translocation: tunnel vision. Cell. 1998;92:381–390. doi: 10.1016/s0092-8674(00)80930-7. [DOI] [PubMed] [Google Scholar]
- 21.Bernardi KM, et al. Derlin-1 Facilitates the Retro-Translocation of Cholera Toxin. Mol Biol Cell. 2008;19:877–884. doi: 10.1091/mbc.E07-08-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bernardi KM, et al. The E3 Ubiquitin Ligases Hrd1 and gp78 Bind to and Promote Cholera Toxin Retro-Translocation. Molecular Biology of the Cell. 21:140–151. doi: 10.1091/mbc.E09-07-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dixit G, et al. Cholera Toxin Up-Regulates Endoplasmic Reticulum Proteins That Correlate with Sensitivity to the Toxin. Experimental Biology and Medicine. 2008;233:163–175. doi: 10.3181/0705-RM-132. [DOI] [PubMed] [Google Scholar]
- 24.Tarrago-Trani MT, et al. Shiga-like toxin subunit B (SLTB)-enhanced delivery of chlorin e6 (Ce6) improves cell killing. Photochem Photobiol. 2006;82:527–537. doi: 10.1562/2005-06-20-RA-583. [DOI] [PubMed] [Google Scholar]
- 25.Chong DC, et al. Clathrin-dependent trafficking of subtilase cytotoxin, a novel AB5 toxin that targets the endoplasmic reticulum chaperone BiP. Cell Microbiol. 2008;10:795–806. doi: 10.1111/j.1462-5822.2007.01085.x. [DOI] [PubMed] [Google Scholar]
- 26.Smith RD, et al. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic. 2009;10:1502–1517. doi: 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kitov PI, et al. In vivo supramolecular templating enhances the activity of multivalent ligands: a potential therapeutic against the Escherichia coli O157 AB5 toxins. Proc Natl Acad Sci U S A. 2008;105:16837–16842. doi: 10.1073/pnas.0804919105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kitov PI, et al. Shiga-like toxins are neutralized by tailored multivalent carbohydrate ligands. Nature. 2000;403:669–672. doi: 10.1038/35001095. [DOI] [PubMed] [Google Scholar]
- 29.Merritt EA, et al. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci. 1994;3:166–175. doi: 10.1002/pro.5560030202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fukuta S, et al. Comparison of the carbohydrate-binding specificities of cholera toxin and Escherichia coli heat-labile enterotoxins LTh-I, LT-IIa, and LT-IIb. Infect Immun. 1988;56:1748–1753. doi: 10.1128/iai.56.7.1748-1753.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Byres E, et al. Incorporation of a non-human glycan mediates human susceptibility to a bacterial toxin. Nature. 2008;456:648–652. doi: 10.1038/nature07428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Soltyk AM, et al. A mutational analysis of the globotriaosylceramide-binding sites of verotoxin VT1. J Biol Chem. 2002;277:5351–5359. doi: 10.1074/jbc.M107472200. [DOI] [PubMed] [Google Scholar]
- 33.Merritt EA, et al. Galactose-binding site in Escherichia coli heat-labile enterotoxin (LT) and cholera toxin (CT) Mol Microbiol. 1994;13:745–753. doi: 10.1111/j.1365-2958.1994.tb00467.x. [DOI] [PubMed] [Google Scholar]
- 34.MacKenzie CR, et al. Quantitative analysis of bacterial toxin affinity and specificity for glycolipid receptors by surface plasmon resonance. J Biol Chem. 1997;272:5533–5538. doi: 10.1074/jbc.272.9.5533. [DOI] [PubMed] [Google Scholar]
- 35.Karlsson KA, et al. Unexpected carbohydrate cross-binding by Escherichia coli heat-labile enterotoxin. Recognition of human and rabbit target cell glycoconjugates in comparison with cholera toxin. Bioorg Med Chem. 1996;4:1919–1928. doi: 10.1016/s0968-0896(96)00174-5. [DOI] [PubMed] [Google Scholar]
- 36.Galvan EM, et al. Functional interaction of Escherichia coli heat-labile enterotoxin with blood group A-active glycoconjugates from differentiated HT29 cells. FEBS J. 2006;273:3444–3453. doi: 10.1111/j.1742-4658.2006.05368.x. [DOI] [PubMed] [Google Scholar]
- 37.Holmner A, et al. Novel binding site identified in a hybrid between cholera toxin and heat-labile enterotoxin: 1.9 A crystal structure reveals the details. Structure. 2004;12:1655–1667. doi: 10.1016/j.str.2004.06.022. [DOI] [PubMed] [Google Scholar]
- 38.Holmner A, et al. Blood group antigen recognition by Escherichia coli heat-labile enterotoxin. J Mol Biol. 2007;371:754–764. doi: 10.1016/j.jmb.2007.05.064. [DOI] [PubMed] [Google Scholar]
- 39.Obara Y, et al. Betagamma subunits of G(i/o) suppress EGF-induced ERK5 phosphorylation, whereas ERK1/2 phosphorylation is enhanced. Cell Signal. 2008;20:1275–1283. doi: 10.1016/j.cellsig.2008.02.016. [DOI] [PubMed] [Google Scholar]
- 40.Nakamura K, et al. G(i)-coupled GPCR signaling controls the formation and organization of human pluripotent colonies. PLoS One. 2009;4:e7780. doi: 10.1371/journal.pone.0007780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Regard JB, et al. Probing cell type-specific functions of Gi in vivo identifies GPCR regulators of insulin secretion. J Clin Invest. 2007;117:4034–4043. doi: 10.1172/JCI32994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanchez J, Holmgren J. Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci. 2008;65:1347–1360. doi: 10.1007/s00018-008-7496-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanden Broeck D, et al. Vibrio cholerae: cholera toxin. Int J Biochem Cell Biol. 2007;39:1771–1775. doi: 10.1016/j.biocel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 44.Boyaka PN, et al. Chimeras of labile toxin one and cholera toxin retain mucosal adjuvanticity and direct Th cell subsets via their B subunit. J Immunol. 2003;170:454–462. doi: 10.4049/jimmunol.170.1.454. [DOI] [PubMed] [Google Scholar]
- 45.Stanford M, et al. Oral tolerization with peptide 336-351 linked to cholera toxin B subunit in preventing relapses of uveitis in Behcet’s disease. Clin Exp Immunol. 2004;137:201–208. doi: 10.1111/j.1365-2249.2004.02520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Smits HH, et al. Cholera toxin B suppresses allergic inflammation through induction of secretory IgA. Mucosal Immunol. 2009;2:331–339. doi: 10.1038/mi.2009.16. [DOI] [PubMed] [Google Scholar]
- 47.Tarrago-Trani MT, Storrie B. Alternate routes for drug delivery to the cell interior: pathways to the Golgi apparatus and endoplasmic reticulum. Adv Drug Deliv Rev. 2007;59:782–797. doi: 10.1016/j.addr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lass A, et al. Decreased ER-associated degradation of alpha-TCR induced by Grp78 depletion with the SubAB cytotoxin. Int J Biochem Cell Biol. 2008;40:2865–2879. doi: 10.1016/j.biocel.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang T, et al. Downregulation of gap junction expression and function by endoplasmic reticulum stress. J Cell Biochem. 2009;107:973–983. doi: 10.1002/jcb.22202. [DOI] [PubMed] [Google Scholar]
- 50.Buchkovich NJ, et al. Human cytomegalovirus specifically controls the levels of the endoplasmic reticulum chaperone BiP/GRP78, which is required for virion assembly. J Virol. 2008;82:31–39. doi: 10.1128/JVI.01881-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wati S, et al. Dengue virus infection induces upregulation of GRP78, which acts to chaperone viral antigen production. J Virol. 2009;83:12871–12880. doi: 10.1128/JVI.01419-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Du S, et al. Suppression of NF-kappaB by cyclosporin a and tacrolimus (FK506) via induction of the C/EBP family: implication for unfolded protein response. J Immunol. 2009;182:7201–7211. doi: 10.4049/jimmunol.0801772. [DOI] [PubMed] [Google Scholar]
- 53.Hayakawa K, et al. Blunted activation of NF-kappaB and NF-kappaB-dependent gene expression by geranylgeranylacetone: involvement of unfolded protein response. Biochem Biophys Res Commun. 2008;365:47–53. doi: 10.1016/j.bbrc.2007.10.115. [DOI] [PubMed] [Google Scholar]
- 54.Hu CC, et al. Subtilase cytotoxin cleaves newly synthesized BiP and blocks antibody secretion in B lymphocytes. J Exp Med. 2009;206:2429–2440. doi: 10.1084/jem.20090782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yamazaki H, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. 2009;183:1480–1487. doi: 10.4049/jimmunol.0900017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harama D, et al. A subcytotoxic dose of subtilase cytotoxin prevents lipopolysaccharide-induced inflammatory responses, depending on its capacity to induce the unfolded protein response. J Immunol. 2009;183:1368–1374. doi: 10.4049/jimmunol.0804066. [DOI] [PubMed] [Google Scholar]
- 57.Distler U, et al. Shiga toxin receptor Gb3Cer/CD77: tumor-association and promising therapeutic target in pancreas and colon cancer. PLoS One. 2009;4:e6813. doi: 10.1371/journal.pone.0006813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Falguieres T, et al. Human colorectal tumors and metastases express Gb3 and can be targeted by an intestinal pathogen-based delivery tool. Mol Cancer Ther. 2008;7:2498–2508. doi: 10.1158/1535-7163.MCT-08-0430. [DOI] [PubMed] [Google Scholar]
- 59.Johansson D, et al. Expression of verotoxin-1 receptor Gb3 in breast cancer tissue and verotoxin-1 signal transduction to apoptosis. BMC Cancer. 2009;9:67. doi: 10.1186/1471-2407-9-67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ishitoya S, et al. Verotoxin induces rapid elimination of human renal tumor xenografts in SCID mice. J Urol. 2004;171:1309–1313. doi: 10.1097/01.ju.0000100110.11129.85. [DOI] [PubMed] [Google Scholar]
- 61.Janssen KP, et al. In vivo tumor targeting using a novel intestinal pathogen-based delivery approach. Cancer Res. 2006;66:7230–7236. doi: 10.1158/0008-5472.CAN-06-0631. [DOI] [PubMed] [Google Scholar]
- 62.Amessou M, et al. Retrograde delivery of photosensitizer (TPPp-O-beta-GluOH)3 selectively potentiates its photodynamic activity. Bioconjug Chem. 2008;19:532–538. doi: 10.1021/bc7003999. [DOI] [PubMed] [Google Scholar]
- 63.El Alaoui A, et al. Shiga toxin-mediated retrograde delivery of a topoisomerase I inhibitor prodrug. Angew Chem Int Ed Engl. 2007;46:6469–6472. doi: 10.1002/anie.200701270. [DOI] [PubMed] [Google Scholar]
- 64.Li J, Lee AS. Stress induction of GRP78/BiP and its role in cancer. Curr Mol Med. 2006;6:45–54. doi: 10.2174/156652406775574523. [DOI] [PubMed] [Google Scholar]
- 65.Hedlund M, et al. Evidence for a human-specific mechanism for diet and antibody-mediated inflammation in carcinoma progression. Proc Natl Acad Sci U S A. 2008;105:18936–18941. doi: 10.1073/pnas.0803943105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Backer JM, et al. Chaperone-targeting cytotoxin and endoplasmic reticulum stress-inducing drug synergize to kill cancer cells. Neoplasia. 2009;11:1165–1173. doi: 10.1593/neo.09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schwarzkopf M, et al. Sialylation is essential for early development in mice. Proc Natl Acad Sci U S A. 2002;99:5267–5270. doi: 10.1073/pnas.072066199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Varki NM, Varki A. Diversity in cell surface sialic acid presentations: implications for biology and disease. Lab Invest. 2007;87:851–857. doi: 10.1038/labinvest.3700656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chou HH, et al. A mutation in human CMP-sialic acid hydroxylase occurred after the Homo-Pan divergence. Proc Natl Acad Sci U S A. 1998;95:11751–11756. doi: 10.1073/pnas.95.20.11751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Muchmore EA, et al. A structural difference between the cell surfaces of humans and the great apes. Am J Phys Anthropol. 1998;107:187–198. doi: 10.1002/(SICI)1096-8644(199810)107:2<187::AID-AJPA5>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 71.Martin MJ, et al. Evolution of human-chimpanzee differences in malaria susceptibility: relationship to human genetic loss of N-glycolylneuraminic acid. Proc Natl Acad Sci U S A. 2005;102:12819–12824. doi: 10.1073/pnas.0503819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rich SM, et al. The origin of malignant malaria. Proc Natl Acad Sci U S A. 2009;106:14902–14907. doi: 10.1073/pnas.0907740106. [DOI] [PMC free article] [PubMed] [Google Scholar]