

Abstract
Bioassay-guided fractionation of an EtOH extract obtained from the roots of the Madagascan plant Dodonaea viscosa led to the isolation of two new antiproliferative oleanane-type triterpenoid saponins, dodoneasides A and B (1 and 2). The structures of these two new compounds were elucidated using 1D and 2D NMR experiments and mass spectrometry. Compounds 1 and 2 showed antiproliferative activity against the A2780 human ovarian cancer cell line with IC50 values of 0.79 and 0.70 μM, respectively.
In our continuing search for bioactive molecules from the Madagascar rainforests as part of an International Cooperative Biodiversity Group (ICBG) program, we obtained an extract of the roots of Dodonaea viscosa (L.) Jacq. (Sapindaceae). This extract, designated MG 3397, showed reproducible cytotoxicity to the A2780 ovarian cancer cell line, with an IC50 value of 6.0 μg/mL. The extract was selected for bioassay-guided fractionation based on this activity. Previous work on Dodonaea viscosa revealed the presence of flavonoids, 2 fatty acids, 3 and cyanolipids. 4 Some ent-clerodane diterpenoids were obtained from D. boroniaefolia. 5 A southern Brazilian outbreak of acute hepatic insufficiency in which 14 dairy animals died after consumption of Dodonaea viscosa has been reported.6 In this paper, we report the isolation, structure elucidation, and antiproliferative activity of two new triterpenoid saponins (1 and 2) obtained from the roots of Dodonaea viscosa.
Liquid-liquid partitioning of a portion of an EtOH extract of the roots of Dodonaea viscosa into hexane, CH2Cl2 and aqueous MeOH fractions indicated that the CH2Cl2 fraction (326.5 mg) was the most active fraction, with an IC50 value of 1.0 μg/mL. Purification of the CH2Cl2 fraction using a C18 open column, followed by preparative C18 HPLC, led to the isolation of antiproliferative compounds 1 and 2.
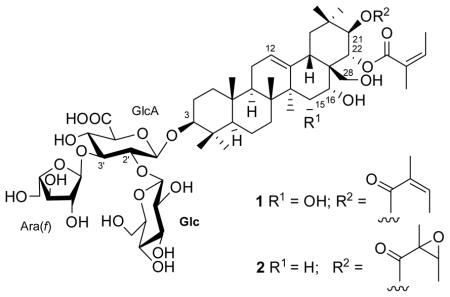
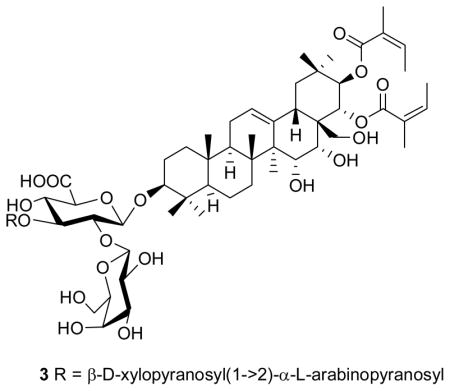
Compound 1 was obtained as white amorphous solid. HRFABMS (positive-ion mode) analysis suggested that the molecular formula 1 was C57H88O23. Its 1D NMR spectra revealed seven tertiary methyl groups between δH 0.87 and 1.42 and a double bond with typical 13C NMR resonances at δC 127.0 and 143.6, indicating an olean-12-ene triterpene derivative since H3-27 (δH 1.42, s) showed a 3J HMBC correlation to C-13 (δC 143.6). The HMBC spectrum (Figure 1) also exhibited correlations between H3-23/H3-24 (δH 1.09, s/δH 0.87, s) and C-3 (δC 92.3), H3-27 (δH 1.42, s) and C-15 (δC 68.5), H2-28 (δH 3.00 & 3.20, d, J = 9.6 Hz) and C-16/C-22 (δC 74.5/δC 73.9), and H3-29/H3-30 (δH 0.89, s/δH 1.06, s) and C-21 (δC 79.7), indicating that the aglycone was 3,15,16,21,22,28-hexaoxygenated olean-12-ene. Signals for three anomeric protons [δH 5.24 (1H, d, J = 2.2 Hz, H-1‴), 4.72 (1H, d, J = 7.7 Hz, H-1″), 4.55 (1H, d, J = 8.0 Hz, H-1′)] were observed in the 1H NMR spectrum. The 1H and 13C NMR data of the sugar moieties were completely assigned on the basis of the 1H-1H COSY, TOCSY, ROESY, HSQC, HSQC-TOCSY and HMBC spectra and by a comparison of their NMR data with those of aesculioside IIe.7 These three sugar moieties were identified as β-glucuronopyranosyl [GlcA-1′–6′ (δH/δC): 4.55, d, J = 8.0 Hz/105.5; 3.78, dd, J = 8.0, 8.2 Hz/78.1; 3.69, dd, J = 8.2, 8.2 Hz/86.1; 3.58, dd, J = 8.2 Hz/72.4; 3.61, d, J = 8.2 Hz/76.8; 174 (C-6′)], β-glucopyranosyl [Glc-1″–6″ (δH/δC): 4.72, d, J = 7.7 Hz/103.7; 3.19, dd, J = 7.7, 8.8 Hz/76.0; 3.36, dd, J = 8.8, 8.8 Hz/77.9; 3.09, dd, J = 8.8, 8.8 Hz/72.0; 3.29, m/77.9; 3.25 & 3.80, m/63.8], and α-arabinofuranosyl [Ara-1‴–5‴ (δH/δC): 5.24, d, J = 2.2 Hz/110.7; 4.14, m/83.4; 3.86, m/78.6; 4.10, m/85.3; 3.64 & 3.76, m/62.8]. The relative stereochemistry of the arabinofuranosyl moiety was assigned based on a comparison of its 13C NMR chemical shifts with those of the arabinofuranose ring of aesculioside IIe.7,8 H-1′, H-1″ and H-1‴ showed 3J HMBC correlations to C-3, C-2′ (δC 78.1) and C-3′ (δC 86.1), respectively, which established the connectivities between these sugar moieties and the aglycone. The 1H NMR spectrum also had signals for two olefinic protons at δH 6.06 (2H, qq, J = 7,3, 1.4 Hz, H-A3 and H-A3′) and four olefinic methyl groups [δH 1.82 (3H, q, J = 1.4 Hz, H3-A5′), 1.84 (3H, q, J = 1.4 Hz, H3-A5), 1.91 (3H, dq, J = 7.3, 1.4 Hz, H3-A4′), 1.91 (3H, dq, J = 7.3, 1.4 Hz, H3-A4)]; these signals were attributed to two angeloyl moieties. The locations of these two angeloyl moieties were determined on the basis of HMBC correlations between C-A1 (δC 169.4) and H-A3/H3-A4/H-21 (δH 5.94, d, J = 10.2 Hz), and C-A1′ (δC 169.2) and H-A3′/H3-A4′/H-22 (δH 5.61, d, J = 10.2 Hz). The double bonds in the angeloyl moieties were determined as E by the ROESY correlations between H-A3 and H3-A5, and H-A3′ and H3-A5′, and also by a comparison of their NMR data with those of floratheasaponin B.9 A ROESY correlation between H-3 and H-5 indicated the α-orientation of H-3. A 10.2 Hz coupling constant between H-21 and H-22 was compatible with a 21–22 diaxial orientation of the hydrogens. The β-axial orientation of H-15, and β-equatorial for H-16, were deduced by the ROESY correlations between H-16 and H-28b, and H-15 and H3-26 (Figure 2). The above results were confirmed by comparing the 13C NMR data of the aglycone of 1 with those of the floratheasaponin B (3) (Table 1).8 The identity of these signals confirmed the structure of compound 1 as shown.10
Figure 1.

Key HMBC correlations for compound 1
Figure 2.

Key ROESY correlations for compound 1
Table 1.
| position | 1 | 2 | 3 | ||
|---|---|---|---|---|---|
| 1H | 13C | 1H | 13C | 13C | |
| 1 | 1.05; 1.70 | 40.0 | 1.10; 1.65 | 39.9 | 39.1 |
| 2 | 1.77; 1.85 | 27.1 | 1.77; 1.85 | 27.0 | 26.6 |
| 3 | 3.24 | 92.3 | 3.21 | 92.3 | 89.6 |
| 4 | 40.4 | 40.4 | 39.6 | ||
| 5 | 0.80 br d (11.0) | 56.6 | 0.79 br d (11.0) | 56.9 | 55.7 |
| 6 | 1.45; 1.55 | 19.5 | 1.45; 1.55 | 19.3 | 18.9 |
| 7 | 1.75 | 37.2 | 1.75 | 33.9 | 36.7 |
| 8 | 42.3 | 40.8 | 41.5 | ||
| 9 | 1.62 | 48.4 | 1.62 | 47.7 | 47.2 |
| 10 | 37.9 | 37.7 | 37.0 | ||
| 11 | 1.95 | 24.8 | 1.95 | 24.7 | 24.0 |
| 12 | 5.47 br t (3.5) | 127.0 | 5.39 br t (3.5) | 125.3 | 125.5 |
| 13 | 143.6 | 142.9 | 143.7 | ||
| 14 | 48.1 | 41.0 | 48.5 | ||
| 15 | 3.80 | 68.5 | 1.65 | 34.9 | 73.1 |
| 16 | 3.84 | 74.5 | 3.99 | 69.7 | 67.5 |
| 17 | 48.5 | 49.0 | 47.8 | ||
| 18 | 2.63 | 41.6 | 2.85 | 42.3 | 40.9 |
| 19 | 1.20; 2.54 | 47.5 | 1.20; 2.65 | 47.9 | 46.9 |
| 20 | 36.8 | 37.1 | 36.4 | ||
| 21 | 5.94 d (10.2) | 79.7 | 6.01 d (10.2) | 81.9 | 78.7 |
| 22 | 5.61 d (10.2) | 73.9 | 5.60 d (10.2) | 73.7 | 73.3 |
| 23 | 1.09 s | 28.2 | 1.07 s | 28.3 | 28.1 |
| 24 | 0.87 s | 16.9 | 0.87 s | 16.8 | 16.9 |
| 25 | 0.99 s | 16.2 | 0.98 s | 16.3 | 15.8 |
| 26 | 1.02 s | 17.9 | 0.94 s | 17.3 | 17.6 |
| 27 | 1.42 s | 21.0 | 1.47 s | 27.8 | 21.2 |
| 28 | 3.00 d (9.6) | 63.6 | 2.92 d (9.6) | 64.4 | 63.1 |
| 29 | 0.89 s | 29.6 | 0.90 s | 29.8 | 29.5 |
| 30 | 1.06 s | 20.2 | 1.11 s | 20.2 | 20.2 |
| 21-angeloyl | |||||
| 1 | 169.4 | 171.34 | 167.6 | ||
| 2 | 129.3 | 61.2 | 128.7 | ||
| 3 | 6.06 qq (7,3, 1.4) | 139.2 | 3.05 qq (5.5, 1.4) | 61.1 | 138.4 |
| 4 | 1.91 dq (7.3, 1.4) | 16.0 | 1.15 dq (5.5, 1.4) | 13.9 | 16.0 |
| 5 | 1.84 q (1.4) | 20.9 | 1.50 br s | 19.8 | 21.0 |
| 22-angeloyl | |||||
| 1 | 169.2 | 169.1 | 168.2 | ||
| 2 | 129.3 | 128.9 | 129.1 | ||
| 3 | 6.06 qq (7,3, 1.4) | 139.1 | 6.18 qq (7,3, 1.4) | 141.4 | 136.6 |
| 4 | 1.91 dq (7.3, 1.4) | 15.9 | 2.02 dq (7.3, 1.4) | 16.2 | 15.7 |
| 5 | 1.82 q (1.4) | 20.8 | 1.86 q (1.4) | 21.0 | 20.6 |
| 3-β-glcA | |||||
| 1′ | 4.55 d (8.0) | 105.5 | 4.48 d (8.0) | 105.4 | 105.6 |
| 2′ | 3.78 dd (8.0, 8.2) | 78.1 | 3.78 dd (8.0, 8.2) | 78.1 | 79.1 |
| 3′ | 3.69 dd (8.2, 8.2) | 86.1 | 3.69 dd (8.2, 8.2) | 86.2 | 84.0 |
| 4′ | 3.58 dd (8.2, 8.2) | 72.4 | 3.58 dd (8.2, 8.2) | 72.4 | 71.1 |
| 5′ | 3.61 d (8.2) | 76.8 | 3.61 d (8.2) | 76.8 | 77.2 |
| 6′ | 172.0 | 172.0 | 172.0 | ||
| 2′-β-Glc | |||||
| 1″ | 4.72 d (7.7) | 103.7 | 4.72 d (7.7) | 103.7 | |
| 2″ | 3.19 dd (7.7, 8.8) | 76.0 | 3.19 dd (7.7, 8.8) | 75.9 | |
| 3″ | 3.36 dd (8.8, 8.8) | 77.9 | 3.36 dd (8.8, 8.8) | 77.8 | |
| 4″ | 3.09 dd (8.8, 8.8) | 72.0 | 3.09 dd (8.8, 8.8) | 72.1 | |
| 5″ | 3.29 m | 77.9 | 3.29 m | 77.8 | |
| 6″ | 3.80 m | 63.8 | 3.80 m | 63.6 | |
| 3′-α-Ara(f) | |||||
| 1‴ | 5.24 d (2.2) | 110.7 | 5.27 br s | 110.7 | |
| 2‴ | 4.14 m | 83.4 | 4.14 m | 83.3 | |
| 3‴ | 3.86 m | 78.6 | 3.86 m | 78.7 | |
| 4‴ | 4.10 m | 85.3 | 4.10 m | 85.4 | |
| 5‴ | 3.64 m, 3.76 m | 62.8 | 3.64 m, 3.76 m | 62.8 | |
δ (ppm) 500 MHz for 1H and 125 MHz for 13C; multiplicities; J values (Hz) in parentheses.
The signals of the sugar carbons were assigned by HSQC-TOCSY and 13C NMR.
In CD3OD.
Compound 2 was also obtained as a white amorphous solid. Comparison of the NMR data (Table 1) of 1 and 2 in CD3OD indicated that there was no substituent at the C-15 position of 2 and that the angeloyl group at the C-21 position of 1 was replaced by an epoxyangeloyl group in 2. The NMR spectra indicated that the other parts of 2 were identical to those of 1. The NMR data of the aglycone and the two substitutents at both the C-21 and C-22 positions of 2 were compatible with those of 22-angeloyl-21-epoxyangeloylbarringtogenol.11 Therefore, the structure of 2 was determined as shown.
Compounds 1 and 2 are oleanane-type triterpenoid saponins with a double bond at the 12-position, an OH group at the 16-position, and substituents at the 3-, 21-, and 28-positions, like gummiferaosides A-C. 12 The triterpenoid sapogenin portion of 1 and 2 is similar to 3β,15α,21β,22α,28-pentahydroxy-16α-angeloyloxy-12-oleanene isolated from Dodonaea viscosa.13
Compounds 1 and 2 were evaluated for antiproliferative activity against the A2780 human ovarian cancer cell line, and compound 1 was also evaluated in the breast cancer BT-549, prostate cancer DU 145, NSCLC NCI-H460, and colon cancer HCC-2998 cell lines (Table 2). The activities against the A2780 cell line were similar to those shown by gummiferaosides AC,12 which suggests that the structural features noted are important for their activity. This finding is similar to that of a recent study that showed that acylation with diangeloyl groups at the C-21 and C-22 positions in triterpenoid saponins is essential for cytotoxicity.14
Table 2.
Antiproliferative activity of compounds 1 and 2.
| Cell line | Cancer type | IC50 (μM) | |
|---|---|---|---|
| 1 | 2 | ||
| A2780 | ovarian | 0.79 | 0.70 |
| BT-549 | breast | >5 | NTa |
| DU 145 | prostate | >5 | NT |
| NCI-H460 | NSCLC | >5 | NT |
| HCC-2998 | colon | >5 | NT |
NT = not tested
Experimental Section
General Experimental Procedures
Optical rotations were recorded on a JASCO P-2000 polarimeter. IR and UV spectra were measured on MIDAC M-series FTIR and Shimadzu UV-1201 spectrophotometers, respectively. NMR spectra were obtained on a JEOL Eclipse 500 for 1H, 13C, HMQC, and HMBC and an INOVA 400 spectrometer for TOCSY, COSY, ROESY, and HSQC-TOCSY. Chemical shifts are given in δ (ppm), and coupling constants are reported in Hz. Mass spectra were obtained on a JEOL JMS-HX-110 instrument, in the positive-ion mode. HPLC was performed on a Shimadzu LC-10AT instrument with a preparative C18 Varian Dynamax column (8 μm, 250 × 21.4 mm) and a semi-preparative C18 Varian Dynamax column (5 μm, 250 × 10 mm).
Antiproliferative Bioassays
Cytotoxicity measurements were performed at Virginia Polytechnic Institute and State University against the A2780 ovarian cancer cell line, as described previously.15 The A2780 cell line is a drug-sensitive human ovarian cancer cell line.16
Plant Material
Dodonaea viscosa Jacq. (Sapindaceae) was collected on the eastern side of the Montagne des Français in littoral forest on sand at Ivovona, Antsiranana Province, Madagascar, elevation: ca. 5 m, co-ordinates: 12.21.40 S, 49.29.42 E, on July 19, 2005. Its assigned collection number is Randrianaivo et al. 1208. The collection was made from a shrub on the seashore. The genus Dodonaea Mill. consists of ca. 50 species, 2 of which occur in Madagascar. One is endemic (Dodonaea madagascariensis Radlk.) and the second one Dodonaea viscosa has a large distribution throughout the tropics near the sea. Voucher specimens have been deposited at herbaria of the Centre National d’Application des Recherches Pharmaceutiques, Madagascar (CNARP); the Parc Botanique et Zoologique de Tsimbazaza, Madagascar (TAN); the Missouri Botanical Garden, St. Louis, Missouri (MO); and the Muséum National d’Histoires Naturelles, Paris, France (P).
Extraction and Isolation
Dried roots of Dodonaea viscosa (253 g) were ground in a hammer mill, then extracted with EtOH by percolation for 24 h at rt to give the crude extract MG 3397 (8.9 g), of which 2.6 g was made available to Virginia Polytechnic Institute and State University (VPISU). Extract MG 3397 (2 g, IC50 6.0 μg/mL) was suspended in aqueous MeOH (MeOH-H2O, 9:1, 100 mL) and extracted with hexane (3 × 100 mL portions). The aqueous layer was then diluted to 70% MeOH with H2O and extracted with CH2Cl2 (3 × 100 mL portions). The CH2Cl2 extract (326.5 mg) was active with an IC50 1.0 μg/mL, while the hexane extract (153.5 mg) was inactive and the aqueous MeOH extract (1.5 g) was much less active than the CH2Cl2 extract. The CH2Cl2 extract was chromatographed on an open C18 column (50 × 10 mm) using H2O-MeOH (80:20 to 20:80, then 0:100) to yield the three fractions A [40.2 mg (polar, inactive)], B [198.7 mg, IC50: 0.5 μg/mL], and C [59.7 mg, IC50: 4.3 μg/mL]. Fraction B furnished 19 subfractions after HPLC separation on a C18 column (0-25-30-60-70 min:50-50-70-70-100% MeOH/H2O, 10 mL/min). Subfraction 18 yielded compound 1 (tR 66 min, 5.0 mg). Compound 2 (tR 21 min, 0.9 mg) was obtained by HPLC of subfraction 16 using C18 HPLC (0-30-40 min:70-70-100% MeOH/H2O, 2 mL/min).
Dodonaeaside A (1)
white solid; [α]25D -44.4 (c 0.18, MeOH); UV (MeOH) λmax (log ε) 209 (4.3) nm; IR (film) νmax 3389, 1727, 1152, 1033 cm−1; 1H NMR (500 MHz, CD3OD) and 13C NMR (125 MHz, CD3OD) see Table 1; HRFABMS m/z 1163.5595 (calcd for C57H88O23Na, 1163.5614).
Dodonaeaside B (2)
white solid; [α]25D -80 (c 0.08, MeOH); UV (MeOH) λmax (log ε) 207 (4.1) nm; IR (film) νmax 3390, 1698, 1076, 1033 cm−1; 1H NMR (500 MHz, CD3OD) and 13C NMR (125 MHz, CD3OD) see Table 1; HRFABMS m/z 1163.5580 (calcd for C57H88O23Na, 1163.5614).
Supplementary Material
Acknowledgments
This project was supported by the Fogarty International Center, the National Cancer Institute, the National Science Foundation, the National Heart, Lung and Blood Institute, the National Institute of Mental Health, the Office of Dietary Supplements, and the Office of the Director of NIH, under Cooperative Agreement U01 TW000313 with the International Cooperative Biodiversity Groups. This project was also supported by the National Research Initiative of the Cooperative State Research, Education and Extension Service, USDA, Grant #2008-35621-04732. These supports are gratefully acknowledged. We thank Mr. B. Bebout for obtaining the mass spectra and Mr. T. Glass for assistance with the NMR spectra. Field work essential for this project was conducted under a collaborative agreement between the Missouri Botanical Garden and the Parc Botanique et Zoologique de Tsimbazaza and a multilateral agreement between the ICBG partners, including the Centre National d’Applications des Recherches Pharmaceutiques. We gratefully acknowledge courtesies extended by the Government of Madagascar (Ministère des Eaux et Forêts).
Footnotes
Supporting Information Available: Spectroscopic data, consisting of 1H NMR spectra of compounds 1 and 2, are available as Supporting Information. This material is available free of charge via the internet at http://pubs.acs.org
References and Notes
- 1.Biodiversity Conservation and Drug Discovery in Madagascar, Part 38. For Part 37, see Rasamison VE, Ranaivo-Harimanana L, Cao S, Pan E, Ratovoson F, Randriantafika F, Rakotondrajaona R, Rakotonandrasana S, Andriantsiferana R, Kingston DGI. Fitoterapia. 2009 doi: 10.1016/j.fitote.2009.07.007. in press.
- 2.Getie M, Gebre-Mariam T, Rietz R, Neubert RHH. Pharmazie. 2002;57:320–322. [PubMed] [Google Scholar]
- 3.Kapur KK, Gupta AS. J Sci & Ind Res. 1959;18B:528–530. [Google Scholar]
- 4.Sherwani MRK, Hasan SQ, Ahmad I, Ahmad F, Osman SM. Chem & Ind. 1979:523–524. [Google Scholar]
- 5.Jefferies PR, Knox JR, Scaf B. Aust J Chem. 1973;26:2199–2211. [Google Scholar]
- 6.Colodel EM, Traverso SD, Seitz AL, Correa A, Oliveira FN, Driemeier D, Gave A. Vet Human Toxicol. 2003;45:147–148. [PubMed] [Google Scholar]
- 7.Zhang Z, Li S. Phytochemistry. 2007;68:2075–2086. doi: 10.1016/j.phytochem.2007.05.020. [DOI] [PubMed] [Google Scholar]
- 8.The 1H NMR chemical shifts of 1 differed from those of the arabinofuranose ring of aesculioside IIe because the latter spectrum was obtained in pyridine-d5. The 1H chemical shifts of carbohydrates are sensitive to solvents, and our 1H NMR shifts for the arabinofuranose ring of 1 in MeOH-d4 are intermediate between the values for aesculioside IIe in pyridine-d5 (reference 7) and those of the arabinofuranose ring of annulatophenonoside in acetone-d6; Nedialkov PT, Kitanov GM. Phytochemistry. 2002;59:867–871. doi: 10.1016/s0031-9422(01)00484-8.
- 9.Yoshikawa M, Morikawa T, Yamamoto K, Kato Y, Nagatomo A, Matsuda H. J Nat Prod. 2005;68:1360–1365. doi: 10.1021/np0580614. [DOI] [PubMed] [Google Scholar]
- 10.The recorded signals for C-15 and C-16 appear to have been reversed in reference 9. Our assignment is consistent with the assignment of these signals in similar compounds such as hippocaesculin: Konoshima T, Lee KH. J Nat Prod. 1986;49:650–6. doi: 10.1021/np50046a015.
- 11.Li ZL, Li X, Li LH, Li N, Yu M, Meng DL. Planta Med. 2005;71:1068–1070. doi: 10.1055/s-2005-873108. [DOI] [PubMed] [Google Scholar]
- 12.Cao S, Norris A, Miller JS, Ratovoson F, Razafitsalama J, Andriantsiferana R, Rasamison VE, TenDyke K, Suh T, Kingston DGI. J Nat Prod. 2007;70:361–366. doi: 10.1021/np060506g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Azam A. Indian J Chem, Sect B. 1993;32B:513–514. [Google Scholar]
- 14.Chan PK. Biochem Pharmacol. 2007;73:341–350. doi: 10.1016/j.bcp.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Cao S, Brodie PJ, Randrianaivo R, Ratovoson F, Callmander M, Andriantsiferana R, Rasamison VE, Kingston DGI. J Nat Prod. 2007;70:679–681. doi: 10.1021/np060627g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Louie KG, Behrens BC, Kinsella TJ, Hamilton TC, Grotzinger KR, McKoy WM, Winker MA, Ozols RF. Cancer Res. 1985;45:2110–2115. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.