

Abstract
Cell fate determination depends in part on the establishment of specific transcriptional programs of gene expression. These programs result from the interpretation of the genomic cis-regulatory information by sequence-specific factors. Decoding this information in sequenced genomes is an important issue. Here, we developed statistical analysis tools to computationally identify the cis-regulatory elements that control gene expression in a set of coregulated genes. Starting with a small number of validated and/or predicted cis-regulatory modules (CRMs) in a reference species as a training set, but with no a priori knowledge of the factors acting in trans, we computationally predicted transcription factor binding sites (TFBSs) and genomic CRMs underlying coregulation. This method was applied to the gene expression program active in Drosophila melanogaster sensory organ precursor cells (SOPs), a specific type of neural progenitor cells. Mutational analysis showed that four, including one newly characterized, out of the five top-ranked families of predicted TFBSs were required for SOP-specific gene expression. Additionaly, 19 out of the 29 top-ranked predicted CRMs directed gene expression in neural progenitor cells, i.e., SOPs or larval brain neuroblasts, with a notable fraction active in SOPs (11/29). We further identified the lola gene as the target of two SOP-specific CRMs and found that the lola gene contributed to SOP specification. The statistics and phylogeny-based tools described here can be more generally applied to identify the cis-regulatory elements of specific gene regulatory networks in any family of related species with sequenced genomes.
Keywords: cis-regulation, gene regulatory network, neural development, phylogenomics, transcription factor
Transcriptional regulatory networks play a central role in many developmental and physiological processes. Mapping the cis-regulatory information underlying transcriptional regulation is therefore of key importance. Experimental (1, 2) and computational methods (3, 4) provide complementary approaches to address this problem. One primary goal is the determination of cis-regulatory modules (CRMs) that often take the form of 500–1,000 nucleotides (nt) long sequences with multiple binding sites for several transcription factors. This has been most successfully achieved when searching for experimentally well-characterized binding sites (5–11) for known transcription factors (TF) usually under the form of position weight matrices (PWMs) (12). CRM determination without prior knowledge of cis-binding information is clearly a much more difficult problem. Several algorithms have tried to differentiate CRMs from nonregulatory sequences by analyzing the distributions of their entire content in small word frequency (13–17). These approaches, however, do not provide direct information on cis-binding motifs. With the advent of multiple sequenced genomes, phylogenetic conservation can also be used to identify regulatory motifs in a dataset on the basis of numerous conserved instances across the genomes (18–20). However, in the absence of expression and/or binding data, the spatio-temporal pattern of activity of the predicted CRMs cannot be inferred.
Here, we consider the specific task of determining the cis-regulatory motifs and associated CRMs that regulate gene expression in a cell-specific manner (21). A frequently encountered instance of this problem is the prediction of novel CRMs based on sequence information given by a small collection of putative and/or validated CRMs in one species. Previous works that addressed this problem showed the usefulness of using conservation between different genomes (9, 11, 22–25). We present an algorithm that combines new statistical tools with phylogenetic information to first discover short, i.e., ≃10 nt, conserved cis-regulatory motifs within a training set of CRMs without prior knowledge on the transcription factors acting via these CRMs and, second, predict previously undescribed CRMs. We applied this method to the discovery of previously undescribed cis-regulatory motifs and genomic CRMs that direct gene expression in Drosophila melanogaster SOPs and neural progenitor cells.
Outline of the Algorithm
The goal of the algorithm described here is to identify TF PWMs from a small number of CRMs that define a training set with no a priori knowledge of the TFs acting via these CRMs. The key steps of our method are summarized in Fig. 1A (see SI Appendix for a complete description). The training set consists in sequences for a given species (D. melanogaster in the present work). Conservation with other species (the 11 other sequenced Drosophilae species here) is used both to enrich the training set with orthologous sequences and to focus on PWMs that have conserved binding sites in different species. Once PWMs specific to the training set are obtained, they are used to predict CRMs genome-wide.
Fig. 1.

Genome-wide, pattern-specific motif and CRM discovery approach. (A) General outline. Training set definition (pink) precedes in silico analysis (blue) and experimental validation (orange). (B) Identification of PWMs. Starting from all D. melanogaster CRM sequences of the training set, a list of nonranked motifs is generated in several steps. First, at each base position in the training set, a 10-mer sequence is extracted and an initial approximative matrix is built using this unique sequence. The training set is then exhaustively scanned for sites corresponding to this approximative matrix, i.e., for sites that have a score higher than Sth. For each site of the training set that has been detected, orthologous sites are searched in the 11 other sequenced Drosophilae species. These orthologous sites are combined to obtain a refined frequency matrix using phylogenetic information and a model of transcription factor binding site evolution. The procedure is iterated to converge on a final frequency matrix. (C) Selection and ranking of PWMs. Starting from the set of PWMs generated in B, PWMs that correspond to repeated sequences and redundant PWMs are removed. Remaining PWMs are ranked to generate a list of predicted TFBSs.
The first step of our algorithm is to infer PWMs (Fig. 1B). To determine the ensemble S of PWMs specific to the training set, we attribute to each possible PWM an a priori probability to belong to S solely based on its information content (see section 2.2 in SI Appendix). The information content of a PWM reflects its binding specificity (12). We therefore require an a priori information content of the PWMs of S, set as the threshold score Sth. Namely, we choose the a priori probability for a PWM to belong to S so that the average information content of a random PWM of S is Sth. With Sth ≃ 13, a random PWM typically binds one site every ≃10 kb.
The algorithm then proceeds as follows. Each n-mer (n = 10 in Fig. 1 B and C) present in the training set is considered as a putative site for a PWM specific to the training set. The a priori probabilities of all possible PWMs to belong to S are modified according to the probability that they recognize the considered n-mer. These biased probabilities provide a first approximate PWM specific to the considered n-mer that is obtained by averaging all possible PWMs according to their probabilities. This first approximate PWM is then used to identify similar n-mers in the training set, i.e., with a score value above the a priori defined Sth. For each n-mer found in the training set, n-mers recognized by the first approximate PWM are then searched in the orthologous set around the same position (a shift of ± 20 nt is allowed to correct for possible alignment errors) with a score value 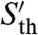 inferior to Sth. At this stage, conservation is enforced by considering for PWM refinement only the sites that are also detected in relatively distant species (Fig. 1B) (see section 2.4 in SI Appendix). Detected sites are then used to modify the probabilities in the set of PWMs using a Bayesian approach as for the initial n-mer. In order to properly combine sites in the training set (reference species) and in the orthologous set (related species), an explicit evolution model for TFBS should be used (9, 26, 27). We chose to use here a simple previously proposed model (26). The evolution model allows the algorithm to weigh sequence differences in different species according to their evolutionary distance and to construct the most likely PWM that binds the set of obtained sites. This refined PWM is again used to search for binding sites in the training set. The process is iterated until convergence is reached. Combining PWMs with an evolution model distinguishes the present algorithm from previous ones that used less flexible binding requirements (11, 23, 25) or used conservation but not phylogeny (22–24).
inferior to Sth. At this stage, conservation is enforced by considering for PWM refinement only the sites that are also detected in relatively distant species (Fig. 1B) (see section 2.4 in SI Appendix). Detected sites are then used to modify the probabilities in the set of PWMs using a Bayesian approach as for the initial n-mer. In order to properly combine sites in the training set (reference species) and in the orthologous set (related species), an explicit evolution model for TFBS should be used (9, 26, 27). We chose to use here a simple previously proposed model (26). The evolution model allows the algorithm to weigh sequence differences in different species according to their evolutionary distance and to construct the most likely PWM that binds the set of obtained sites. This refined PWM is again used to search for binding sites in the training set. The process is iterated until convergence is reached. Combining PWMs with an evolution model distinguishes the present algorithm from previous ones that used less flexible binding requirements (11, 23, 25) or used conservation but not phylogeny (22–24).
In our initial attempts to apply the above procedure to the sensory organ precursor (SOP) training set (see below), PWMs matching repeated sequences were produced. Masking the training set using repeat masker (28) did not solve this problem. This led us to study the distribution of the sites identified by the PWMs within the training set relative to a set of intergenic sequences from the reference species (20 Mb background of D. melanogaster genomic DNA in the present work). This set is defined here as the background set. For each PWM, all sites present in the background set are identified. PWMs corresponding to repeated sequences are then discriminated and eliminated based on the strong non-Poisson distribution of the sites that they recognize (Fig. 1C) (see section 2.5 of SI Appendix). This filtering step based on PWM binding statistics is the automatic repeat pruning step (Fig. 1C). It produces a filtered list of PWMs that have approximately Poisson-distributed binding sites on the background sequences.
The PWMs on this filtered list are then ranked according to the deviation of their site distribution from this background Poisson statistics, on the training set (Fig. 1C) (see section 2.5 of SI Appendix). A PWM is thus ranked high not only if corresponding sites are overrepresented in the training set but also, for instance, if its sites are less evenly distributed in the training set than in the background set.
Finally, the ranked list of PWMs obtained from this procedure contained several PWMs that appeared similar. Therefore, in the last step of the algorithm, PWMs are tested for similarity based on a proximity index defined here based on the overlap between the sets of their binding sites (see section 2.5 of SI Appendix). Duplicates of top-ranked matrices are then removed to eventually generate a list of nonredundant ranked PWMs (Fig. 1C). These PWMs can then be used to predict CRMs genome-wide. Additionally, as described below, CRM prediction can also be used to optimize the parameters of this PWM identification algorithm.
Results and Discussion
A Training Set for CRMs Active in Neural Progenitor Cells of D. Melanogaster.
This method was applied to identify cis-regulatory motifs that regulate gene expression in neural progenitor cells of D. melanogaster. Previous studies have used the formation of adult sensory bristles in D. melanogaster as a model system for neurogenesis (29). The transcriptional logic underlying the specification of SOPs from groups of neuroepthelial cells is relatively well understood (30) (Fig. 2E). In brief, expression of the proneural genes achaete (ac) and scute (sc) confer upon groups of proneural cluster (PNC) cells the competence to become SOPs (29). Inhibitory cell–cell signaling mediated by Notch restricts this competence to regularly spaced cells (31). Proneural genes encode bHLH transcriptional activators that promote both adoption of the SOP fate within each PNC cell and, at the same time, inhibitory Notch signaling between PNC cells. Within each PNC cell, proneural activity is antagonized by Notch via the E(spl)-bHLH repressors. Thus, SOPs emerge as winners of this competition for the SOP fate as PNC cells with both high proneural and low Notch/E(spl)-bHLH activities. In contrast, nonselected PNC cells have low proneural and high Notch/E(spl)-bHLH activities. This suggests a model whereby SOP-specific expression results from activation by proneural factors in SOPs and repression by E(spl)-bHLH repressors in nonSOP cells (Fig. 2E) (30, 32). This transcriptional logic is thought to apply widely to sensory and neural cells from cnidarian to mammals (33, 34).
Fig. 2.

Motif analysis. (A) Top-ranked motifs from the SOP training set. (B–C′) Site-directed mutagenesis of motif 1 in cpoCRM6 (2xm1: two sites were mutated in B′) and motif 5 in spdoCRM4 (2xm5: two sites were mutated in C′) strongly reduced the regulatory activity of these CRMs in SOPs of the pupal thorax. SOPs were marked by Cut (red). CRM activity was revealed by lacZ expression (β-galactosidase, green). Note that some SOPs have divided (as indicated by pairs of Cut-positive nuclei). (D) Top-ranked motifs from the PNC training set. (E) Cis-regulatory logic for SOP-specific (red) and non-SOP (blue) gene expression. Proneural factors positively regulate the expression of SOP (red) and PNC (blue) genes in both SOP and non-SOP cells via E-boxes. Notch activation specifically up-regulates the expression of PNC genes in non-SOP cells. Expression of SOP-specific genes is inhibited in non-SOP cells by E(spl)-bHLH repressors. Conversely, the expression of non-SOP genes is inhibited in SOPs by Su(H).
The genetic program of SOPs is well-suited for our CRM discovery approach. First, only a relatively small number of SOP-specific genes, i.e., expressed in SOPs but not in other PNCs of the pupal thorax, are known (35, 36). Thus, many SOP-specific genes probably remain to be discovered. Second, several CRMs active in SOPs, and not in other cells of the pupal neuroepithelium, have been validated using stringent in vivo assay and can be used as a training set (37–39) (Table S1 in SI Appendix). Several of these SOP-specific CRMs also direct expression in other neural progenitor cells, indicating that the genetic program active in SOPs in part reflects a more general program active in neural progenitor cells. Third, high-quality genomic sequence data are available for 12 Drosophila species.
Our SOP training set consisted in eight CRMs that have previously been shown to be active in SOPs (Table S1 in SI Appendix and references therein), six novel CRMs identified here based on their proximity to SOP-specific genes and shown to direct reporter gene expression in SOPs (Fig. S1 and Table S1 in SI Appendix) as well as 31 other sequences that are positioned close to SOP-specific genes but that did not direct reporter gene expression in SOPs (Table S2 in SI Appendix). These validated and putative CRMs ranged in size from 144 to 2398 nt (Table S1 in SI Appendix). Eleven Drosophila genomes (40) were used to assemble the orthologous set (see section 3.2 in SI Appendix).
Prediction and Validation of Cis-Regulatory Motifs.
We applied the algorithm to these training and orthologous sets to computationally predict SOP-specific PWMs of width 10 using Sth = 13.3 (see Fig. 3A and Fig. S2 in SI Appendix for the choice of these parameters). The five top-ranked motifs are shown in Fig. 2A (see Tables S4 and S5 in SI Appendix for additional PWMs; the five top-ranked motifs corresponding to repeated sequences and that were discarded are also shown in Table S4 in SI Appendix).
Fig. 3.

CRM prediction and analysis. (A) Selection of optimal Sth value and motif number. The number of predicted CRMs associated with a gene annotation related to sensory organs (number of matches in the y axis for the 100 top-ranked fragments; see section 3.3 of SI Appendix) was plotted as a function of the number of motifs (1 to 12; x axis) for different Sth values (from 12.7 to 13.6). Sth = 13.3 and five motifs were chosen for all predictions reported here. (B) Number of predicted CRMs associated with a gene annotation related to sensory organs as a function of their genome-wide ranking using five motifs and Sth = 13.3 (bin size: 100 fragments). In the first bin, 20 predicted CRMs are associated with a gene that has a sensory organ annotation. The mean number of annotations in the other bin is 5.78 (p = 3.2 × 10-7 for five motifs and Sth = 13.3). The training set was excluded from the analysis shown in A and B, as described in the main text and the SI Appendix. (C) Venn diagram showing the in vivo activity of the 29 newly predicted CRMs that were tested for CRM activity in vivo. Thirteen and 11 CRMs were expressed in larval neuroblasts (NB) and SOPs, respectively. (D–E′) Regulatory activity of CRM7 and CRM41 in SOPs of pupal thorax using a lacZ reporter gene. β-galactosidase, green; Cut, red, as a SOP marker; DAPI, blue.
Motif 1 perfectly matched the site α2, previously shown to regulate the SOP-specific expression of the proneural gene scute (32). This motif might correspond to a Rel family factor (41). Site-directed mutagenesis of this motif reduced the activity of cpo CRM6 and neur CRM1 (Fig. 2B and B′ and Fig. S3 in SI Appendix).
Motifs 2 and 4 matched the binding site for proneural activators, or E-box (42–44). The high Sth value selected for our analysis (see Fig. 3A) imposed that differences in the sequence flanking the E-box were sufficient to prevent these PWMs from merging. Indeed, for Sth = 13.3, only 5% of the sites associated with motif 2 were also associated with motif 4. The overlap contains an E-box of the SensCRM3 CRM that matched both motif 2 and 4 at Sth = 13.3. This E-box binds in vitro the heterodimeric factors Achaete/Daughterless (Da), Scute/Da and Atonal/Da. Moreover, mutation of this E-box disrupted the SOP-specific activity of this CRM (35). The identification of functional E-boxes is a good measure of the effectiveness of our algorithm because the core E-box contain relatively low information, and functional TFBSs are notoriously difficult to predict. Here, the predicted E-boxes are more specific and account for the information contained in the flanking bases (45).
Motif 3 was related to the predicted binding site for the E(spl)-bHLH repressors (20, 32) and matched the PWM for Hairy, a related factor, on the Jaspar database (46). However, site-directed mutagenesis of motif 3 in sens CRM3, CG32150 CRM1 and spdo CRM4 did not detectably affect the in vivo activity of these CRMs (Fig. S3 in SI Appendix). Thus, the potential function of this motif remains unknown. Nevertheless, our identification of possible binding sites for proneural activators and E(spl)-bHLH repressors within the training set is consistent with the notion that expression of SOP-specific genes is postively regulated by proneural factors in SOPs and repressed by E(spl)-bHLH factors in non-SOP cells (Fig. 2E).
Motif 5 was not related to known binding sites. Site-directed mutagenesis revealed that this motif was required for the SOP-specific activity of spdo CRM4 (Fig. 2C and C′). Together, these results indicate that our motif discovery algorithm identified both known and novel cis-regulatory motifs required in vivo for SOP-specific gene expression.
To further test our algorithm, we also applied the same motif discovery approach to a distinct training set comprising eight CRMs active in PNCs (Table S3 in SI Appendix) (38, 47). Interestingly, the two best-ranked motifs matched the S-box, i.e., the Suppressor of Hairless (Su(H)) binding site (48, 49) and the E-box, respectively (Fig. 2D) (see Tables S6 and S7 in SI Appendix for an extended list of PWMs). Noticeably, all instances of motifs 1 and 2 detected within our PNC training set were only a subset of the previously identified S- and E-boxes (30, 31). This indicates that the 13.3 Sth value chosen here confered high selectivity to our motif prediction. As proposed earlier, the presence of E- and S-boxes within PNC-specific CRMs supports a model whereby expression of PNC genes in non-SOP cells is positively regulated by proneural factors and activated Notch whereas Su(H) represses the expression of PNC genes in the absence of Notch activity in SOPs (Fig. 2E) (30).
Genome-Wide Identification of CRMs.
We next performed a genome-wide binding site search for the 12 top-ranked PWMs from the SOP training set (Tables S4 and S5 in SI Appendix). To identify conserved sites only, we used the same conservation requirements as those used earlier to identify sites in the training set (see section 2.4 of of SI Appendix). At Sth = 13.3 (see below), 206, 2062, 469, 988, and 378 conserved sites were found for motifs 1–5, respectively (sites that are recognized by motifs 2 and 4 are scored as motif 2 sites here). Motif co-occurrence can efficiently predict CRMs (5, 7, 8, 11, 24). Co-occurrence was first tested by studying the genome-wide distribution of the top five motifs at Sth = 13.3. We found that motif 2 exhibited significative cross-correlation with motif 1 (Fig. S4 in SI Appendix; randomized versions of motifs 1 were used as negative controls). We therefore scored genomic fragments by adding the scores of each conserved sites. Overlapping 1,000 nt genomic fragments covering the whole noncoding repeat-masked D. melanogaster genome were scored and ranked based on occurrence of conserved motifs (see section 2.6 of SI Appendix). Four parameters of our algorithm, including the Sth value and the number of motifs used for CRM ranking (Fig. 3A), were varied. To select appropriate parameters, we scored the overrepresentation, within top-ranked genomic fragments, of the “sensory organ” and “sensory mother cell” terms in the Flybase (50) “phenotype” annotations. To do so, each 1,000 nt fragment was associated with the transcription start site, hence the gene, located closest to the center of this fragment (Fig. 3B). A very significant enrichment (p = 3.2 × 10-7 for five motifs and Sth = 13.3) (Fig. 3B) was observed for a range of stringent Sth values with a minimum number of two motifs (Fig. 3A). For these results, the training set was masked before fragment ranking, so as to avoid biasing the results. When it was reintroduced, at Sth = 13.3, nine of the 14 validated CRMs of the training set ranked within the 100 first-ranked genomic fragments (Table S8 in SI Appendix).
We chose to use five motifs of width 10 and Sth = 13.3, to predict novel SOP-specific CRMs of width 1,000 nt (see Fig. 3A and Fig. S2 in SI Appendix). We assayed the regulatory activity of the top-ranked genomic fragments, with a score > 9.95 (Table S8 in SI Appendix) using a transgenic reporter assay. Transgenic flies were obtained for all of the newly predicted 29 top-ranked CRMs. Immunostaining analysis indicated that 11 of these 29 predicted CRMs (38%) directed gene expression in SOPs of the pupal notum, with three of these 11 CRMs being also expressed in PNCs (Fig. 3 C–E′ and Fig. S5 and Table S8 in SI Appendix). CRM activity was also observed in other neural progenitor cells, including neuroblasts in larval brains (45%, n = 29) (Fig. 3C and Fig. S6 and Table S8 in SI Appendix) and chordotonal SOPs in leg imaginal discs (24%, n = 29) (Fig. 3C and Fig. S7 and Table S8 in SI Appendix). These findings are entirely consistent with the notion that neural progenitor cells, i.e., SOPs and neuroblasts, are specified by a common genetic circuitry.
Finally, four additional fragments, ranked between positions 39 and 100 and chosen based on their proximity to genes putatively involved in neural development, were found to direct SOP-specific and/or neuroblast-specific expression (Fig. 3 E and E′, Fig. 4 B and B′ and Fig. S5 and Table S8 in SI Appendix). In total, 15 novel SOP-specific CRMs were identified. Thus, our algorithm has a high predictive value for CRMs active in SOPs and neural progenitor cells.
Fig. 4.

Functional analysis of the lola gene. (A and B′) Regulatory activity of CRM20 (A and A′) and CRM40 (B and B′) in SOPs. β-galactosidase, green; Cut, red, as a SOP marker; DAPI, blue. (C) Expression of a Lola-GFP protein trap (green) in living 16 hours APF pupae. Higher levels of Lola-GFP were seen in SOPs, identified based on their division pattern, than in non-SOP cells. (D) Genome view showing the lola locus. CRM20 and CRM40 are indicated by blue boxes. (E and F) Immunodetection of endogenous Lola (green): Higher nuclear levels were observed in SOPs (Sens, red, marked SOPs; DAPI in blue). (G) SOP specification (Sens, red) was largely unaffected in lola5D2 mutant clones (marked by loss of nuclear GFP, green). (H and I) lola5D2 mutant clones (marked by loss of nuclear GFP, green) generated in HE31 heterozygous flies resulted in double socket and bristle loss phenotypes, two phenotypes associated with increased Notch activity and/or loss of Hairless activity. These phenotypes were not observed at microchaete position in HE31 heterozygous flies, indicating that loss of lola activity enhanced the Hairless phenotype. (J) Clone border analysis showed that only 29% (n = 117 and n = 121) of the SOPs located along clone borders were lola mutant [red bar; control wild-type clones were equally, 52% (n = 289), found on either side of the border]. This bias was observed for two strong loss of function alleles (p < 0.001 relative to wild-type control clones). Thus, lola mutant cells are less likely to become SOPs than wild-type cells.
Of note, our transgenic assay likely underestimates the real predictive value of our algorithm. Indeed, CRM1 is included within a larger genomic fragment defined as a Peripheral Nervous System (PNS) CRM of the gene string (stg) (51). It is thus possible that the 1,000 nt genomic fragment that corresponds to CRM1 contains some cis-regulatory information but that the latter is not sufficient to direct reporter gene expression in our assay (Table S8 in SI Appendix). Moreover, CRM22, that is also negative in our assay (Table S8 in SI Appendix), is included within a 2.1 kb fragment, CRM22”, that directs reporter gene expression in SOPs and PNCs (Fig. S5 in SI Appendix). Additionally, our algorithm predicted a CRM close to the SOP-specific gene asense, raising the possibility that this predicted CRM, despite being negative in our assay, may contribute to regulate the expression of asense in SOPs.
A potential risk when trying to learn motifs from a small training set is to overfit it, i.e., to obtain PWMs that discriminate the training set from the background by fitting nonrelevant sequence particularities. Three lines of evidence indicate that overfitting was not a major issue here. First, the top-ranked predicted CRMs were associated with the sensory organ annotated genes (Fig. 3B). Second, four of the five top-ranked motifs appeared to be functional in vivo (Fig. 2 A–C′). And third, SOP-specific CRMs were successfully predicted from a larger dataset, i.e., the D. melanogaster genome, using the obtained PWMs (Table S8 in SI Appendix).
Identification and Functional Analysis of the Lola Gene.
This approach should help identifying genes differentially expressed in SOPs. To test this notion, we have studied the expression and function of the gene located near CRM20 and CRM40 that are both active in SOPs (Fig. 4 A–B′). These two CRMs are located 5’ to transcription start sites of the lola gene (Fig. 4D). The lola locus encodes a family of zinc finger DNA-binding proteins that share a common BTB domain (52, 53). BTB domains have been associated with transcriptional repression, and previous studies have shown that Lola acts as a transcriptional repressor (54). While the Lola binding site(s) and target genes are not known, lola is known to antagonize Notch during eye development (55). Using in situ hybridization, anti-Lola antibodies as well as a GFP protein-trap line, we showed that Lola gene products are present in all cells of the pupal thorax and that the lola gene is specifically up-regulated in SOPs (Fig. 4 C–F′ and Fig. S8 A–E in SI Appendix). This pattern of expression differed from the activity patterns seen with CRM20 and CRM40 that were not active outside of SOPs. We interpret this difference to suggest that transcription of the lola gene is regulated by two types of regulatory elements: ubiquituously active CRMs would direct lola expression in all cells whereas CRM20 and CRM40 would up-regulate lola expression in SOPs. We therefore suggest that in silico approaches may perform better than expression-based methods at identifying genes that are both broadly expressed and up-regulated, via discrete cell-type specific CRMs, in a small population of cells. Indeed, this cell-specific up-regulation might easily be masked by uniform low-level expression.
The function of the lola gene was studied in clones using two strong loss of function alleles. While the loss of lola activity did not significantly perturb the specification of SOPs (Fig. 4G), clone border analysis (56) indicated that lola mutant cells have a reduced ability to adopt the SOP fate (Fig. 4I). This suggests that Lola acts in SOPs to promote SOP specification, possibly by repressing Notch target gene expression (54). Consistent with this interpretation, lola genetically interacts with Hairless (Fig. S8 F–J in SI Appendix) and cells that are both mutant lola and heterozygous for Hairless show a double socket and, occasionally, a bristle loss phenotype (Fig. 4 H and I). Together, these results led us to propose that Lola antagonizes Notch in SOPs.
Conclusion
The approach presented here successfully predicted genome-wide TFBSs and CRMs regulating gene expression in D. melanogaster neural progenitor cells. Binding sites for transcription factors known to be important for SOP-specific gene expression were recovered. Six CRMs for genes known to be expressed in SOPs were identified. One previously undescribed cis-regulatory motif required for SOP-specific CRM activity was identified. Fifteen newly predicted CRMs were shown to be active in SOPs. This approach also served to identify lola as a gene up-regulated in SOPs. Future work will test whether large datasets originating from binding (e.g., ChIP-seq) and/or expression data (e.g., RNA-seq) can serve to define training sets and lead to the discovery of new motifs (57). Most importantly, the statistical analysis tools developed here are species-independent and may be applied to the identification of cis-regulatory elements in any family of related species with sequenced genomes including vertebrates.
Materials and Methods
Genome and Statistical Analysis.
Genome sequences were processed through a custom C++ program (see SI Appendix). All statistical operations were performed within the R software environment (ref. (58); www.R-project.org). Genome views were obtained from Flybase (50).
Transgenes.
Wild-type genomic DNA was PCR amplified to generate all DNA fragments tested for CRM activity. PCR products were cloned as EcoRII-NotI or EcorRI-XbaI fragments into pCaSpeR-hs43-lacZ (https://dgrc.cgb.indiana.edu) or pCaSpeRattP-hs43-lacZ (same vector but with an attP site cloned at the NsiI site; cloning details available upon request). QuickChange site-directed mutagenesis was used to mutate the sequence of motifs 1, 3, and 5. The following mutations were introduced: motif 1: CCCC was changed into AAAC; motif 3: CGCG was changed into CTAT; motif 5: GCTGC was changed into GATTA.
Flies.
P-element transformation and phiC31-mediated integration at the attP site located at 68D2 were performed by BestGene (http://www.thebestgene.com). Insertion of the empty integration vector at this location did not result in lacZ expression in the larval and pupal tissues examined in this study. RNAi-mediated inactivation of the lola gene was performed at 29 °C using the VDRC lines ID12573 and ID12574 (http://www.vdrc.at/). P[GAL4-Hsp70.PB]l(3)Eq1 (Eq-GAL4) was used as a driver in combination with a pTub-GAL80ts transgene. Two lola loss of function alleles, 5D2 and e76, were used for clone analysis. The lola protein trap was lola[CB02888] Mitotic clones were induced by a 45 min heat shock at 36.5 °C in hs-flp ; FRT42D ubi-nlsGFP/FRT42D lola first and second instar larvae.
Immunostaining.
Pupal nota and larval tissues were dissected and immunostained using standard protocols. Notum stainings were performed at 17 h after puparium formation (APF). When transgenesis involved P-element, 3-5 independent lines per transgene were analyzed. The following primary antibodies were used: rabbit anti-Lola (1∶200; purified polyclonal antibodies from E. Giniger), rabbit anti-βgalactosidase (1∶1,000; Cappel), mouse anti-Cut (1∶500; 2B10, DHSB), and guinea-pig anti-Sensless (1∶2,000; from H. Bellen). Cy2, Cy3, and Cy5-coupled secondary antibodies were from Jackson’s Immunoresearch. In situ hybridization was performed as described in (38). Images were acquired on Leica SPE confocal microscope. Images were processed using ImageJ and Photoshop.
Supplementary Material
Acknowledgments.
We thank H. Bellen, E. Giniger, E. Lai, T. Southall, A. Spradling, the Bloomington and Szeged Stock Centers, the Vienna Drosophila RNAi Center (VDRC), the Drosophila Genomics Resource Center, and the Developmental Studies Hybridoma Bank (DSHB) for flies, antibodies, and plasmids. We thank N. Vodovar, M. Santolini, and all members of the Schweisguth’s lab for discussion and comments on the manuscript. This work was funded by core funding from the Institut Pasteur, École Normale Supérieure, and Centre National de la Recherche Scientifique and by a specific grant from the ANR (08-BLAN-0235).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1002876107/-/DCSupplemental.
References
- 1.Wyrick J, Young R. Deciphering gene expression regulatory networks. Curr Opin Genet Dev. 2002;12:130–136. doi: 10.1016/s0959-437x(02)00277-0. [DOI] [PubMed] [Google Scholar]
- 2.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457:854–858. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siggia E. Computational methods for transcriptional regulation. Curr Opin Genet Dev. 2005;15:214–221. doi: 10.1016/j.gde.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 4.Vingron M, et al. Integrating sequence, evolution and functional genomics in regulatory genomics. Genome Biol. 2009;10:202. doi: 10.1186/gb-2009-10-1-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berman B, et al. Exploiting transcription factor binding site clustering to identify cis-regulatory modules involved in pattern formation in the Drosophila genome. Proc Natl Acad Sci USA. 2002;99:757–762. doi: 10.1073/pnas.231608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halfon M, Grad Y, Church G, Michelson A. Computation-based discovery of related transcriptional regulatory modules and motifs using an experimentally validated combinatorial model. Genome Res. 2002;12:1019–1028. doi: 10.1101/gr.228902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rebeiz M, Reeves N, Posakony J. SCORE: A computational approach to the identification of cis-regulatory modules and target genes in whole-genome sequence data. Proc Natl Acad Sci USA. 2002;99:9888–9893. doi: 10.1073/pnas.152320899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schroeder M, et al. Transcriptional control in the segmentation gene network of Drosophila. PLoS Biol. 2004;2:e271. doi: 10.1371/journal.pbio.0020271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siddharthan R, Siggia E, van Nimwegen E. PhyloGibbs: A Gibbs sampling motif finder that incorporates phylogeny. PLoS Comput Biol. 2005;1:e67. doi: 10.1371/journal.pcbi.0010067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hallikas O, et al. Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell. 2006;124:47–59. doi: 10.1016/j.cell.2005.10.042. [DOI] [PubMed] [Google Scholar]
- 11.Pierstorff N, Bergman C, Wiehe T. Identifying cis-regulatory modules by combining comparative and compositional analysis of DNA. Bioinformatics. 2006;22:2858–2864. doi: 10.1093/bioinformatics/btl499. [DOI] [PubMed] [Google Scholar]
- 12.Stormo G, Fields D. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- 13.Nazina A, Papatsenko D. Statistical extraction of Drosophila cis-regulatory modules using exhaustive assessment of local word frequency. BMC Bioinformatics. 2003;4:65. doi: 10.1186/1471-2105-4-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abnizova I, te Boekhorst R, Walter K, Gilks W. Some statistical properties of regulatory DNA sequences, and their use in predicting regulatory regions in the Drosophila genome: The fluffy-tail test. BMC Bioinformatics. 2005;6:109. doi: 10.1186/1471-2105-6-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan B, Kibler D. Using hexamers to predict cis-regulatory motifs in Drosophila. BMC Bioinformatics. 2005;6:262. doi: 10.1186/1471-2105-6-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung G, Eisen M, Provart N. Identifying cis-regulatory sequences by word profile similarity. PLoS ONE. 2009;4:e6901. doi: 10.1371/journal.pone.0006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kantorovitz M, et al. Motif-blind, genome-wide discovery of cis-regulatory modules in Drosophila and mouse. Dev Cell. 2009;17:568–579. doi: 10.1016/j.devcel.2009.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xie X, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434:338–345. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ettwiller L, et al. The discovery, positioning and verification of a set of transcription-associated motifs in vertebrates. Genome Biol. 2005;6:R104. doi: 10.1186/gb-2005-6-12-r104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stark A, et al. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature. 2007;450:219–232. doi: 10.1038/nature06340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hughes J, Estep P, Tavazoie S, Church G. Computational identification of cis-regulatory elements associated with groups of functionally related genes in Saccharomyces cerevisiae. J Mol Biol. 2000;296:1205–1214. doi: 10.1006/jmbi.2000.3519. [DOI] [PubMed] [Google Scholar]
- 22.Wang T, Stormo G. Combining phylogenetic data with co-regulated genes to identify regulatory motifs. Bioinformatics. 2003;19:2369–2380. doi: 10.1093/bioinformatics/btg329. [DOI] [PubMed] [Google Scholar]
- 23.Grad Y, Roth F, Halfon M, Church G. Prediction of similarly acting cis-regulatory modules by subsequence profiling and comparative genomics in Drosophila melanogaster and D. pseudoobscura. Bioinformatics. 2004;20:2738–2750. doi: 10.1093/bioinformatics/bth320. [DOI] [PubMed] [Google Scholar]
- 24.Zhao G, Schriefer L, Stormo G. Identification of muscle-specific regulatory modules in Caenorhabditis elegans. Genome Res. 2007;17:348–357. doi: 10.1101/gr.5989907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sosinsky A, Honig B, Mann R, Califano A. Discovering transcriptional regulatory regions in Drosophila by a nonalignment method for phylogenetic footprinting. Proc Natl Acad Sci USA. 2007;104:6305–6310. doi: 10.1073/pnas.0701614104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinha S, van Nimwegen E, Siggia E. A probabilistic method to detect regulatory modules. Bioinformatics 19 suppl. 2003;1:i292–301. doi: 10.1093/bioinformatics/btg1040. [DOI] [PubMed] [Google Scholar]
- 27.Moses A, Chiang D, Pollard D, Iyer V, Eisen M. MONKEY: Identifying conserved transcription-factor binding sites in multiple alignments using a binding site-specific evolutionary model. Genome Biol. 2004;5:R98. doi: 10.1186/gb-2004-5-12-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao Z, Eddy S. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12:1269–1276. doi: 10.1101/gr.88502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cubas P, De Celis J, Campuzano S, Modolell J. Proneural clusters of achaete-scute expression and the generation of sensory organs in the Drosophila imaginal wing disc. Gene Dev. 1991;5:996–1008. doi: 10.1101/gad.5.6.996. [DOI] [PubMed] [Google Scholar]
- 30.Castro B, Barolo S, Bailey A, Posakony J. Lateral inhibition in proneural clusters: Cis-regulatory logic and default repression by suppressor of Hairless. Development. 2005;132:3333–3344. doi: 10.1242/dev.01920. [DOI] [PubMed] [Google Scholar]
- 31.Singson A, Leviten M, Bang A, Hua X, Posakony J. Direct downstream targets of proneural activators in the imaginal disc include genes involved in lateral inhibitory signaling. Gene Dev. 1994;8:2058–2071. doi: 10.1101/gad.8.17.2058. [DOI] [PubMed] [Google Scholar]
- 32.Culí J, Modolell J. Proneural gene self-stimulation in neural precursors: An essential mechanism for sense organ development that is regulated by Notch signaling. Gene Dev. 1998;12:2036–2047. doi: 10.1101/gad.12.13.2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- 34.Richards GS, et al. Sponge genes provide new insight into the evolutionary origin of the neurogenic circuit. Curr Biol. 2008;18:1156–1161. doi: 10.1016/j.cub.2008.06.074. [DOI] [PubMed] [Google Scholar]
- 35.Jafar-Nejad H, et al. Senseless acts as a binary switch during sensory organ precursor selection. Gene Dev. 2003;17:2966–2978. doi: 10.1101/gad.1122403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wildonger J, Mann R. Evidence that nervy, the Drosophila homolog of ETO/MTG8, promotes mechanosensory organ development by enhancing Notch signaling. Dev Biol. 2005;286:507–520. doi: 10.1016/j.ydbio.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 37.Pi H, Huang S, Tang C, Sun Y, Chien C. phyllopod is a target gene of proneural proteins in Drosophila external sensory organ development. Proc Natl Acad Sci USA. 2004;101:8378–8383. doi: 10.1073/pnas.0306010101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reeves N, Posakony J. Genetic programs activated by proneural proteins in the developing Drosophila PNS. Dev Cell. 2005;8:413–425. doi: 10.1016/j.devcel.2005.01.020. [DOI] [PubMed] [Google Scholar]
- 39.Gomes JE, Corado M, Schweisguth F. Van Gogh and Frizzled act redundantly in the Drosophila sensory organ precursor cell to orient its asymmetric division. PLoS ONE. 2009;4:e4485. doi: 10.1371/journal.pone.0004485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clark A, et al. Evolution of genes and genomes on the Drosophila phylogeny. Nature. 2007;450:203–218. doi: 10.1038/nature06341. [DOI] [PubMed] [Google Scholar]
- 41.Ayyar S, et al. NF-κB/Rel-mediated regulation of the neural fate in Drosophila. PLoS ONE. 2007;2:e1178. doi: 10.1371/journal.pone.0001178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cabrera C, Alonso M. Transcriptional activation by heterodimers of the achaete-scute and daughterless gene products of Drosophila. EMBO J. 1991;10:2965–2973. doi: 10.1002/j.1460-2075.1991.tb07847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Doren M, Ellis H, Posakony J. The Drosophila extramacrochaetae protein antagonizes sequence-specific DNA binding by daughterless/achaete-scute protein complexes. Development. 1991;113:245–255. doi: 10.1242/dev.113.1.245. [DOI] [PubMed] [Google Scholar]
- 44.Powell L, zur Lage P, Prentice D, Senthinathan B, Jarman A. The proneural proteins Atonal and Scute regulate neural target genes through different E-box binding sites. Mol Cell Biol. 2004;24:9517–9526. doi: 10.1128/MCB.24.21.9517-9526.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maerkl S, Quake S. Experimental determination of the evolvability of a transcription factor. Proc Natl Acad Sci USA. 2009;106:18650–18655. doi: 10.1073/pnas.0907688106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bryne J, et al. JASPAR, the open access database of transcription factor-binding profiles: New content and tools in the 2008 update. Nucleic Acids Res. 2007 doi: 10.1093/nar/gkm955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nellesen D, Lai E, Posakony J. Discrete enhancer elements mediate selective responsiveness of enhancer of split complex genes to common transcriptional activators. Dev Biol. 1999;213:33–53. doi: 10.1006/dbio.1999.9324. [DOI] [PubMed] [Google Scholar]
- 48.Lecourtois M, Schweisguth F. The neurogenic suppressor of hairless DNA-binding protein mediates the transcriptional activation of the enhancer of split complex genes triggered by Notch signaling. Gene Dev. 1995;9:2598–2608. doi: 10.1101/gad.9.21.2598. [DOI] [PubMed] [Google Scholar]
- 49.Bailey A, Posakony J. Suppressor of hairless directly activates transcription of enhancer of split complex genes in response to Notch receptor activity. Gene Dev. 1995;9:2609–2622. doi: 10.1101/gad.9.21.2609. [DOI] [PubMed] [Google Scholar]
- 50.Tweedie S, et al. FlyBase: Enhancing Drosophila gene ontology annotations. Nucleic Acids Res. 2009;37:D555–D559. doi: 10.1093/nar/gkn788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lehman D, et al. Cis-regulatory elements of the mitotic regulator, string/Cdc25. Development. 1999;126:1793–1803. doi: 10.1242/dev.126.9.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Giniger E, Tietje K, Jan L, Jan Y. lola encodes a putative transcription factor required for axon growth and guidance in Drosophila. Development. 1994;120:1385–1398. doi: 10.1242/dev.120.6.1385. [DOI] [PubMed] [Google Scholar]
- 53.Goeke S, et al. Alternative splicing of lola generates 19 transcription factors controlling axon guidance in Drosophila. Nat Neurosci. 2003;6:917–924. doi: 10.1038/nn1105. [DOI] [PubMed] [Google Scholar]
- 54.Ferres-Marco D, et al. Epigenetic silencers and Notch collaborate to promote malignant tumours by Rb silencing. Nature. 2006;439:430–436. doi: 10.1038/nature04376. [DOI] [PubMed] [Google Scholar]
- 55.Zheng L, Carthew R. Lola regulates cell fate by antagonizing Notch induction in the Drosophila eye. Mech Develop. 2008;125:18–29. doi: 10.1016/j.mod.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Heitzler P, Simpson P. The choice of cell fate in the epidermis of Drosophila. Cell. 1991;64:1083–1092. doi: 10.1016/0092-8674(91)90263-x. [DOI] [PubMed] [Google Scholar]
- 57.Zinzen R, Girardot C, Gagneur J, Braun M, Furlong E. Combinatorial binding predicts spatio-temporal cis-regulatory activity. Nature. 2009;462:65–70. doi: 10.1038/nature08531. [DOI] [PubMed] [Google Scholar]
- 58.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; 2009. ISBN3-900051-07-0. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.