

Abstract
Studies in cystic fibrosis patients and mice overexpressing the epithelial Na+ channel β-subunit (βENaC-Tg) suggest that raised airway Na+ transport and airway surface liquid (ASL) depletion are central to the pathogenesis of cystic fibrosis lung disease. However, patients or mice with Liddle gain-of-function βENaC mutations exhibit hypertension but no lung disease. To investigate this apparent paradox, we compared the airway phenotype (nasal versus tracheal) of Liddle with CFTR-null, βENaC-Tg, and double mutant mice. In mouse nasal epithelium, the region that functionally mimics human airways, high levels of CFTR expression inhibited Liddle epithelial Nat channel (ENaC) hyperfunction. Conversely, in mouse trachea, low levels of CFTR failed to suppress Liddle ENaC hyperfunction. Indeed, Na+ transport measured in Ussing chambers (“flooded” conditions) was raised in both Liddle and βENaC-Tg mice. Because enhanced Na+ transport did not correlate with lung disease in these mutant mice, measurements in tracheal cultures under physiologic “thin film” conditions and in vivo were performed. Regulation of ASL volume and ENaC-mediated Na+ absorption were intact in Liddle but defective in βENaC-Tg mice. We conclude that the capacity to regulate Na+ transport and ASL volume, not absolute Na+ transport rates in Ussing chambers, is the key physiologic function protecting airways from dehydration-induced lung disease.
Keywords: Cystic Fibrosis, Epithelium, Genetic Diseases, Lung, Sodium Channels, Sodium Transport
Introduction
The amiloride-sensitive epithelial Na+ channel (ENaC)3 is a heteromultimeric protein composed of three subunits (α, β, and γ). ENaC is limiting for the absorption of salt and water from airway and other epithelia, including renal and colonic epithelia (1–3). Increased ENaC-mediated airway Na+ absorption is a hallmark of cystic fibrosis (CF) (4, 5), a genetic form of chronic obstructive lung disease caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene (6, 7). Numerous studies have indicated that CFTR functions as a Cl− channel regulated by cAMP-dependent phosphorylation and as a regulator of ENaC in airway epithelia (1, 8–11). Previous in vitro studies indicated that the increased Na+ absorption in CF airways is caused by increased apical membrane ENaC activity, reflecting deficient inhibition by mutant CFTR, and that Na+ hyperabsorption is associated with airway surface liquid (ASL) volume depletion and impaired mucus clearance (12–14).
The concept that increased airway Na+ absorption and ASL volume depletion play a critical role in the in vivo pathogenesis of CF lung disease, however, remains controversial. For example, a recent hypothesis proposes that an acidic ASL pH, due to deficient HCO3− secretion by mutant CFTR, rather than deficient ASL volume regulation, produces increased mucus viscosity, reduced mucus clearance, and CF airway disease (15). The role of ASL volume depletion in CF pathogenesis was buttressed by a recently developed mouse model with airway-specific overexpression of βENaC under the control of the Clara cell secretory protein promoter (16). In this mouse model, selective airway overexpression of βENaC was sufficient to increase airway Na+ absorption, cause ASL volume depletion, reduce mucus clearance, and produce a spontaneous lung disease that shared key features with CF and other chronic obstructive pulmonary diseases, including substantial pulmonary mortality, airway mucus obstruction, goblet cell metaplasia, chronic airway inflammation, and slowed clearance of bacterial pathogens (16, 17). Taken together, the results from these in vitro and in vivo studies suggest that increased airway Na+ absorption, produced either by deficient CFTR-mediated inhibition of ENaC in human CF airways or by transgenic overexpression of wild-type βENaC in murine airways, causes ASL volume depletion, which initiates a series of pathogenetic steps that cause chronic obstructive lung disease.
Importantly, a third mechanism exists that can increase epithelial Na+ absorption. Gain-of-function mutations have been identified in the genes encoding β- and γENaC (Scnn1b and Scnn1g) that cause a constitutive increase in Na+ transport in the distal nephron of the kidney, resulting in an autosomal dominant form of salt-sensitive hypertension known as Liddle syndrome or pseudoaldosteronism (18–20). The molecular pathogenesis of Liddle syndrome has been studied in detail, and it has been demonstrated that the disease is caused by mutations that delete a proline-rich (PY) motif in the C termini of β- and γENaC that is critical for binding of the ubiquitin-protein ligase Nedd4-2. Nedd4-2 binding leads to ubiquitination and retrieval of ENaC from the plasma membrane. In Liddle syndrome, abrogation of the ENaC/Nedd4-2 interaction results in an increased number of ENaC channels in the plasma membrane, producing increased renal sodium absorption and salt sensitive hypertension (21–23). Because Nedd4-2 binding to ENaC is under control of Sgk1 (serum- and glucocorticoid-induced kinase 1), both Nedd4-2 and Sgk1 are key regulators of ENaC in the kidney (24–26).
Little is known about the pulmonary phenotype in Liddle syndrome, but Liddle patients do not develop overt lung disease (27, 28). In a mouse model that carries the classical Liddle mutation (i.e. a premature stop codon in the Scnn1b gene corresponding to the R556X mutation in Liddle patients), it has been shown that this gain-of-function mutation causes increased Na+ absorption in the distal nephron of the kidney, the colon, and the alveolar air spaces of the lung, but the phenotype of the conducting airways has not been studied (29–33). The aim of the present study was to elucidate the effects of this endogenously expressed gain-of-function mutation in βENaC on airway Na+ and volume transport and correlate each parameter with phenotypes in the upper (nasal) and lower airways. Studies of the lower airways included (i) Ussing chamber measurements of transepithelial Na+ transport under “flooded” conditions in native tissues, (ii) confocal ASL volume measurements under physiologic “thin film” conditions in cultured preparations, and (iii) a morphologic search for evidence of abnormal regulation of Na+ transport in vivo.
EXPERIMENTAL PROCEDURES
Experimental Animals
All animal studies were approved by the animal care and use committee of the University of North Carolina at Chapel Hill and the Regierungspräsidium Karlsruhe, Germany. The generation and genotyping of Liddle mice, βENaC-Tg mice, and CF (Cftr−/−) mice has been described previously (16, 29, 34, 35). Liddle mice were originally generated on a mixed genetic background (129Ola × C57BL/6J) and were backcrossed for >5 generations to the C57BL/6J background. βENaC-Tg mice (line 6608) were generated on a mixed genetic background (C3H/HeN × C57BL/6N) and were backcrossed (N10) to the C57BL/6J background. CF mice were on a mixed genetic background (BALB/c, C57BL/6, DBA/2, and 129/SvEv). Mice heterozygous for the Liddle mutation (L/+) were interbred, and homozygous Liddle mice (L/L) and wild-type (WT) littermates were studied at the age of 12–16 weeks. CF mice were intercrossed with Liddle mice and double mutant CF mice homozygous for the Liddle mutation (CF-Liddle), and CF, L/L, and WT littermate controls were studied at the age of 16–54 weeks. βENaC-Tg mice and WT littermates were 6–8 weeks of age. Airway epithelial necrosis was studied in 3-day-old mice of respective genotypes. Mice were housed in specific pathogen-free animal facilities. CF mice received Colyte instead of drinking water to prevent intestinal obstruction. All other animals had free access to regular chow and water.
In Vivo Potential Difference (PD) Measurements
For in vivo studies of transepithelial PD, mice were anesthetized via intraperitoneal injection of avertin (0.4 g/kg tribromoethanol, 0.4 ml/kg amyl alcohol), and the body temperature was continually monitored with a rectal thermocouple (Physitemp) and maintained at 37 °C with a heat lamp. Nasal and rectal PD measurements were performed, and the basal PD and the amiloride-sensitive PD were determined as described previously (29, 36).
Ussing Chamber Measurements
Mice were killed with 100% CO2, nasal and tracheal tissues were removed and mounted on modified Ussing chambers, and bioelectric measurements were taken as described previously (13, 16, 37). In most studies, we used recirculating chambers to measure basal short circuit current (Isc) and the sequential effects of amiloride (100 μmol/liter; luminal), forskolin (10 μmol/liter; luminal), and UTP (100 μmol/liter; luminal). Studies on protease regulation of ENaC-mediated Na+ transport and amiloride dose-response studies were performed in continuously perfused chambers under open circuit conditions, and the equivalent short circuit current (Ieq) was calculated from the PD and the transepithelial resistance (Rte) according to Ohm's law (i.e. Ieq = PD/Rte) as described previously (13). Dose-response curves were obtained by measuring the change in Ieq induced by exposing tissues to increasing concentrations of amiloride (10−9 to 10−3 mol/liter) and plotted as remaining amiloride-sensitive Ieq (Iamil) normalized to the maximal amiloride-sensitive Ieq (Imax). IC50 values were determined by fitting dose-response data to the Hill equation with the Hill coefficient constrained to 1.0 as follows,
where A is the apical bath amiloride concentration. Additionally, data were fitted with a two-component Hill model,
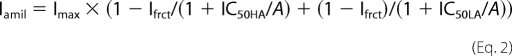 |
where subscripts HA and LA denote the high affinity and low affinity IC50, respectively, and Ifrct is the fractional HA ENaC population. The LA ENaC population = 1 − Ifrct. The models that best fit the dose-response data were judged based on comparison of S.E. using an F-test distribution,
where S is the residual sum of squares, n is the number of data points, k is the number of free parameters, and subscripts 1 and 2 depict parameters of the one- and two-component Hill model, respectively (38). For studies of ENaC regulation by extracellular serine proteases, tissues were equilibrated in the presence of the protease inhibitor aprotinin (100 μg/ml, ∼3.8 trypsin inhibitory units/mg; Sigma) added to the apical bath, and the effect of amiloride (100 μmol/liter; luminal) was either measured in the presence of aprotinin alone or after adding a ∼2-fold excess of trypsin (550 μg/ml, ∼14,000 N-benzoyl-l-arginine ethyl ester units/mg; Sigma) on the basis of activity (1 trypsin unit = 9000 N-benzoyl-l-arginine ethyl ester units).
Real-time RT-PCR
Mice were killed with 100% CO2 and nasal and tracheal tissues were immediately removed and stored in RNAlater (Ambion, Austin, TX). Total RNA was isolated using the RNeasy kit (Qiagen Inc., Valencia, CA) and reverse transcribed into cDNA using Superscript II (Invitrogen). Quantification of αENaC, βENaC, γENaC, CFTR, Nedd4-2, and Sgk1 mRNA expression was performed using SYBR Green detection in a LightCycler PCR machine according to the manufacturer's instructions (Roche Applied Science) and the following custom made primers (MWG Biotech, High Point, NC): αENaC, 5′-CGG AGT TGC TAA ACT CAA CATC-3′ (sense) and 5′-TGG AGA CCA GTA CCG GCT-3′ (antisense); βENaC, 5′-TAA TGG AGG CAG TCC TGG-3′ (sense) and 5′- GTT GGC AGA AGG AAG TGT CT-3′ (antisense); γENaC, 5′-CAG GCG CAT AGC AGA GGT A-3′ (sense) and 5′-ACC TGG CCA AGC TCT TGA TA-3′ (antisense); CFTR, 5′-ACC ACA GGC ATA ATC ATG G-3′ (sense) and 5′-AGC AGA ATG AAA CTC TTC CAC-3′ (antisense); Nedd4-2, 5′-GAT ACC CTT TCC AAT CCA CA-3′ (sense) and 5′-AGG ACG TCA GGT CTC TTT ACA-3′ (antisense); Sgk1, 5′-ACA ACA TCT ACC TTC TGT GGC-3′ (sense) and 5′- CAT AAA GTC ATC CTT GGC AC-3′ (antisense). Relative -fold changes in target gene expression were calculated from the efficiency of the PCR and the crossing point deviation between samples from experimental groups and determined by normalization to expression of the reference gene 18 S or β-actin, as described previously (17).
Histology and Airway Morphometry
Adult mice were killed with 100% CO2, and neonatal mice were killed by decapitation. Lungs were removed through a median sternotomy, immersion-fixed in 4% buffered formalin, and embedded in paraffin. Sections were cut at 5 μm and stained with hematoxylin and eosin (H&E) or Alcian blue periodic acid-Schiff as described previously (16). Necrotic airway epithelial cells were identified by morphologic criteria (i.e. cell swelling with cytoplasmic vacuolization), and numeric cell densities were quantitated by counting epithelial cells/mm of the basement membrane as described previously (17).
Primary Tracheal Epithelial Cultures
Mice were euthanized with 100% CO2, the tracheas were removed, and epithelial cells were isolated and cultured on membranes (T-Col, Costar, Cambridge, MA) under air-liquid interface conditions and studied after reaching confluence (10 days), and some cultures were subjected to Ussing chamber studies to determine amiloride-sensitive Na+ transport, as described previously (16).
ASL Height Measurements
Primary tracheal epithelial cultures were washed, and 20 μl of PBS containing 0.2% (v/v) Texas Red-dextran (10 kDa; Molecular Probes) was added to the lumen to visualize the ASL layer. Adding this volume of PBS results in an initial ASL height of ∼20–30 μm, as described previously (39). Images of the Texas Red-labeled ASL were acquired by laser-scanning confocal microscopy (LSM 510, Carl Zeiss, Jena, Germany) using the appropriate filters for Texas Red (540 nm excitation/630 nm emission). To avoid evaporation of the thin ASL layer, 100 μl of immiscible perfluorocarbon (Fluorinert-77, 3M Corp.) was added to the airway surface following the addition of the labeling dye (39). The height of the ASL was measured by averaging the heights obtained from xz scans of five predetermined points on the culture. In the experiments described here, ASL height was measured 15 min following the addition of the Texas Red-dextran and at designated time points over a period of 24 h in primary tracheal epithelial cultures from Liddle mice, βENaC-Tg mice, and their respective WT littermates.
Statistics
All data were analyzed with SigmaStat version 3.1 (Systat Software, Erkrath, Germany) and are reported as mean ± S.E. Statistical analyses were performed using Student's t test, the Mann-Whitney rank sum test, one-way analysis of variance, and Kruskal-Wallis analysis of variance on ranks as appropriate, and p < 0.05 was accepted to indicate statistical significance.
RESULTS
The Liddle Mutation Does Not Cause Gain of Na+ Transport Function in Nasal Epithelia
To study the effect of the Liddle mutation on airway Na+ transport, we first performed in vivo measurements of nasal PD in Liddle mice and WT littermate controls. Basal and amiloride-sensitive nasal PD values in WT mice were similar to those in previous studies (36), and neither value was different in Liddle mice (Fig. 1A). In contrast, rectal PD measurements demonstrated that both basal and amiloride-sensitive rectal PD were significantly increased in Liddle mice compared with WT controls (supplemental Fig. 1), as described previously (29). The absence of increased Na+ absorption in nasal epithelia of Liddle mice was confirmed by in vitro measurements of basal and amiloride-sensitive transepithelial short circuit current (Isc) across freshly excised nasal epithelia (Fig. 1B). Furthermore, following amiloride pretreatment, cAMP-stimulated Cl− secretion was the same in Liddle and WT tissues (supplemental Fig. 2A). Ca2+-activated (UTP) Cl− secretion was absent in both genotypes (supplemental Fig. 2A), as described previously for WT mice (36).
FIGURE 1.

Effect of the Liddle mutation on airway Na+ absorption. A–C, in vivo measurements of basal and amiloride-sensitive nasal PD (A) and ex vivo measurements of basal and amiloride-sensitive Isc in nasal tissues (B) and tracheal tissues (C) from Liddle (L/L) and WT mice. n = 5–17 mice/group. *, p < 0.05; **, p < 0.001 compared with WT. Error bars, S.E.
The Liddle Mutation Does Cause Gain of Na+ Transport Function in Tracheal Epithelia
Because previous studies from CF mice suggested that the regulation of ENaC function differs in mouse upper and lower airways (36, 37, 40), we also studied the effect of the Liddle mutation on ion transport in freshly excised tracheal epithelia. The magnitude of amiloride-sensitive Na+ absorption in WT mice was substantially smaller in tracheal than nasal epithelia (Fig. 1, B and C). However, basal and amiloride-sensitive Isc were significantly increased in tracheal tissues from Liddle compared with WT tracheas (Fig. 1C). Following amiloride pretreatment, the magnitude of Cl− secretion stimulated by either cAMP or Ca2+ agonists did not differ between Liddle and WT mice (supplemental Fig. 2B). This indicates that agonist-mediated Cl− secretory capacity was not altered by the Liddle mutation under conditions of amiloride block.
Expression of ENaC Subunits and ENaC Regulators in Upper and Lower Airway Epithelia
To identify mechanisms regulating Na+ transport rates in nasal versus tracheal epithelia in WT and Liddle mice, we measured transcript levels of individual α-, β-, and γENaC subunits and several known ENaC regulators, including CFTR, the ubiquitin protein ligase Nedd4-2, and the serum and glucocorticoid-activated kinase Sgk1 in freshly excised tissues from both genotypes (10, 11, 23, 24, 26).
To investigate the differences in Na+ transport rates in WT versus Liddle mouse airways, transcript levels in Liddle mice were normalized to levels in WT mice. Expression of the mutated βENaC was significantly reduced in both Liddle nasal and tracheal tissues, as described previously for kidney and colon, probably reflecting RNA instability caused by the residual loxP site in the targeting construct (29) (Fig. 2). However, expression levels of αENaC, γENaC, Nedd4-2, Sgk1, and CFTR were not different in Liddle mice compared with WT controls in either nasal or tracheal epithelia (Fig. 2, A and B).
FIGURE 2.

Expression of ENaC subunits and ENaC regulators in nasal and tracheal epithelia. A–C, transcript levels of αENaC, βENaC, γENaC, CFTR, Nedd4-2, and Sgk1 in freshly excised nasal and tracheal tissues from L/L and WT mice. A and B, comparison of transcript expression levels in nasal (A) and tracheal (B) tissues from L/L versus WT mice. Data are expressed as -fold changes from WT. n = 5–6 mice/group; *, p < 0.05; **, p < 0.01. C, comparison of expression levels in tracheal tissues (open bars) versus nasal tissues (closed bars) from WT mice. Data are expressed as -fold changes from tracheal tissues. n = 5–6 mice/group; **, p < 0.01; ***, p < 0.001. Error bars, S.E.
To determine the differences in nasal versus tracheal Na+ transport, we normalized transcript levels in nasal to tracheal epithelia of WT mice. Consistent with higher Na+ transport in nasal versus tracheal epithelia (Fig. 1, B and C), these studies revealed that expression levels of α-, β-, and γENaC were all significantly increased in the nose compared with the trachea (Fig. 2C). Although Nedd4-2 and Sgk1 were also relatively increased in nasal epithelia, the highest -fold change in expression was for CFTR mRNA, which was ∼20-fold higher in nasal compared with tracheal epithelia (Fig. 2C).
CFTR Inhibits Gain of ENaC Function in Nasal Epithelia from Liddle Mice
To more directly investigate the role of CFTR in regulation of the Liddle mutated ENaC in native airway tissues, we generated double mutant mice that were deficient in CFTR (CF) and homozygous for either the Liddle mutation (CF-Liddle) or the wild-type Scnn1b allele and performed in vivo nasal PD measurements and transepithelial Ussing chamber measurements on nasal and tracheal epithelia (Fig. 3, A–C). In vivo nasal PD measurements demonstrated that basal and amiloride-sensitive PD were significantly increased in double mutant CF-Liddle, in a stepwise relationship, compared with (i) the raised PDs of CF littermates, (ii) “normal” PDs of Liddle mice with a wild-type CFTR genotype, and (iii) WT mice (Fig. 3A). Further, in vitro measurements of transepithelial Isc revealed similar stepwise relationships for basal and amiloride-sensitive Isc in nasal tissues from CF-Liddle compared with CF mice, Liddle mice expressing wild-type CFTR, or WT mice (Fig. 3B). Conversely, in tracheal tissues, the lack of CFTR had no effect on basal and amiloride-sensitive Isc in CF-Liddle compared with Liddle mice (Fig. 3C). Similar to Liddle mice expressing wild-type CFTR (supplemental Fig. 2), Cl− secretory capacity following activation with forskolin or UTP was not different in amiloride-pretreated nasal or tracheal tissues from CF-Liddle mice compared with CF controls (supplemental Fig. 3). Taken together, these data indicate that wild-type CFTR inhibits the Liddle ENaC gain of Na+ transport function when both proteins are co-expressed in the nasal epithelium. Conversely, these data are consistent with the hypothesis that low levels of tracheal CFTR expression were insufficient to inhibit the Liddle ENaC gain of function in the trachea.
FIGURE 3.

Effect of the Liddle mutation on airway Na+ absorption in CFTR-deficient mice. A–C, in vivo measurements of basal and amiloride-sensitive nasal PD (A) and ex vivo measurements of basal and amiloride-sensitive Isc in nasal tissues (B) and tracheal tissues (C) from WT mice, L/L mice, single mutant CFTR-deficient (CF) mice, and double mutant CFTR-deficient Liddle (CF + L/L) mice. A, nasal PD measurements. n = 8–12 mice/group. *, p < 0.001 compared with WT and L/L mice; †, p < 0.001 compared with WT, L/L, and CF mice. B, nasal Isc measurements. n = 5–7 mice/group. *, p < 0.05 compared with WT and L/L mice; †, p < 0.02 compared with L/L mice; ‡, p < 0.01 compared with WT and L/L mice; §, p < 0.05 compared with CF mice. C, tracheal Isc measurements. n = 6–9 mice/group. *, p < 0.05 compared with WT mice; †, p < 0.01 compared with L/L mice. ‡, p ≤ 0.02 compared with CF mice; §, p < 0.001 compared with WT and CF mice. Error bars, S.E.
Morphologic Evaluation of Lungs from Liddle Mice
Liddle mice exhibited normal survival and no spontaneous airway disease phenotype, including absence of histological evidence of mucus obstruction, goblet cell metaplasia, or airway inflammation, compared with WT controls (Fig. 4). Similar to the findings in Liddle mice, airways pathology was also absent in double mutant CF-Liddle mice (Fig. 4). In contrast, βENaC-Tg mice with a comparable level of increased tracheal Na+ absorption at 4–8 weeks after birth exhibit airway mucus plugging, airway epithelial remodeling with goblet cell hyperplasia, neutrophilic inflammation, and emphysema (16, 17).
FIGURE 4.

Effect of the Liddle mutation on airway morphology. Lung histology from adult WT mice, L/L mice, CF mice, double mutant CFTR-deficient Liddle (CF + L/L) mice, and βENaC-Tg mice stained with H&E and Alcian blue periodic acid-Schiff (AB-PAS). Scale bars, 100 μm. Results shown are representative for n = 6–14 mice/group.
ASL Volume Regulation by WT, Liddle, and βENaC-Tg Cultured Airway Epithelia
Airway epithelial volume homeostasis requires the capacity to reciprocally regulate Na+ absorption and Cl− secretion (39, 41). Our previous studies demonstrated that airway Na+ hyperabsorption induced by tissue-specific overexpression of βENaC causes ASL volume depletion and deficient mucus clearance in vivo, indicating that overexpression of βENaC produced an unregulated ENaC channel that produced lung disease via airway surface dehydration (16, 17). To test the hypothesis that the absence of pulmonary pathology in Liddle mice reflected “normal” regulation of Liddle ENaC in vivo in the lung, we generated the primary epithelial culture preparations required for studies of ASL volume regulation under physiologic thin film conditions. Similar to freshly excised tracheal tissues, Ussing chamber studies under flooded conditions demonstrated that amiloride-sensitive Na+ transport was significantly increased in tracheal cultures from Liddle mice (ΔIsc = 21.8 ± 5.4 μA/cm2, n = 12 for WT; ΔIsc = 60.7 ± 15.3 μA/cm2, n = 12 for Liddle; p < 0.05) and βENaC-Tg mice (ΔIsc = 13.9 ± 3.4 μA/cm2, n = 6 for WT; ΔIsc = 45.9 ± 7.9 μA/cm2, n = 5 for βENaC-Tg; p < 0.01) compared with WT controls.
For ASL measurements under thin film conditions, a small volume of liquid (20 μl) was added to the luminal compartment of confluent tracheal cultures grown at an air-liquid interface, and ASL height was monitored sequentially over a period of 24 h by confocal laser-scanning microscopy (39, 41). In WT cultures, the added liquid was largely absorbed within a period of ∼4 h. Subsequently, ASL height was maintained at ∼6 μm, corresponding to the height of extended cilia in mice (Fig. 5). This volume homeostatic response probably reflected the inhibition of Na+ transport and acceleration of Cl− secretion reported during ASL volume homeostasis in cultured normal human airway epithelia (39, 41). In cultures from βENaC-Tg mice, the initial rate of liquid absorption was significantly increased compared with WT controls, consistent with increased Na+ absorption. Importantly, these cultures failed to slow absorption when ASL approached the height of outstretched cilia and removed all of the available liquid from airway surfaces (Fig. 5A). This pattern of ASL volume hyperabsorption reflects increased and unregulated airway Na+ absorption, a pattern also observed in human CF airway cultures (14, 39). In tracheal cultures from Liddle mice, similar to βENaC-Tg mice, initial ASL volume absorption was also accelerated, and ASL was almost entirely removed from culture surfaces within 2 h following the volume challenge. However, in contrast to βENaC-Tg cultures, where ASL remained depleted, Liddle epithelia added ASL to their surfaces during the 7–24 h phase and regulated ASL height to levels similar to WT controls (i.e. ∼6 μm) (Fig. 5B).
FIGURE 5.

ASL volume regulation in airway epithelia from Liddle mice and βENaC-Tg mice. A–C, Confocal images (A) and summary of measurements of ASL height (B and C) at 0, 2, 4, 8, and 24 h after the mucosal addition of 20 μl of PBS containing Texas Red dextran to primary tracheal epithelial cultures from βENaC-Tg (A and B), L/L mice (A and C), and the respective WT littermates. Scale bars, 7 μm. n = 3–6 mice/group. *, p < 0.05 compared with WT. **, p < 0.01 compared with WT. Error bars, S.E.
The Liddle Mutation Does Not Cause Airway Epithelial Cell Necrosis
The confocal ASL data suggest that Liddle ENaC is regulated in the lower airways at physiologic ASL volumes, whereas βENaC-Tg channels are not. To search for in vivo correlates of Na+ channel regulation, we quantitated the number of Clara cells undergoing hydropic degeneration. Hydropic degeneration has been reported in neurons in Caenorhabditis elegans that express gain-of-function mutations in a channel homologous to ENaC, the degenerin channel (42, 43). The gain-of-function mutations in the degenerin channel appear to lock the channel into an unregulated open mode with an open probability (Po) approaching 1.0. Necrotic (hydropic degenerated) cells were detected in airway epithelia from neonatal βENaC-Tg mice (Fig. 6, A and B) but not in adult βENaC-Tg mice (data not shown), as reported previously (17). In contrast to βENaC-Tg mice, we detected no necrotic cells in airways of Liddle mice at any time period (Fig. 6, A and B).
FIGURE 6.

Regulation of ENaC in airway epithelia is abnormal in βENaC-Tg mice but preserved in Liddle mice. A, airway histology from neonatal (3-day-old) βENaC-Tg mice, L/L mice, and their respective WT littermates. Sections were stained with H&E and evaluated for degenerative airway epithelial cells (arrows). Scale bars, 20 μm (upper panels) and 10 μm (lower panels). B, summary of airway epithelial necrosis as determined from the number of degenerative epithelial cells per mm of the basement membrane. n = 3–5 mice for each group. *, p ≤ 0.01 compared with WT. C–E, amiloride dose-response curves (C and D) and summary of IC50 values (E) obtained from tracheal tissues of βENaC-Tg mice, L/L mice, and respective WT littermates. n = 6–12 mice/group. *, p < 0.01 compared with WT mice; †, p = 0.001 compared with L/L mice. Error bars, S.E.
ENaC Sensitivity to Amiloride in Airways of WT, Liddle, and βENaC-Tg Mice
The presence of necrotic cells in the airway epithelium of βENaC-Tg mice, but not Liddle mice, suggested a defect in ENaC regulation in βENaC-Tg but not Liddle murine airway epithelia. We therefore searched for a mechanism that may predict abnormal regulation of βENaC-Tg channels. In the βENaC-Tg mice, βENaC is expressed >20–100-fold greater than the endogenous βENaC, raising the possibility that αβ ENaC channels may be formed in vivo. Compared with αβγ ENaC channels, heterologous expression of α- and βENaC subunits in Xenopus oocytes produces reduced whole cell Na+ currents, probably due to inefficient processing of αβ ENaC (1). However, αβ ENaC channels that are inserted into the plasma membrane have a resting open probability of ∼1.0, compared with ∼0.5 for heterologously expressed αβγ ENaC channels (44). This finding suggests that αβ ENaC channels are abnormally regulated in oocytes so that they are maximally activated under resting conditions. Another important characteristic of the αβ ENaC channel is that it is less sensitive to amiloride than the αβγ ENaC channel in oocytes, with the amiloride IC50 shifted about 1 log to the right of αβγ ENaC (1, 11, 45). In contrast to alveolar epithelial cells (46), it was not possible to obtain cell-attached membrane patches from the apical membrane of well differentiated mouse airway epithelial cells to measure Po directly. We therefore tested the hypothesis that βENaC-Tg may express abnormally regulated αβ ENaC channels by characterizing the amiloride sensitivity of freshly excised tracheas from βENaC-Tg, Liddle, and WT mice.
Analysis of amiloride dose-response curves using a single component Hill model demonstrated that the amiloride IC50 for Liddle tracheas was slightly shifted (∼1.5-fold) to the right of WT littermate controls. However, in βENaC-Tg tracheas, the shift in IC50 to the right of WT tissues was significantly larger (∼4.8-fold), suggesting that the sensitivity to amiloride was significantly reduced in βENaC-Tg compared with Liddle tracheas (Fig. 6, C–E). Modeling of dose-response curves with a single component versus a two-component Hill model demonstrated that the amiloride dose-response curve in βENaC-Tg tracheas was described significantly better by the two-component model compared with the single component model (p < 0.025). This finding is consistent with two channel populations (i.e. a high affinity channel population (IC50 = 0.6 ± 0.1 μmol/liter) and a second population of low affinity channels (IC50 = 30.9 ± 10.7 μmol/liter)), which constituted ∼35% of the total ENaC channel population (Table 1). In contrast, IC50 values determined by the single component versus the two-component Hill model did not differ statistically in WT and Liddle tracheae (Table 1), consistent with expression of a single high affinity ENaC channel population in WT and Liddle mice. Thus, these data suggest that the necrotic cells in βENaC-Tg mouse airways may reflect excessive Na+ absorption via a population of unregulated αβ ENaC channels, whereas the absence of necrotic cells suggests that the Liddle αβγ ENaC channel is sufficiently regulated in vivo to protect airway epithelia from necrosis.
TABLE 1.
Hill model dose-response fit parameters ± S.E. for ENaC channels in native tracheal epithelia of WT, L/L, and βENaC-Tg mice
Parameters were obtained from averaged amiloride dose-response data (see Fig. 6, C and D) fitted with single and two-component Hill equations (see “Experimental Procedures”). Results are consistent with tracheal tissues from βENaC-Tg mice expressing a subpopulation of ENaC channels with low affinity toward amiloride. NS, not significantly different.
| Single component Hill model IC50 | Two-component Hill model |
Statistical comparison single versus two-component modela | |||
|---|---|---|---|---|---|
| High affinity IC50 | High affinity relative population Ifrct | Low affinity IC50 | |||
| μm | μm | % | μm | ||
| WT | 0.3 ± 0.0 | 0.3 ± 0.2 | 86 ± 105 | 0.9 ± 4.4 | NS |
| L/L | 0.6 ± 0.0 | 0.2 ± 0.2 | 22 ± 31 | 0.8 ± 0.3 | NS |
| βENaC-Tg | 1.8 ± 0.4 | 0.6 ± 0.1 | 64 ± 3.2 | 30.9 ± 0.7 | p < 0.025 |
a βENaC-Tg Fcalc(3, 7) = 59.3; WT Fcalc(3, 5) = 0.2; Liddle Fcalc(3, 5) = 2.5.
ENaC Regulation by Extracellular Proteases Is Abnormal in Airways of βENaC-Tg Mice
Recent studies demonstrated that proteolytic activation of αβγ ENaC channels by extracellular serine proteases plays an important role in regulating ENaC-mediated Na+ absorption in airway epithelia. However, protease-dependent activation is not required for αβ ENaC channels, suggesting that the γENaC subunit is essential in this process (47–49). To further test the hypothesis that βENaC-Tg mice express abnormally regulated αβ ENaC channels, we compared ENaC regulation by both block of protease activation (aprotinin) and application of trypsin as a prototypic serine protease in freshly excised WT and βENaC-Tg tracheal tissues. To maximize the effect of the block of protease activation of ENaC, these experiments were performed in the presence of the serine protease inhibitor aprotinin from the time immediately after tracheal excision to a measure of steady state current 30 min after mounting of the tissues in Ussing chambers. Amiloride-sensitive Na+ transport was measured both under steady state (aprotinin) conditions and after the addition of luminal trypsin. Under steady state aprotinin conditions, the amiloride-sensitive Isc was significantly (∼5.5-fold) increased in trachea from βENaC-Tg compared with WT mice (Fig. 7). The subsequent addition of trypsin to the luminal bath increased the amiloride-sensitive Isc by a similar magnitude in WT and βENaC-Tg tracheae (ΔIsc = 26.0 ± 5.6 μA/cm2, n = 10 for WT; ΔIsc = 37.5 ± 12.4 μA/cm2, n = 14 for βENaC-Tg; p < 0.93), reducing the ratio of amiloride-sensitive Isc in βENaC versus WT mice to ∼2.7-fold, typical for βENaC-Tg and WT tissues in the absence of aprotinin (16). Taken together, these data indicate that WT and βENaC-Tg airway epithelia express a similar number of apical membrane αβγ ENaC channels that are largely inhibited by aprotinin and reactivated by trypsin. The βENaC-Tg mice express a population of αβ channels that are not regulated by proteases, accounting for the proportionately reduced effects of aprotinin and trypsin in βENaC-Tg mice as compared with WT mice.
FIGURE 7.

Regulation of ENaC by extracellular proteases is abnormal in airway epithelia from βENaC-Tg mice. Freshly excised tracheal tissues from WT and βENaC-Tg mice were pretreated with luminal aprotinin, and amiloride-sensitive Isc was measured either in the presence of aprotinin alone (Aprotinin) or after the addition of luminal trypsin (+Trypsin). n = 9–14 mice/group. *, p < 0.05; **, p < 0.001 compared with mice of same genotype; †, p < 0.001 compared with WT mice; §, p < 0.001 compared with βENaC-Tg mice. Error bars, S.E.
DISCUSSION
Proper regulation of ASL volume, a function dependent on coordinate regulation of ENaC-mediated Na+ absorption and CFTR/CaCC-mediated Cl− secretion, plays a critical role in maintaining the normal mucociliary clearance component of lung defense (50, 51). Therefore, defects in either process (i.e. active Cl− secretion or Na+ absorption) are predicted to produce ASL depletion, mucociliary dysfunction, and lung disease. The aim of this study was to elucidate the role of abnormal ENaC function and ASL volume regulation in the in vivo pathogenesis of chronic airway disease. We therefore studied a mouse model of a gain-of-function mutation in ENaC (Liddle) reported to exhibit increased Na+ absorption in renal and colonic epithelia (29–32). Our investigations led us to study both (i) why Liddle-mediated increased Na+ transport was expressed in the tracheal but not nasal epithelium and (ii) why, despite Ussing chamber measured increases in Na+ transport in the Liddle trachea, there was no associated lung disease.
First, we sought the mechanisms to explain the apparent discordance of the Liddle Na+ transport phenotype in the two respiratory regions (Fig. 1). The expression of all three ENaC subunits (α, β, and γ) is required to obtain maximal Na+ currents (1), and CFTR, Nedd4-2, and Sgk1 have been reported to regulate apical membrane ENaC number and activity (10, 12, 23–26). We therefore hypothesized that absence of increased Na+ absorption in nasal epithelia from Liddle compared with WT mice was caused by reduced expression of α- or γENaC subunits or increased expression of ENaC regulators in nasal epithelia from Liddle compared with WT mice. Comparison of ENaC subunit expression levels between WT and Liddle mice confirmed a ∼50% reduction in Liddle βENaC transcripts in nasal epithelia, as previously described for kidney and colon (29). However, no differences in expression levels of α- or γENaC or other ENaC regulators between WT and Liddle nasal tissues were detected that could compensate for the Liddle gain-of-function mutation (Fig. 2).
However, we noted that the level of CFTR expression in mice was markedly higher in nasal compared with tracheal epithelia in both WT and Liddle mice (Fig. 2). The importance of CFTR in regulation of Na+ transport rates in mice has been suggested previously by observations from CF mice. In CF mice, the amiloride-sensitive Isc is raised in the nasal epithelia, but not trachea, reflecting the likelihood that CFTR is expressed at levels sufficient to regulate ENaC in the nasal cavity but that other regulatory paths dominate in the lower airways, where CFTR expression is very low (36, 37, 40). The role of CFTR in regulation of Liddle ENaC in native airway tissues has not been studied, and co-expression studies in heterologous cells produced conflicting results. Whereas one study suggested that CFTR inhibits ENaC channels carrying Liddle mutations, including stop mutations that produce truncations of the C termini of β- and γENaC (52), another study indicated that C termini of β- and γENaC are required for functional interaction between CFTR and ENaC (53).
To genetically study the role of CFTR in regulation of Liddle ENaC in native nasal versus tracheal epithelia, we crossed CF mice with Liddle mice and observed a ∼2-fold increase in PD in CF-Liddle nasal as compared with CF nasal epithelia. The additivity in rates of Na+ transport observed in the CF-Liddle mice compared with CF mice is consistent with the absence of CFTR-mediated inhibition of ENaC in CF mice (10, 11, 13) and gain of ENaC function in the Liddle mice (21, 54). In contrast, the rates of Na+ transport in CF-Liddle and Liddle tracheas were not different (Fig. 3), consistent with the relative absence of CFTR expression, and hence CFTR regulatory function, in either the Liddle or WT tracheas.
Thus, these genetic and functional studies demonstrate that CFTR plays a critical role in the regulation of both WT and Liddle ENaC in nasal epithelial cells that co-express ENaC and CFTR. Similar to Liddle patients expressing the R556X mutation, the C terminus of the βENaC subunit is truncated in Liddle mice. Therefore, our studies suggest that the functional interaction between CFTR and ENaC in native nasal tissues did not rely on the presence of the βENaC C terminus. The gain of Na+ transport function in the tracheas of Liddle mice is consistent with data reporting Liddle mediated gain of Na+ transport function in other epithelia in which CFTR is expressed at low levels (e.g. the cortical collecting duct of the kidney or the surface epithelium of the colon) (3, 55). Because in humans, unlike mice, CFTR is co-expressed with ENaC at relatively high levels in the surface epithelium of both upper and lower airways (56), our findings also may explain why patients with Liddle syndrome do not develop CF-like lung disease (27, 28).
Second, we investigated why increased Na+ absorption in the lower airways of Liddle mice did not cause CF-like lung disease. In contrast to βENaC-Tg mice, Liddle mice exhibited an absence of airway mucus obstruction, goblet cell hyperplasia, or inflammation (Fig. 4) (29, 33). We hypothesized that it may not be an increase in the absolute rate of Na+ transport but rather the lack of regulation of Na+ transport required for normal ASL volume homeostasis that caused lung disease.
We tested the hypothesis that βENaC-Tg mice cannot regulate ENaC sufficiently for ASL volume homeostasis in vitro, whereas Liddle mice can, by measuring ASL homeostasis in primary tracheal cultures under thin film conditions in response to a volume challenge. Normal mouse airway epithelia exhibited ASL volume regulation similar to human epithelia, with a key feature being inhibition of ENaC-mediated Na+ transport as excess liquid is absorbed from the airway surface (Fig. 5). The volume response of βENaC-Tg tracheal cultures was strikingly similar to the response observed in human CF airway cultures (i.e. initially accelerated absorption, failure to slow absorption at low ASL volume, and steady state depletion of ASL volume from epithelial surfaces) (Fig. 5) (39, 41). This physiology is consistent with a failure to regulate (inhibit) ENaC at low ASL volumes.
In tracheal cultures from Liddle mice, consistent with increased Na+ absorption observed in Ussing chamber experiments under flooded conditions, the rate of absorption of the added liquid was accelerated. However, in contrast to βENaC-Tg epithelia, “normal” ASL volume homeostasis was achieved in ∼6 h by Liddle tracheal cultures following the removal of excess fluid, and ASL remained at a steady state thereafter. With respect to the delay of normal homeostatic ASL regulation in Liddle compared with WT mice, we speculate that Liddle (and WT) ENaC may be regulated by soluble inhibitor(s) that were diluted after the volume challenge and reconcentrated under thin film conditions. For example, previous studies demonstrated that ENaC is activated by proteolytic cleavage and blocked by protease inhibitors, suggesting that an endogenous protease/antiprotease system plays an important role in CFTR-independent regulation of ENaC on airway surfaces (49, 57–59). Further, recent evidence suggested that ENaC-activating proteases are membrane-anchored, whereas protease inhibitors are soluble and can therefore act as ASL sensors to inhibit ENaC activity under thin film conditions (60). Because the Liddle mutation results in an increased number of active ENaC channels on the plasma membrane, we speculate that (i) the early rapid phase of volume absorption reflected the activity of a relatively (to WT) large number of active ENaC channels on the membrane, and (ii) the return to normal ASL homeostatic volumes reflected the gradual accumulation of potentially higher concentrations of inhibitor required to prevent activation of Liddle channels newly inserted into the plasma membrane. Thus, “normal” ASL regulation under steady state conditions in Liddle epithelia suggests the preservation of regulation (inhibition) of Liddle ENaC under thin film conditions (Fig. 5).
A finding consistent with the key role of Na+ transport (ENaC) inhibition in ASL regulation in βENaC-Tg versus Liddle mice in vitro was observed in vivo (i.e. necrotic cells were detected in neonatal βENaC-Tg but not in Liddle mouse airways). Like degenerin mutation-expressing neurons in C. elegans (42, 43), the presence of necrotic cells suggests unregulated entry of Na+ into βENaC-Tg-expressing Clara cells, leading to hydropic degeneration and necrosis. In a previous study, we demonstrated that hydropic degeneration in neonatal βENaC-Tg airways was reduced by preventive treatment with amiloride (61). Interestingly, a link between Na+ channel dysregulation and cell death was also established in an animal model for Duchenne muscular dystrophy. In mdx mice, the absence of dystrophin causes dysregulation of the skeletal muscle isoforms of the voltage-gated Na+ channel Nav1.4, producing intracellular Na+ overload and cell death, which were both prevented by a specific Nav1.4 blocker (62). Taken together, these data indicate that degeneration and cell death were a direct consequence of dysregulated airway Na+ absorption in βENaC-Tg mice. In contrast, necrotic cells were not detected in neonatal Liddle airway epithelia, suggesting that there was sufficient regulation of Liddle ENaC channels in vivo to prevent this event (Fig. 6).
Of note, hydropic degeneration was only observed in neonatal and not in adult βENaC-Tg airways (17). We speculate that several factors may explain why Clara cells in adult βENaC-Tg mice were protected from degeneration and cell death. Previous studies demonstrated that the activity of the Clara cell secretory protein promoter peaks in newborn mice and wanes thereafter (63). Consistent with this finding, Clara cell secretory protein-driven induction of βENaC transcripts and amiloride-sensitive Isc was significantly greater in neonatal compared with adult βENaC-Tg mice (supplemental Fig. 4). These results suggest that lower levels of Na+ hyperabsorption may protect Clara cells in adult βENaC-Tg airways from undergoing hydropic degeneration. Additionally, Clara cells may be more susceptible to Na+ influx in the neonatal period. For example, in contrast to adult airways, Clara cells in neonatal airways store large amounts of glycogen, which were depleted by Na+ hyperabsorption in βENaC-Tg mice, suggesting that differences in energy metabolism in neonatal versus adult Clara cells may contribute to susceptibility to cell death (17). Further, our previous studies demonstrated that mucus plugging of central airways is more severe in neonatal versus adult βENaC-Tg mice. Therefore, airway hypoxia resulting from severe airway mucus plugging may also contribute to cell death triggered by Na+ hyperabsorption in neonatal βENaC-Tg airways (17).
We hypothesized that the failure to regulate Na+ transport in mice overexpressing the βENaC subunit (βENaC-Tg) reflected the expression of a population of protease activation-independent αβ ENaC channels in the luminal membrane of airway epithelial cells. Indeed, we detected evidence for αβ ENaC channels, as reflected in the relative insensitivity of Na+ transport to amiloride inhibition and the detection of a subpopulation of low affinity ENaC channels in native tracheal tissues from βENaC-Tg mice (Fig. 6 and Table 1) (44). Further, our data suggest that tracheal epithelia from βENaC-Tg mice express a pool of αβ ENaC channels that remained constitutively active during aprotinin treatment and were not activated by subsequent trypsin addition (Fig. 7). Taken together with previous studies demonstrating that (i) proteolytic cleavage by extracellular proteases regulates ENaC activity by increasing its Po and (ii) that the γENaC subunit plays an essential role in this process (47–49), our results are consistent with the hypothesis that airways from βENaC-Tg mice express αβ ENaC channels that exhibit a basal Po of ∼1.0 (44) and are insensitive to normal inhibitors of ENaC activity, including endogenous protease inhibitors (59, 60) present in ASL in vitro and in the vicinity of necrotic cells in vivo.
An important hypothesis generated by our comparisons of Na+ transport in Ussing chambers versus thin film conditions with the development of lung disease in Liddle versus βENaC-Tg mice is that it is the regulation of Na+ transport, not the maximal rate of Na+ transport, that determines whether airway surfaces are adequately hydrated for health. This hypothesis is consistent with recent data that suggest that bypassing the normal regulation of ENaC at low ASL volumes with an unregulated “exogenous channel” (e.g. nystatin) can produce a phenotype of ASL depletion and mucus adhesion to airway surfaces (64). In the mouse experiments described here and in human bronchial epithelial cells treated with mucosal nystatin (64), the persistence of the capacity to secrete Cl− is not sufficient to protect the hydration status of the airway surface. This result probably reflects the inability of airway epithelia to generate driving forces sufficient to secrete Cl− in the absence of ENaC inhibition (65).
In summary, our study demonstrated that CFTR inhibits the increase in Na+ absorption caused by Liddle ENaC when the two proteins are endogenously co-expressed in nasal epithelia. Because humans also co-express both CFTR and ENaC in the lower airways, we suggest that this mechanism protects Liddle patients from CF-like lung disease (28, 56). Importantly, our studies in mice indicate that, under physiological thin film conditions, non-CFTR-mediated regulatory factors inhibit the raised Na+ absorption intrinsic to the Liddle mutation in ENaC and thus mediate normal ASL homeostasis and protect Liddle mice from lower airway disease. The importance of ASL volume regulation was buttressed by the observation that transgenic overexpression of wild-type βENaC escaped the ENaC regulation required for ASL homeostasis and produced airway dehydration and CF-like lung disease. Further elucidation of the pathways that regulate ENaC under thin film conditions may produce novel therapeutic targets for pulmonary diseases caused by increased ENaC-mediated Na+ absorption, such as CF.
Supplementary Material
Acknowledgments
We gratefully acknowledge the expert technical assistance of Kim Burns, Elizabeth Hudson, Stephanie Hirtz, Jolanthe Schatterny, and Troy Rogers. We thank Dr. Beverly Koller for providing CF mice.
This work was supported, in whole or in part, by National Institutes of Health Grants SCOR P50 HL60280 and P01 HL 34322 (to R. C. B.). This work was also supported by Deutsche Forschungsgemeinschaft Grants DFG MA 2081/3-2, MA 2081/3-3, and MA 2081/4-1 (to M. A. M.) and European Commission Grant MEXT-CT-2004-013666 (to M. A. M.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. 1–4.
- ENaC
- epithelial sodium channel
- ASL
- airway surface liquid
- CFTR
- cystic fibrosis transmembrane conductance regulator
- CF
- cystic fibrosis
- L/L
- mice homozygous for the Liddle mutation
- CF mice
- mice that are deficient in CFTR
- CF-Liddle mice
- CF mice homozygous for the Liddle mutation
- βENaC-Tg
- mice overexpressing the epithelial Na+ channel β-subunit
- PD
- potential difference
- Isc
- short circuit current
- Ieq
- equivalent short circuit current
- Iamil
- amiloride-sensitive Ieq
- Imax
- maximal amiloride-sensitive Ieq
- Ifrct
- fractional HA ENaC population
- Rte
- transepithelial resistance.
REFERENCES
- 1.Canessa C. M., Schild L., Buell G., Thorens B., Gautschi I., Horisberger J. D., Rossier B. C. (1994) Nature 367, 463–467 [DOI] [PubMed] [Google Scholar]
- 2.Rossier B. C., Pradervand S., Schild L., Hummler E. (2002) Annu. Rev. Physiol. 64, 877–897 [DOI] [PubMed] [Google Scholar]
- 3.Kunzelmann K., Mall M. (2002) Physiol. Rev. 82, 245–289 [DOI] [PubMed] [Google Scholar]
- 4.Knowles M. R., Stutts M. J., Spock A., Fischer N., Gatzy J. T., Boucher R. C. (1983) Science 221, 1067–1070 [DOI] [PubMed] [Google Scholar]
- 5.Boucher R. C., Stutts M. J., Knowles M. R., Cantley L., Gatzy J. T. (1986) J. Clin. Invest. 78, 1245–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerem B., Rommens J. M., Buchanan J. A., Markiewicz D., Cox T. K., Chakravarti A., Buchwald M., Tsui L. C. (1989) Science 245, 1073–1080 [DOI] [PubMed] [Google Scholar]
- 7.Welsh M. J., Ramsey B. W., Accurso F., Cutting G. R. (2001) in The Metabolic and Molecular Bases of Inherited Disease (Scriver C. R., Beaudet A. L., Sly W. S., Valle D. eds) pp. 5121–5188, McGraw-Hill, New York [Google Scholar]
- 8.Anderson M. P., Gregory R. J., Thompson S., Souza D. W., Paul S., Mulligan R. C., Smith A. E., Welsh M. J. (1991) Science 253, 202–205 [DOI] [PubMed] [Google Scholar]
- 9.Sheppard D. N., Welsh M. J. (1999) Physiol. Rev. 79, S23–S45 [DOI] [PubMed] [Google Scholar]
- 10.Stutts M. J., Canessa C. M., Olsen J. C., Hamrick M., Cohn J. A., Rossier B. C., Boucher R. C. (1995) Science 269, 847–850 [DOI] [PubMed] [Google Scholar]
- 11.Mall M., Hipper A., Greger R., Kunzelmann K. (1996) FEBS Lett. 381, 47–52 [DOI] [PubMed] [Google Scholar]
- 12.Willumsen N. J., Boucher R. C. (1991) Am. J. Physiol. 261, C332–C341 [DOI] [PubMed] [Google Scholar]
- 13.Mall M., Bleich M., Greger R., Schreiber R., Kunzelmann K. (1998) J. Clin. Invest. 102, 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui H., Grubb B. R., Tarran R., Randell S. H., Gatzy J. T., Davis C. W., Boucher R. C. (1998) Cell 95, 1005–1015 [DOI] [PubMed] [Google Scholar]
- 15.Quinton P. M. (2008) Lancet 372, 415–417 [DOI] [PubMed] [Google Scholar]
- 16.Mall M., Grubb B. R., Harkema J. R., O'Neal W. K., Boucher R. C. (2004) Nat. Med. 10, 487–493 [DOI] [PubMed] [Google Scholar]
- 17.Mall M. A., Harkema J. R., Trojanek J. B., Treis D., Livraghi A., Schubert S., Zhou Z., Kreda S. M., Tilley S. L., Hudson E. J., O'Neal W. K., Boucher R. C. (2008) Am. J. Respir. Crit. Care Med. 177, 730–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liddle G. W., Bledsoe T., Coppage W. S. (1963) Trans. Assoc. Am. Physicians 76, 199–213 [Google Scholar]
- 19.Shimkets R. A., Warnock D. G., Bositis C. M., Nelson-Williams C., Hansson J. H., Schambelan M., Gill J. R., Jr., Ulick S., Milora R. V., Findling J. W. (1994) Cell 79, 407–414 [DOI] [PubMed] [Google Scholar]
- 20.Hansson J. H., Nelson-Williams C., Suzuki H., Schild L., Shimkets R., Lu Y., Canessa C., Iwasaki T., Rossier B., Lifton R. P. (1995) Nat. Genet. 11, 76–82 [DOI] [PubMed] [Google Scholar]
- 21.Snyder P. M., Price M. P., McDonald F. J., Adams C. M., Volk K. A., Zeiher B. G., Stokes J. B., Welsh M. J. (1995) Cell 83, 969–978 [DOI] [PubMed] [Google Scholar]
- 22.Abriel H., Loffing J., Rebhun J. F., Pratt J. H., Schild L., Horisberger J. D., Rotin D., Staub O. (1999) J. Clin. Invest. 103, 667–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kamynina E., Debonneville C., Bens M., Vandewalle A., Staub O. (2001) FASEB J. 15, 204–214 [DOI] [PubMed] [Google Scholar]
- 24.Chen S. Y., Bhargava A., Mastroberardino L., Meijer O. C., Wang J., Buse P., Firestone G. L., Verrey F., Pearce D. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 2514–2519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Debonneville C., Flores S. Y., Kamynina E., Plant P. J., Tauxe C., Thomas M. A., Münster C., Chraïbi A., Pratt J. H., Horisberger J. D., Pearce D., Loffing J., Staub O. (2001) EMBO J. 20, 7052–7059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kamynina E., Staub O. (2002) Am. J. Physiol. Renal Physiol. 283, F377–F387 [DOI] [PubMed] [Google Scholar]
- 27.Baker E., Jeunemaitre X., Portal A. J., Grimbert P., Markandu N., Persu A., Corvol P., MacGregor G. (1998) J. Clin. Invest. 102, 10–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stutts M. J., Homolya V., Robinson J., Zhou J., Boucher R. C., Knowles M. R. (1998) Ped. Pulmonol. 217, Suppl. 17 (abstract) [Google Scholar]
- 29.Pradervand S., Wang Q., Burnier M., Beermann F., Horisberger J. D., Hummler E., Rossier B. C. (1999) J. Am. Soc. Nephrol. 10, 2527–2533 [DOI] [PubMed] [Google Scholar]
- 30.Dahlmann A., Pradervand S., Hummler E., Rossier B. C., Frindt G., Palmer L. G. (2003) Am. J. Physiol. Renal Physiol. 285, F310–F318 [DOI] [PubMed] [Google Scholar]
- 31.Pradervand S., Vandewalle A., Bens M., Gautschi I., Loffing J., Hummler E., Schild L., Rossier B. C. (2003) J. Am. Soc. Nephrol. 14, 2219–2228 [DOI] [PubMed] [Google Scholar]
- 32.Bertog M., Cuffe J. E., Pradervand S., Hummler E., Hartner A., Porst M., Hilgers K. F., Rossier B. C., Korbmacher C. (2008) J. Physiol. 586, 459–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Randrianarison N., Escoubet B., Ferreira C., Fontayne A., Fowler-Jaeger N., Clerici C., Hummler E., Rossier B. C., Planès C. (2007) J. Physiol. 582, 777–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pradervand S., Barker P. M., Wang Q., Ernst S. A., Beermann F., Grubb B. R., Burnier M., Schmidt A., Bindels R. J., Gatzy J. T., Rossier B. C., Hummler E. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 1732–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Snouwaert J. N., Brigman K. K., Latour A. M., Malouf N. N., Boucher R. C., Smithies O., Koller B. H. (1992) Science 257, 1083–1088 [DOI] [PubMed] [Google Scholar]
- 36.Grubb B. R., Vick R. N., Boucher R. C. (1994) Am. J. Physiol. 266, C1478–C1483 [DOI] [PubMed] [Google Scholar]
- 37.Grubb B. R., Paradiso A. M., Boucher R. C. (1994) Am. J. Physiol. 267, C293–C300 [DOI] [PubMed] [Google Scholar]
- 38.Dempster J. (1993) Computer Analysis of Electrophysiological Signals, Academic Press, London [Google Scholar]
- 39.Tarran R., Button B., Picher M., Paradiso A. M., Ribeiro C. M., Lazarowski E. R., Zhang L., Collins P. L., Pickles R. J., Fredberg J. J., Boucher R. C. (2005) J. Biol. Chem. 280, 35751–35759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grubb B. R., Boucher R. C. (1999) Physiol. Rev. 79, S193–S214 [DOI] [PubMed] [Google Scholar]
- 41.Tarran R., Trout L., Donaldson S. H., Boucher R. C. (2006) J. Gen. Physiol. 127, 591–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García-Añoveros J., Ma C., Chalfie M. (1995) Curr. Biol. 5, 441–448 [DOI] [PubMed] [Google Scholar]
- 43.Syntichaki P., Tavernarakis N. (2002) EMBO Rep. 3, 604–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fyfe G. K., Canessa C. M. (1998) J. Gen. Physiol. 112, 423–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McNicholas C. M., Canessa C. M. (1997) J. Gen. Physiol. 109, 681–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Helms M. N., Self J., Bao H. F., Job L. C., Jain L., Eaton D. C. (2006) Am. J. Physiol. Lung Cell Mol. Physiol. 291, L610–L618 [DOI] [PubMed] [Google Scholar]
- 47.Caldwell R. A., Boucher R. C., Stutts M. J. (2005) Am. J. Physiol. Lung Cell Mol. Physiol. 288, L813–L819 [DOI] [PubMed] [Google Scholar]
- 48.Diakov A., Bera K., Mokrushina M., Krueger B., Korbmacher C. (2008) J. Physiol. 586, 4587–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleyman T. R., Carattino M. D., Hughey R. P. (2009) J. Biol. Chem. 284, 20447–20451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knowles M. R., Boucher R. C. (2002) J. Clin. Invest. 109, 571–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mall M. A. (2008) J. Aerosol Med. Pulm. Drug Deliv. 21, 13–24 [DOI] [PubMed] [Google Scholar]
- 52.Hopf A., Schreiber R., Mall M., Greger R., Kunzelmann K. (1999) J. Biol. Chem. 274, 13894–13899 [DOI] [PubMed] [Google Scholar]
- 53.Ji H. L., Chalfant M. L., Jovov B., Lockhart J. P., Parker S. B., Fuller C. M., Stanton B. A., Benos D. J. (2000) J. Biol. Chem. 275, 27947–27956 [DOI] [PubMed] [Google Scholar]
- 54.Firsov D., Schild L., Gautschi I., Mérillat A. M., Schneeberger E., Rossier B. C. (1996) Proc. Natl. Acad. Sci. U.S.A. 93, 15370–15375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mall M., Kreda S. M., Mengos A., Jensen T. J., Hirtz S., Seydewitz H. H., Yankaskas J., Kunzelmann K., Riordan J. R., Boucher R. C. (2004) Gastroenterology 126, 32–41 [DOI] [PubMed] [Google Scholar]
- 56.Kreda S. M., Mall M., Mengos A., Rochelle L., Yankaskas J., Riordan J. R., Boucher R. C. (2005) Mol. Biol. Cell 16, 2154–2167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vallet V., Chraibi A., Gaeggeler H. P., Horisberger J. D., Rossier B. C. (1997) Nature 389, 607–610 [DOI] [PubMed] [Google Scholar]
- 58.Donaldson S. H., Hirsh A., Li D. C., Holloway G., Chao J., Boucher R. C., Gabriel S. E. (2002) J. Biol. Chem. 277, 8338–8345 [DOI] [PubMed] [Google Scholar]
- 59.Myerburg M. M., Butterworth M. B., McKenna E. E., Peters K. W., Frizzell R. A., Kleyman T. R., Pilewski J. M. (2006) J. Biol. Chem. 281, 27942–27949 [DOI] [PubMed] [Google Scholar]
- 60.Garcia-Caballero A., Rasmussen J. E., Gaillard E., Watson M. J., Olsen J. C., Donaldson S. H., Stutts M. J., Tarran R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 11412–11417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhou Z., Treis D., Schubert S. C., Harm M., Schatterny J., Hirtz S., Duerr J., Boucher R. C., Mall M. A. (2008) Am. J. Respir. Crit. Care Med. 178, 1245–1256 [DOI] [PubMed] [Google Scholar]
- 62.Hirn C., Shapovalov G., Petermann O., Roulet E., Ruegg U. T. (2008) J. Gen. Physiol. 132, 199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Perl A. K., Tichelaar J. W., Whitsett J. A. (2002) Transgenic Res. 11, 21–29 [DOI] [PubMed] [Google Scholar]
- 64.Livraghi A., Mall M., Paradiso A. M., Boucher R. C., Ribeiro C. M. (2008) Am. J. Respir. Cell Mol. Biol. 38, 423–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Willumsen N. J., Davis C. W., Boucher R. C. (1989) Am. J. Physiol. 256, C1033–C1044 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.