

Abstract
The sodium ion-translocating NADH:quinone oxidoreductase (Na+-NQR) from the human pathogen Vibrio cholerae is a respiratory membrane protein complex that couples the oxidation of NADH to the transport of Na+ across the bacterial membrane. The Na+-NQR comprises the six subunits NqrABCDEF, but the stoichiometry and arrangement of these subunits are unknown. Redox-active cofactors are FAD and a 2Fe-2S cluster on NqrF, covalently attached FMNs on NqrB and NqrC, and riboflavin and ubiquinone-8 with unknown localization in the complex. By analyzing the cofactor content and NADH oxidation activity of subcomplexes of the Na+-NQR lacking individual subunits, the riboflavin cofactor was unequivocally assigned to the membrane-bound NqrB subunit. Quantitative analysis of the N-terminal amino acids of the holo-complex revealed that NqrB is present in a single copy in the holo-complex. It is concluded that the hydrophobic NqrB harbors one riboflavin in addition to its covalently attached FMN. The catalytic role of two flavins in subunit NqrB during the reduction of ubiquinone to ubiquinol by the Na+-NQR is discussed.
Keywords: Membrane/Proteins, Respiration, Transport/Sodium, Vitamins and Cofactors/Riboflavin, Protein Assembly, <I>Vibrio cholerae</I>, Na<SUP>+</SUP>-NQR
Introduction
Pathogenic strains of the water-borne bacterium Vibrio cholerae cause the diarrheal disease cholera. By the help of a respiratory Na+ pump, V. cholerae maintains an electrochemical Na+ gradient across the inner membrane to drive central processes like flagellar rotation, nutrient uptake, and detoxification. The respiratory Na+ pump, also called Na+-translocating NADH:quinone oxidoreductase (Na+-NQR),2 is a membrane-bound enzyme complex composed of six subunits (NqrABCDEF) (1, 2). NqrA is a peripheral subunit, NqrC and NqrF are predominantly hydrophilic yet anchored to the membrane, whereas the hydrophobic NqrB, NqrD, and NqrE subunits each comprise several transmembrane helices. The Na+-NQR contains at least five distinct redox centers participating in electron transfer from NADH to substrate quinone: noncovalently bound FAD, a 2Fe-2S cluster, covalently bound FMNs, and noncovalently bound riboflavin (3–5). In addition, ubiquinone-8 (Q8) was co-purified with the Na+-NQR (6). Binding sites for FAD, 2Fe-2S cluster, and substrate NADH are located on NqrF, which does not directly participate in Na+-transport but represents the electron input module of the complex catalyzing the NADH dehydrogenase reaction (7, 8). The covalently bound FMN residues are attached via phosphodiester bonds to Thr-236 in subunit NqrB and Thr-225 in subunit NqrC, respectively (9, 10). The subunit(s) responsible for riboflavin and quinone binding have not been identified yet. Riboflavin was detected in a subcomplex composed of NqrA, -B, -C, and -F but was not present in the isolated A and F subunits (3). Furthermore, there is evidence that NqrB participates in the quinone reduction step (11).
By analyzing the flavin cofactors in defined subcomplexes of the Na+-NQR from V. cholerae, we show that riboflavin is localized on subunit NqrB. Analysis of the molar size and subunit composition of the holo-complex reveals that the Na+-NQR contains one copy of each subunit and a total of four flavins, with one FAD on NqrF, one covalently bound FMN on NqrC, and one covalently bound FMN and one noncovalently bound riboflavin on NqrB. Thus, the Na+-NQR represents a unique example for a respiratory complex, which relies on the redox activity of two flavins located in its membrane-embedded NqrB subunit. The possible role of these membrane-bound flavins during the two-electron reduction of quinone to quinol by the Na+-NQR is discussed.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, and Growth Conditions
Escherichia coli DH5α (12) was used as a host for cloning and propagation of plasmids. V. cholerae O395 N1 Δnqr (13), a streptomycin-resistant strain lacking the entire nqr operon, was used as expression host. The plasmid pNQR1 bearing the entire nqr operon from V. cholerae under the control of an arabinose promoter (3) was used as expression vector for the Na+-NQR complex and as a cloning template. E. coli was grown aerobically in Luria-Bertani medium at 37 °C. V. cholerae was grown aerobically at 37 °C in Luria-Bertani medium supplemented with 50 mm potassium phosphate pH 8.5, 10 mm glucose, and 50 μg ml−1 streptomycin. For plasmid maintenance, ampicillin was added to growth medium at concentrations of 150 μg ml−1 (E. coli) or 200 μg ml−1 (V. cholerae).
Purification of Na+-NQR
Production and purification of wild-type and mutant forms of the Na+-NQR from V. cholerae as recombinant proteins containing a His6 tag at the N terminus of subunit NqrA was performed as described elsewhere (3). Briefly, His6-tagged Na+-NQR was expressed in V. cholerae Δnqr. Membranes were isolated, solubilized with n-dodecyl-β-d-maltoside (DDM), and Na+-NQR was purified via nickel affinity chromatography followed by gel filtration chromatography in gel filtration buffer (10 mm HEPES·NaOH, pH 8.0, 5% (v/v) glycerol, 300 mm NaCl, and 0.05% (w/v) DDM).
Reconstitution of Na+-NQR with Riboflavin
Na+-NQR from the nickel affinity chromatographic step (4.3–6.5 mg ml−1) was mixed with 0.1 mm riboflavin from a 1 mm riboflavin stock solution in N,N-dimethylformamide. After 10 min at 25 °C, excess riboflavin was removed by gel filtration as described above.
Analytical Methods
Protein was determined by the bicinchoninic acid method using the reagent from Pierce (14). Bovine serum albumin served as standard. SDS-PAGE was performed with 10% polyacrylamide gels in the presence of 6 m urea (1, 15). Aliquots of purified Na+-NQR were solubilized with SDS prior to loading on the gel (3). Membranes were resuspended in 2% SDS, 50 mm Tris·HCl, pH 6.8, 5.8% (w/v) glycerol, and 125 mm β-mercaptoethanol, agitated at room temperature for 1 h, and centrifuged for 5 min at 16,100 × g prior to loading on the gel. Gel electrophoresis was performed at room temperature at a constant current of 15 mA. Proteins were stained with Coomassie Brilliant Blue G-250. Prior to staining, flavins were detected by in gel fluorography (excitation, 457 nm; emission, 526 nm; Typhoon 9400 scanner, Molecular Dynamics). To detect covalently bound FMN in membrane-bound NQR, the gels were washed for 2 h in 10% acetic acid and 50% methanol prior to scanning to remove fluorescent low molecular weight compounds present in the crude membrane fractions. His6-tagged NqrA subunit was detected immunochemically by use of an anti-His6-peroxidase conjugate according to the procedures of the supplier (Roche). The peroxidase was detected using the ECL detection kit (GE Healthcare) with a chemiluminescence reader (Fujifilm, LAS-3000 CCD) equipped with a blue 515 nm emission filter. Thin layer chromatography (TLC) of noncovalently bound flavins was performed on plates of silica gel 60 (SIL G-25, Macherey-Nagel) using a developing solution of 2-butanol:glacial acetic acid:water (2:1:1) (16). TLC spots were visualized by UV illumination (excitation, 302 nm; emission, 520–620 nm; GeneGenius Bioimaging system, Syngene). Extraction of noncovalently bound flavins from purified Na+-NQR or isolated membranes of V. cholerae with trichloroacetic acid (TCA), and the identification and quantification of flavins by HPLC was performed as described previously (3). Standards for FAD and FMN were purchased from Sigma-Aldrich. Riboflavin was from Fluka.
Analytical Ultracentrifugation
Sedimentation velocity experiments for Na+-NQR were performed at 4 °C in a Beckman ProteomeLab XL-I analytical ultracentrifuge with an An-50 Ti analytical rotor (Beckman) at a speed of 30,000 rpm. All data acquired from these experiments were obtained using the UV/Vis absorbance detection system on the ultracentrifuge at 280 nm using double sector 12 mm charcoal-filled Epon centerpieces. Protein concentration was ∼0.2 mg ml−1 in 50 mm potassium phosphate, 300 mm NaCl, 5% (w/v) glycerol, 0.05% (w/v) DDM, pH 8.0. Analysis of the continuous sedimentation coefficient distribution was performed with the software Sedfit version 11.8 (17, 18) using the Lamm equation. The partial specific volume of Na+-NQR, buffer viscosity, and buffer density were calculated using software UltraScan II (19). Membrane protein and detergent form a complex which has a different density than the protein alone. Therefore, sedimentation behavior is dependent on the nature of the detergent, and amount of detergent bound to protein. In such a case the buoyant mass of the particle is determined. Knowing the ratio of detergent bound per protein, the mass of the protein without micelle is calculated according to Equation 1,
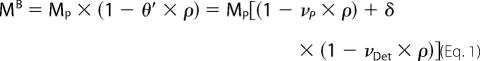 |
(20, 21), where MB is the buoyant mass, θ′ is the buoyant term that is composed of the different components of the analyzed complex, MP is the molecular mass of the protein, νP is the partial specific volume of the protein, νDet is the partial specific volume of the detergent (22), δ is the ratio of detergent bound per protein in g g−1, and ρ is the density of the buffer.
Static Light Scattering
For size determination by static light scattering, size exclusion chromatography was coupled with continuous laser light scattering, refractive index, and ultraviolet detection of the eluent as described (23). Na+-NQR was separated on a Superdex 200 10/300 column (GE Healthcare) connected to an Agilent 1100 HPLC system at 20 °C, and protein was eluted with a flow rate of 0.5 ml min−1 using gel filtration buffer. Protein elution was followed online by absorption at 280 nm (Agilent), by refractive index at 690 nm using an Optilab rEX detector (Wyatt Technology), and by static light scattering at 690 nm using a miniDAWN instrument (Wyatt Technology). Static light scattering data were analyzed using ASTRA software (Wyatt Technology). A specific refractive index increment (dn/dc) of 0.185 ml g−1 for protein (23) and 0.143 ml g−1 for DDM (24) was used. The experimentally determined specific absorptions of Na+-NQR and DDM at 280 nm were 1.540 ml mg−1 cm−1 and 2.32 ml g−1 cm−1, respectively. Bovine serum albumin was used as standard.
DDM Binding to Na+-NQR
The amount of DDM bound to purified Na+-NQR was determined using the phenol-sulfuric acid method (25) in combination with anionic exchange chromatography as described previously (26). Na+-NQR was applied at 4 °C to a HiTrap ANX FF column (GE Healthcare) and washed with start buffer (10 mm HEPES, pH 8.0, 5% (w/v) glycerol, 0.05% (w/v) DDM). Na+-NQR was eluted with a 0 to 1 m linear gradient of NaCl in start buffer. Fractions of 1 ml were analyzed for protein and DDM content. Protein-bound DDM was determined by subtracting the DDM concentration of the elution buffer from the DDM concentration of fractions containing Na+-NQR.
Recombinant DNA Techniques
Standard protocols were used for plasmid preparation and purification, agarose gel electrophoresis, dephosphorylation, and ligation of DNA (27). Restriction enzymes (New England Biolabs) and DNA polymerase (Finnzymes) were used as recommended by the manufacturers. E. coli was transformed chemically (28), V. cholerae was transformed by electroporation (29). Expected sequences of expression vectors were confirmed by DNA sequencing (Microsynth).
Generation of Expression Plasmids for Production of Na+-NQR Devoid of Individual Subunits
Fig. 1 depicts the plasmids encoding for subcomplexes of the Na+-NQR. Primers are listed in Table 1. Plasmid pNA1 was obtained by replacing the nqr operon in pNQR1 with the nqrA gene. The PCR primers MC044-NqrA-F and MC045-NqrA-R were designed to amplify nqrA from pNQR1 and to introduce an EcoRI site with the reverse primer. The PCR product was finally subcloned into pNQR1 using EcoRI and NdeI.
FIGURE 1.

Plasmids for expression of NQR subcomplexes. Only regions of plasmids downstream of the arabinose promoters are shown. Restriction sites on the parent plasmid pNQR1 that were used for cloning of truncated expression plasmids (black arrows) and introduced restriction sites (gray arrows) are indicated. DNA fragments that were deleted from the corresponding parent plasmid are shown as striped bars.
TABLE 1.
DNA primers used for construction of plasmids
| Primer | Sequencea | Restriction enzyme |
|---|---|---|
| Primers for gene deletions | ||
| MC044-NqrA-F | AAGAAGGAGATATACCATGGGCAGCAGCCATC | NcoI |
| MC045-NqrA-R | CAGAATTCTTACCCTTCTTTCTCGATCTTG | EcoRI |
| MC046-NqrF-F | TTCCCGGGATGTCTACTATTATTTTTGG | XmaI |
| MC048-NqrEF-F | TTCCCGGGATGGAACATTATATTAGTCT | XmaI |
| MC050-NqrC-R | TTCCCGGGTTAGTTAAGACCTCCGTCAC | XmaI |
| RD-NQRb-Vc | CGATATACATATGGGCCTTAAAAAGTTTCTTG | NdeI |
| Seq-Vc-NQRc-R | CAGACGTGGTTCAATGCTTTTA | |
| TH001-deltaNqrC-F | ATTTGGTACCATGTCTAGCGCAAAAGAGCT | KpnI |
| TH002-deltaNqrC-R | ATTTGGTACCTTATTGCTTGCCGTAGCGCG | KpnI |
| TH003-deltaNqrB-F | GGTACCATGGCAAGCAATAACGATAG | KpnI |
| TH004-deltaNqrB-R | GGTACCTTACCCTTCTTTCTCGATCT | KpnI |
| Flanking primers for site-directed mutagenesis | ||
| NQR-F | GTATGAAGCAGCGGCGACGCTGTTCTATAC | |
| NQR-R | TTGGTATCTGAGCCAATCACGTTAAACAGT | |
| NqrB-Trp227Leu | ||
| NQR3-F | CTCAGGTGACCTAGTActGACgGCcGCAGATGGCTACTCAGGC | EagI |
| NQR3-R | GCCTGAGTAGCCATCTGCgGCcGTCagTACTAGGTCACCTGAG | EagI |
| NqrB-Phe186Ala | ||
| NQR5-1F | GCACTGGGTATTACCgcgGGTGTTGTGGTTGCC | |
| NQR5-1R | GGCAACCACAACACCcgcGGTAATACCCAGTGC | |
| NQR5-2F | TGACCTAGTATGGACgGCcGCAGATGGCTACTCAGGC | EagI |
| NQR5-2R | GCCTGAGTAGCCATCTGCgGCcGTCCATACTAGGTCA | EagI |
a Sequences are given from 5′ to 3′. Restriction sites are underlined, and nucleotides substituted for mutagenesis are designated by lower case letters.
Plasmid pNmA1 was obtained by deleting the nqrA gene from pNQR1. The PCR primers RD-Nqrb-Vc and Seq-Vc-Nqrc-R were designed to amplify a sequence downstream of the nqrA gene in pNQR1 and to introduce an NdeI site with the forward primer. The PCR product was subcloned into pNQR1 using NdeI and SalI.
Plasmid pNmB1 was obtained by deleting the nqrB gene from pNQR1. The PCR primers TH003-deltaNqrB-F and TH004-deltaNqrB-R were designed to amplify the entire pNQR1 plasmid excluding nqrB. Both primers introduced a KpnI site and thereby allowed self-ligation of the PCR product after digestion with KpnI to form plasmid pNmB1*. A fragment containing the newly joined KpnI site was excised from pNmB1* with MluI and SbfI and subcloned into pNQR1.
Plasmids pNmC1, pNmD1, and pNmDE1 (Fig. 1) were cloned following the same strategy as for pNmB1. For pNmC1, primers TH001-deltaNqrC-F and TH002-deltaNqrC-R were used to eliminate nqrC and to introduce a KpnI site for circularization of the PCR product under formation of pNmC1*. The KpnI-containing fragment was excised from pNmC1* with SalI and NheI and subcloned into pNQR1, generating plasmid pNmC1. For pNmD1, primers MC048-NqrEF-F and MC050-NqrC-R were used to eliminate nqrD and to introduce an XmaI site for circularization of the PCR product under formation of pNmD1*. The XmaI containing fragment was excised from pNmD1* with MluI and HindIII and subcloned into pNQR1, generating plasmid pNmD1. For pNmDE1, primers MC046-NqrF-F and MC050-NqrC-R were used to eliminate nqrD and nqrE and to introduce an XmaI site for circularization of the PCR product under formation of pNmDE1*. The XmaI-containing fragment was excised from pNmD1* with MluI and HindIII and subcloned into pNQR1, generating plasmid pNmDE1.
Site-directed Mutagenesis
The plasmid pNQR1 was used as template for mutagenesis. Amino acid substitutions in subunit NqrB were made by overlapping PCR using individual primer pairs and the flanking primers NQR-F and NQR-R (Table 1). The PCR products harboring the mutations were subcloned into pNQR1 using SexAI and SalI, and confirmed by DNA sequencing. An additional EagI restriction site was introduced with the primers by silent nucleotide substitution to facilitate identification of mutated plasmids by restriction enzyme digestion.
Preparation of Membranes Containing Subcomplexes of Na+-NQR
Expression of Na+-NQR lacking individual subunits and preparation of membranes was performed as described (3), and modified as follows. A pre-culture of V. cholerae Δnqr transformed with the chosen expression plasmid was diluted 1:100 with fresh, prewarmed medium and growth was continued at 37 °C. At an A600 of 0.8, protein production was induced by the addition of 10 mm l-arabinose. The temperature was decreased to 30 °C and incubation was continued for 20 h. Cells were harvested, washed with buffer (50 mm sodium phosphate, pH 8.0, 500 mm NaCl), flash-frozen in liquid nitrogen, and stored at −80 °C until use.
Approximately 4 g of cells were resuspended in 25 ml of buffer supplemented with 1 mm phenylmethylsulfonyl fluoride, 0.1 mm diisopropyl-fluorophosphate, 5 mm dithiothreitol, 5 mm MgCl2, and traces of DNase I. The cell suspension was passed through a French pressure cell at 7.58 MPa. Unbroken cells and large debris were removed by centrifugation at 27,000 × g for 30 min. Soluble proteins were separated from the membrane fraction by ultracentrifugation (183,000 × g, 1 h, 4 °C). The membranes were washed with buffer (50 mm sodium phosphate, pH 8.0, 300 mm NaCl, and 5% (w/v) glycerol), resuspended in 10 ml of the same buffer, flash-frozen in liquid nitrogen, and stored at −80 °C.
Enzymatic Activity
Na+-NQR activity was determined in buffer containing 20 mm Tris·H2SO4, pH 7.5, 50 mm Na2SO4, and 50 μg ml−1 bovine serum albumin in the presence of 0.1 mm NADH and 0.1 mm ubiquinone-1 using a diode array spectrophotometer (Agilent 8453). Ubiquinone-1, a ubiquinone-side chain homologue with one isoprene unit in the polyprenyl side chain, was used in replacement of the native substrate ubiquinone-8 for reasons of solubility. The reaction was started by the addition of Na+-NQR diluted in buffer. NADH oxidation and quinol formation were followed simultaneously as previously described (30). NADH:ubiquinone-1 oxidoreduction activity by alternative NADH dehydrogenases was determined in the presence of 1 μm AgNO3, which specifically inhibited the Na+-NQR (31, 32). Statistical significance was evaluated by using a two-tailed, unpaired Student's t test. Differences were determined to be significant when p < 0.001.
Edman Degradation
The subunit stoichiometry of Na+-NQR was determined as described (33), using a Procise-cLC protein sequencer (Applied Biosystems). Twelve cycles of automated Edman degradation of the intact Na+-NQR complex were performed, and uncorrected peak integrals of the separated phenylthiohydantion (PTH) amino acids were used for determination of the relative amount of subunits in the Na+-NQR.
Sequence Alignments
Amino acid sequences for NqrB homologues were obtained from UniProtKB (34) with the query string “gene:NqrB”. Redundant entries were removed from the dataset. Of the remaining 73 sequences, a multiple sequence alignment was performed using the ClustalW algorithm (35) implemented in the BioEdit Sequence Alignment Editor (Version 7.0.9.0, (36)) with default settings. Entries A9NGU6, Q73PG2, Q1Q257, and A6AT93 were removed from the dataset due to large length deviations at the N- or C termini. Identical residues and similar residues were identified with the BioEdit program using an identity and similarity threshold of 85% and the BLOSUM62 scoring matrix (37). The sequences in the alignment were used to construct an average distance tree using Jalview (38). To reduce the number of sequences while maintaining the sequence diversity high, sequences with identity distance scorings below 18 were grouped and all but one sequence was deleted. This allowed minimizing the alignment to the 6 sequences of NqrB from Chlamydia pneumoniae (Q9Z8B6), Protochlamydia amoebophila (Q6MEH4), Shewanella oneidensis (Q8EID9), Porphyromonas gingivalis (Q7MT18), Rhodopirellula baltica (Q7UWS4), and V. cholerae (A6XUU9).
Membrane Topology Prediction
The amino acid sequence of V. cholerae NqrB (A6XUU9) was obtained from UniProtKB. Eleven different topology prediction methods were used to obtain putative topologies of NqrB:TMHMM (39), TMpred (40), SOSUI (41), DAS (42), HMMTOP (43, 44), TopPred (45), MEMSAT (46), Split 4.0 (47), PHDhtm (48), TMAP (49), and PolyPhobius (50). All methods were used with default settings. A histogram from the individual prediction results was constructed giving each residue a score of 1 if predicted to be located in a transmembrane helix (TMH) and a value of 0.5 for residues from a predicted low probability TMH or aliphatic helix. A consensus prediction model was constructed by assigning a TMH to regions where the summed score exceeded 7.33, which is ⅔ of the maximal score. Furthermore, larger regions with a summed score above 3 were considered as hydrophobic stretches. The C terminus of NqrB was assumed to be located in the cytoplasm, as reported by Duffy et al. (51).
RESULTS
Riboflavin Content of the Na+-NQR
In addition to the noncovalently bound FAD and the covalently attached FMN cofactors, riboflavin was reported to be a noncovalently bound cofactor of the Na+-NQR from V. cholerae and the highly conserved enzyme from V. harveyi (52). Protocols for the analysis of noncovalently bound flavins in the Na+-NQR include a precipitation step using TCA, which might result in the cleavage of FMN under formation of riboflavin. This riboflavin could be erroneously assigned as intrinsic, noncovalently bound flavin cofactor of the Na+-NQR, as pointed out by Bogachev et al. (53). We analyzed the noncovalently bound flavins in purified Na+-NQR from V. cholerae by thin layer chromatography (TLC), which allowed application of the purified protein without any pretreatment. Thereby, the presence of noncovalently bound FAD and riboflavin was confirmed but no noncovalently bound FMN was observed (Fig. 2). These results indicated that riboflavin in purified Na+-NQR is not liberated from FMN but represents an intrinsic component of the Na+-NQR complex. We then quantified the endogenous amount of noncovalently bound flavins by HPLC after their release from Na+-NQR by TCA precipitation (3). With a calculated overall protein size of 213 kDa for the His6-tagged Na+-NQR complex (assuming that every subunit is present in a single copy), 0.88 ± 0.04 mol of FAD and 0.56 ± 0.08 mol of riboflavin were extracted per mol of Na+-NQR (n = 9 measurements). Compared with FAD, the riboflavin content of purified Na+-NQR was lower, resulting in a molar ratio of riboflavin:FAD of 0.5–0.8. Incubation of purified Na+-NQR with exogenous riboflavin increased the amount of bound riboflavin to riboflavin:FAD ratios of 1.10 ± 0.10, while the amount of bound FAD was not affected. These results indicated that V. cholerae Na+-NQR contained one FAD and one riboflavin per complex. FAD was previously assigned to subunit NqrF (8). The localization of riboflavin in the Na+-NQR is described in the following section.
FIGURE 2.

Thin layer chromatography of flavins in Na+-NQR. Flavin standards and purified Na+-NQR were spotted on the TLC plate, developed with 2-butanol:glacial acetic acid:water (2:1:1) and analyzed by UV illumination. Approximately 150 pmol of standard or protein complex (Na+-NQR) were applied per spot.
Experimental Strategy to Identify the Riboflavin-binding Subunit(s) of Na+-NQR
To identify the riboflavin-binding subunit(s) of Na+-NQR, we analyzed the membrane-bound riboflavin content of subcomplexes of the Na+-NQR lacking individual subunits. Plasmids for the expression of subcomplexes were derived from plasmid pNQR1 (3), which encodes for all six nqr genes, by deleting genes nqrA, nqrB, nqrC, nqrD, and nqrD to nqrE, respectively. The resulting plasmids were termed pNmA, pNmB, etc., denominating the deleted subunit(s) (Fig. 1). Subunit NqrF does not contain riboflavin, as shown by analysis of isolated NqrF (8, 54, 55). Therefore, an expression plasmid for a subcomplex devoid of NqrF was not included in our study. A mutant strain of V. cholerae, lacking the chromosomal nqr genes, served as expression host. Plasmid pNA1, which encodes for the isolated NqrA subunit, served as control, because NqrA is (i) not attached to the membrane in its isolated state (51) and (ii) does not bind riboflavin (3). Whereas the expression of the isolated NqrB subunit was reported to be toxic for the cells (56), no toxicity effects were observed during the expression of NqrA or any of the subcomplexes of NQR (data not shown).
Detection of NqrA, -B, -C, and -F in Subcomplexes of Na+-NQR
Membranes from V. cholerae Δnqr strains producing Na+-NQR or subcomplexes thereof were analyzed for the presence of NqrA, NqrB, NqrC, and NqrF. To this end, we monitored the binding of FMN to subunits NqrB or NqrC, the binding of FAD to subunit NqrF, and the recruitment of the soluble subunit NqrA to the NQR complex in its native membrane environment. Covalent modification of NqrB and NqrC by FMN and the recruitment of these subunits to the membrane were analyzed by denaturing gel electrophoresis of washed membranes and in gel fluorography. For NqrB and NqrC, a fluorescence signal was observed in all subcomplexes in which their presence was expected, with comparable fluorescence intensities of the bands in all Na+-NQR subcomplexes (Fig. 3, A and B). This observation was consistent with the previous finding that FMN attachment to NqrC does not require assembly of the Na+-NQR complex (56). Note that NqrC in the purified Na+-NQR complex migrated faster than in the solubilized membranes and that the highly hydrophobic NqrB subunit gave diffuse bands on SDS-PAGE. Please note that the migration behavior of membrane proteins in SDS-PAGE depends on the environment (lipids, detergents) and folding state of the proteins subjected to analysis. This was shown for the c subunit of the F1F0 ATPase from Ilyobacter tartaricus (57) where the differences in migration behavior were caused by varying amounts of bound detergent and incomplete denaturation by SDS (9, 58).
FIGURE 3.

Detection of NqrA, NqrB, and NqrC in membranes containing NQR subcomplexes. Washed membranes from V. cholerae Δnqr transformants expressing different sets of Nqr subunits were solubilized with SDS and membrane proteins (30 μg) were separated by SDS-PAGE. FMN covalently bound to subunits NqrB and NqrC was detected by in gel fluorography (A and B). NqrA was detected by Western blot analysis against its N-terminal His6 tag (C).
Expression of subunit NqrA and its recruitment to the membrane was followed by Western blot analysis of washed membranes against the His6 tag fused to NqrA (Fig. 3C). Membrane-bound NqrA was observed with similar intensities in membranes from cells expressing the entire Na+-NQR complex or subcomplexes lacking NqrB, NqrC, NqrD, or NqrD plus NqrE, respectively, but not in membranes from cells producing the individual, soluble NqrA subunit. These results indicated that binding of NqrA to the membrane required the presence of other, membrane-bound Nqr subunits. As observed with NqrC, NqrA from the purified Na+-NQR migrated faster in SDS-PAGE than NqrA from (sub)complexes solubilized from membranes.
The presence of NqrF in membrane-bound subcomplexes of Na+-NQR was confirmed by the specific inhibition of its enzymatic activity in the presence of Ag+. NADH oxidation rates of membrane preparations were determined with ubiquinone-1 (Q1), which acts as substrate for the oxidation of NADH by the holo-Na+-NQR coupled to Na+ translocation. Q1 also accepts electrons in a non-physiological reaction catalyzed by isolated NqrF or subcomplexes containing the NqrF subunit. Both the coupled and the non-physiological modes of NADH oxidation with Q1 require FAD as cofactor (8) and are specifically inhibited by Ag+ (8, 31, 59). In contrast, the non-electrogenic NADH dehydrogenase (NDH-2) present in V. cholerae membranes (UniProt accession number A5F704) is not inhibited by Ag+ in the micromolar concentration range (59). We observed significant inhibition of NADH oxidation in the presence of Ag+ with all membrane-bound subcomplexes of Na+-NQR expected to contain NqrF (Fig. 4). In the absence of the inhibitor, highest specific activities were observed with holo-Na+-NQR, followed by the subcomplex lacking NqrA. This indicated that membranes of subcomplexes contained less NqrF than membranes containing the holo-complex, but we note that the relative inhibition of NADH oxidation activities of subcomplexes in the presence of Ag+ was comparable (75–99% inhibition; Fig. 4). In summary, the results showed that subcomplexes of NQR composed of NqrBCDEF, NqrACDEF, NqrABDEF, NqrABCEF, and NqrABCF were produced and inserted into membranes of a V. cholerae expression host lacking the holo-complex.
FIGURE 4.

Ag+-sensitive NADH oxidation by membranes containing subcomplexes of Na+-NQR. The specific NADH oxidation activity of washed membrane vesicles from V. cholerae Δnqr transformants expressing different sets of Nqr subunits were determined in the absence of Ag+ (black bars) and in the presence of 1 μm AgNO3 (gray bars). Ubiquinone-1 was used as electron acceptor. Membranes from V. cholerae Δnqr and from transformants expressing NqrA served as controls. Mean values and standard deviations from at least two measurements are shown.
Riboflavin Analysis of Membranes Containing NQR Subcomplexes
It was shown by Barquera et al. (52) that membranes from V. cholerae devoid of Na+-NQR do not contain any riboflavin. To assign riboflavin to a distinct Nqr subunit, we analyzed membranes from V. cholerae Δnqr strains producing Na+-NQR or defined subcomplexes thereof for riboflavin. Cells expressing no Nqr subunits (Δnqr) or the isolated NqrA subunit served as controls. Riboflavin was extracted by TCA precipitation of membrane proteins, followed by HPLC analysis (Fig. 5). From cells expressing the entire Na+-NQR 198 ± 58 nmol riboflavin was extracted per mg membrane protein (n = 3), whereas Δnqr cells contained only residual amounts of riboflavin (3 ± 2 nmol mg−1). This confirmed previous findings by Barquera et al. (52). Membranes from cells expressing isolated NqrA contained 3 ± 1 nmol riboflavin per mg membrane protein (Fig. 5).
FIGURE 5.

Flavin analysis of membranes containing NQR subcomplexes. Washed membrane vesicles (∼1 mg of protein) from V. cholerae Δnqr transformants expressing different sets of Nqr subunits were analyzed for flavins by HPLC. Elution of flavins was monitored at 450 nm. Elution times of standards (FAD, 120 pmol; FMN, 37 pmol; riboflavin, 56 pmol) are indicated.
Membranes from cells expressing the subcomplexes of NQR composed of NqrBCDEF (107 ± 9 nmol mg−1), NqrABDEF (299 ± 79 nmol mg−1), NqrABCEF (141 ± 27 nmol mg−1), and NqrABCF (132 ± 53 nmol mg−1) contained riboflavin in amounts that were comparable to the riboflavin content of membranes containing the holo-NQR complex whereas only residual amounts of riboflavin were found in membranes devoid of NqrB (NqrACDEF, 3 ± 2 nmol mg−1). Thus, we concluded that riboflavin is bound to subunit NqrB, which also comprises a covalently linked FMN.
Riboflavin Content of NqrB Mutants
To test for the role of the conserved Trp-227 and Phe-186 residues of NqrB on riboflavin binding, these residues were substituted using site-directed mutagenesis. The wild type and mutant forms of Na+-NQR were expressed in parallel and purified by gel filtration as described above, and riboflavin content of the mutants were compared with the wild-type enzyme. The molar ratio of riboflavin to Na+-NQR complex was 0.40 ± 0.02 (n = 3) for the wild-type Na+-NQR, 0.31 ± 0.03 (n = 9) for the NqrB-Trp227Leu variant and 0.29 ± 0.03 (n = 3) for the NqrB-Phe186Ala variant.
DDM Binding to Na+-NQR
Determination of the molecular mass of the Na+-NQR complex with gel filtration or analytical ultracentrifugation requires an independent estimation of the mass ratio of detergent:protein in the Na+-NQR-DDM complex. This ratio was determined using the equilibrium column desorption method (22, 26). Na+-NQR was applied to a HiTrap ANX-FF column (GE Healthcare), washed, and eluted with a linear gradient of NaCl. An increase in the DDM concentration was observed matching the profile of the protein peak (Fig. 6). The mass ratio of bound DDM to protein was determined using fractions eluting between 18 and 23 ml (Fig. 6). We obtained a value of 0.541 ± 0.096 mg DDM bound per mg of protein (n = 5). If we assume an overall size of 306 kDa as determined by gel filtration in the presence of DDM (3), the Na+-NQR complex consists of 107.4 ± 12.4 kDa DDM and 198.6 ± 12.4 kDa protein, which is in good agreement with the theoretical mass of 213 kDa of a complex containing each Nqr subunit in a single copy.
FIGURE 6.

DDM binding to Na+-NQR. A, separation of 1 mg Na+-NQR on a 1-ml HiTrap ANX-FF column (GE-Healthcare) monitored from the absorbance at 280 nm. B, concentrations of protein (gray bars) and protein-bound DDM (black bars). The concentration of protein-bound DDM was obtained by subtracting the DDM concentration of the buffer (0.639 ± 0.015 mg ml−1) from the DDM concentration of the eluate.
Static Light Scattering of Na+-NQR
By static light scattering, the size of a protein in solution is determined directly, because the scattered light (the Rayleigh ratio) is proportional to the product of the weight-average molar mass and the protein concentration (23, 24). With known UV-extinction coefficient and dn/dc, the concentration of a soluble protein can be determined either with a UV-detector or with a refractive index detector. In the case of Na+-NQR, where the mass contribution of the detergent layer surrounding the protein complex has to be considered as well, both detectors are required, and the UV-extinction coefficient and the dn/dc for the protein and the detergent have to be known to calculate the molecular masses of the detergent fraction and the protein fraction (23, 24). For analyzing Na+-NQR, a gel filtration system (with UV detection) was connected online to a static light scattering detector and a refractive index detector as described under “Experimental Procedures.” It should be noted that gel filtration is used here only for sample fractionation and supply and that the calculated weight-average molar mass is independent of retention time from gel filtration. For Na+-NQR, a dn/dc of 0.185 ml g−1 (23) and a UV-extinction coefficient of 1.543 ml mg−1 cm−1 (at 280 nm), which was determined experimentally from the gel filtration peak fraction, were used. For DDM, a dn/dc of 0.143 ml g−1 (24) and an experimentally determined UV-extinction coefficient of 2.32 ml g−1 cm−1 were used. With Na+-NQR in gel filtration buffer, we detected a monodisperse peak with a total mass (protein and detergent micelle) of 291.9 ± 8.8 kDa and a molar mass of the protein fraction (protein without detergent) of 203.2 ± 6.1 kDa (Fig. 7). The size determined for the protein fraction corresponds well to the theoretical protein size of 213 kDa, which was calculated under the assumption that each subunit in Na+-NQR is present in a single copy.
FIGURE 7.

Determination of the mass of holo-Na+-NQR by static light scattering. The analysis was performed online with Na+-NQR eluting from a Superdex 200 column. The UV absorbance signal (thin solid line) and the differential refractive index signal (thin dashed line) are shown. Na+-NQR eluted at 21.94 min. The calculated molar masses of the detergent fraction (thick light gray line, bottom), the protein fraction (thick dark gray line, middle), and the protein-detergent conjugate (thick black line, top) are shown.
Sedimentation Velocity Analysis of Na+-NQR
Analysis of the sedimentation velocity runs revealed two peaks for Na+-NQR purified by nickel affinity chromatography (Fig. 8). The larger peak comprises 75% of the protein as calculated from the integrated peak area and was assigned to the holo-Na+-NQR complex, whereas the smaller peak was assigned to the NqrA subunit. Because the detergent micelle contributes largely to the overall shape of the protein, a spherical particle was assumed in the analysis. The mass of the holo-Na+-NQR calculated from the buoyant mass of the larger peak was 221 kDa. Assuming the same detergent:protein ratio for the smaller peak a mass of 82 kDa is calculated. However, at a lower detergent:protein ratio the calculated mass would be considerably smaller.
FIGURE 8.

Sedimentation velocity analysis of Na+-NQR. A, sedimentation distributions of Na+-NQR monitored by absorption at 280 nm at different time points are shown as colored dots. The fits of the curves are shown as black lines. B, residuals between experimental data and fits. C, protein molecular mass distribution obtained from the fits in A.
Stoichiometry of Na+-NQR Subunits
The stoichiometry of Na+-NQR subunits was determined by subjecting the holo-Na+-NQR to automated Edman degradation. The N termini of subunits NqrA to NqrD were unblocked and therefore accessible to Edman degradation, while the N termini of subunits NqrE and NqrF were blocked by N-formylation (60). Simultaneous quantification of subunits NqrB, NqrC, and NqrD was possible, although in some cycles a position in two or more subunits was occupied by the same amino acid residue. For example, NqrB and NqrD have a Lys at position 4. As these subunits contained unique amino acids at positions 3, 5, 7, 9, 10, and 12, the ratio of subunits NqrB, NqrC, and NqrD could be determined nevertheless. These results indicated that subunits NqrB, NqrC, and NqrD are present in Na+-NQR at a 1:1:1 ratio (Table 2). Note that Ser, Thr, and His residues are generally prone to decomposition during Edman degradation (61). This prevented the unambiguous quantification of NqrA with its six N-terminal histidines. However, yields of instable residues from NqrA are comparable to yields of the same amino acid residues from NqrB and NqrC at other positions (e.g. Ser-3 and Ser-10 from NqrA compared with Thr-10 from NqrC and His-5, His-7, and His-10 from NqrA compared with His-12 from NqrB, Table 2), supporting a 1:1 ratio of NqrA to NqrB and NqrC. The aggregate molar mass of NqrA, NqrB, NqrC, and NqrD is 146.6 kDa, and only a single copy of this set of subunits fits into the Na+-NQR complex with its overall size of ∼200 kDa (see above).
TABLE 2.
Subunit quantification in holo-Na+-NQR by Edman degradation
| Cycle | NqrA | NqrB | NqrC | NqrD | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| aaa | pmolb | aaNqrA/aaNqrDc | aa | pmol | aaNqrB/aaNqrD | aa | pmol | aaNqrC/aaNqrD | aa | pmol | aaNqrD/aaNqrD | |
| 3 | S | 10.06 | 0.44 | K | 19.29 | 0.85 | N | 12.64 | 0.56 | A | 22.62 | 1.00 |
| 5 | H | 6.67 | 0.39 | F | 23.60 | 1.38 | D | 18.39 | 1.08 | E | 17.07 | 1.00 |
| 7 | H | 6.74 | 0.40 | E | 16.95 | 1.01 | I | 16.14 | 0.96 | K | 16.74 | 1.00 |
| 9 | H | 7.76 | 0.73 | I | 16.74 | 1.58 | K | 25.53 | 2.41 | S | 10.58 | 1.00 |
| 10 | S | 7.06 | 0.30 | E | 16.83 | 0.72 | T | 11.49 | 0.49 | V | 23.40 | 1.00 |
| 12 | G | 11.89 | 0.52 | H | 9.19 | 0.41 | F | 14.95 | 0.66 | A | 22.67 | 1.00 |
| Mean | 0.47 | 0.99 | 1.03 | 1.00 |
a aa, amino acid residue present at that position as derived from the corresponding nqr gene on plasmid pNQR1.
b Amount normalized against the standard PTH amino acid mixture, not corrected for the sequencing yield is shown.
c The ratio between the amount of amino acid at this position of the given subunit and the amount of the amino acid at the corresponding position in subunit NqrD is shown.
Subunit NqrF was shown to contain one FAD-binding site and to harbor one 2Fe-2S cluster (8), being the only subunit carrying a sequence motif for ligation of an iron-sulfur cluster in the Na+-NQR. Our Na+-NQR preparation contained 0.88 ± 0.04 mol FAD per 213 kg of purified Na+-NQR, and 2.5–3.0 mol iron per 215 kg of purified Na+-NQR (62). The contents of FAD and iron are in accord with the presence of a single copy of NqrF in a Na+-NQR complex with an approximate mass of 200 kDa. Here, a calculated molecular mass of the NqrF subunit of 45.1 kDa was assumed.
Taken together, the Na+-NQR harbors one copy of each NqrA, NqrB, NqrC, NqrD, and NqrF, with a calculated total mass of 191.7 kDa. Next, we consider the mass contribution of NqrE subunit with a calculated mass of 21.5 kDa. NqrE was shown to be present in our Na+-NQR preparation (3). By subtracting the 191.7 kDa for NqrABCDF from the sizes of holo-Na+-NQR complex determined by gel filtration, sedimentation velocity analysis, or static light scattering, we calculate a residual mass of 6.9 ± 12.4 kDa, 29.3 kDa, or 11.5 ± 6.1 kDa, respectively, which complies with the presence of only a single copy of NqrE in the holo-Na+-NQR. It is concluded that the subunit composition of the Na+-NQR complex is NqrABCDEF.
DISCUSSION
As a prerequisite for the molecular understanding of an enzymatic system, the elements that contribute to the reaction mechanism have to be identified. In complex enzymes like the Na+-NQR, several membrane-bound subunits and a set of different cofactors have to be considered. We addressed here two open questions concerning the composition of the Na+-NQR: (i) which subunit binds the riboflavin cofactor and (ii) what is the stoichiometry of the Nqr subunits?
Studying the riboflavin content of Na+-NQR subcomplexes revealed that riboflavin binds to membranes of V. cholerae only in the presence of subunit NqrB, and that elimination of neither of the other Nqr subunits abolished riboflavin binding. We conclude that the riboflavin-binding site is localized on subunit NqrB, which is required for riboflavin binding. We do not know if NqrB is sufficient for riboflavin binding because attempts to express the individual subunit in V. cholerae were not successful. In addition to noncovalently bound riboflavin, NqrB contains a covalently attached FMN. However, one should also consider the possibility that the holo-Na+-NQR might house two NqrB subunits, with riboflavin occupying a vacant FMN-binding site on one NqrB copy and a covalently attached FMN in the FMN-binding site of the second copy of NqrB. In this case, the riboflavin associated with (one copy of) NqrB would represent a cleavage product of the previously attached FMN.
To exclude the latter possibility, it was necessary to determine the subunit composition of the Na+-NQR. Only one copy of NqrB is present in the complex, which demonstrates that riboflavin and FMN represent two cofactors found on one NqrB subunit. Our study revealed the subunit and cofactor composition of the Na+-NQR complex, which contains one copy of NqrF with one FAD and one 2Fe-2S cluster, one copy of NqrB with one covalently attached FMN and one riboflavin, one copy of NqrC with one covalently bound FMN, one copy of NqrD, and one copy of NqrE.
We asked if the primary structure of NqrB bears information to predict a putative riboflavin-binding site. Only few high-resolution structures from proteins in complex with riboflavin are available from the RCSB Protein Data Bank (63) (see supplemental Table S1), which are mainly from proteins involved in riboflavin metabolism and storage. The high resolution structure of flavodoxin in complex with riboflavin is one of a few examples for a protein in which riboflavin can act as a redox cofactor (64). However, it is noteworthy that the native cofactor of flavodoxin is FMN (65). Analyzing the mode of riboflavin binding to the proteins listed in supplemental Table S1, e.g. flavodoxin (PDB entry 1BU5), riboflavin kinase (PDB entry 1NB9), chicken riboflavin-binding protein (66), and dodecin (PDB entry 1MOG), showed that riboflavin mainly interacts with the protein backbone. Additionally, a majority of these proteins contain aromatic residues which undergo π-stacking interactions to the isoalloxazine ring (supplemental Table S1). Thus, conserved aromatic residues in NqrB which are either not buried by the membrane or located at the border of transmembrane helices would be promising candidates contributing to an isoalloxazine-binding site. To identify aromatic residues potentially involved in flavin binding, we performed multiple sequence alignments of a set of 69 non-redundant NqrB sequences (Fig. 9 and supplemental Fig. S1). Highest conservation was found in regions 1–78, 125–237, and 264–411, comprising Thr-236, which is covalently modified by FMN, whereas regions 81–121, 243–259, and 297–325 showed a high degree of variability (V. cholerae numbering). The alignment showed several conserved and semi-conserved aromatic residues that could participate in riboflavin binding (Fig. 9). The region between residues 177 to 231 of NqrB from V. cholerae is of special interest, because it shows sequence similarity to flavodoxin (Fig. 10). The structure of flavodoxin from Desulfovibrio vulgaris in complex with riboflavin (64) reveals that riboflavin is mainly bound by two loops comprising residues 59–65 and 94–102 (D. vulgaris numbering). Residues Thr-59, Trp-60, Gly-94, Asp-95, and Tyr-98 in flavodoxin mediate direct side-chain interactions to the riboflavin (64) and are conserved or semi-conserved in NqrB (Fig. 10). Thus, this sequence similarity could indicate that region 177–231 of NqrB comprises a binding site for riboflavin or FMN. This assumption was supported by our finding that substitution of residue Phe-186 and Trp-227 of NqrB, which would correspond to Trp-60 and Tyr-98 of flavodoxin, respectively (Fig. 10), resulted in a decrease in riboflavin content of the mutant variant compared with wild-type Na+-NQR by 27%. It however remains to be investigated whether Phe-186 and Trp-227 of NqrB directly interact with riboflavin, or whether the F186A and the W227L substitutions exert an indirect effect on riboflavin binding.
FIGURE 9.

Sequence alignment of NqrB. Sequences of NqrB from Chlamydia pneumoniae (Q9Z8B6), Protochlamydia amoebophila (Q6MEH4), Shewanella oneidensis (Q8EID9), Porphyromonas gingivalis (Q7MT18), Rhodopirellula baltica (Q7UWS4), and V. cholerae (A6XUU9) were compared. The alignment shown here is a section from a larger alignment of 69 non-redundant sequences of NqrB homologues, which was constructed using the ClustalW algorithm implemented in the BioEdit software (Version 7.0.9.0) with default settings (see “Experimental Procedures” and supplemental Fig. S1). Identical residues and similar residues are underlaid with black and gray bars, respectively, and were derived from the alignment in supplemental Fig. S1 using an identity and similarity threshold of 85%. Black bars indicate transmembrane helices, gray bars indicate hydrophobic stretches, as predicted from a consensus model based on 11 different topology prediction algorithms (see “Experimental Procedures” and supplemental Fig. S2). The FMN phosphorylthreonine (Thr-236) is highlighted by an asterisk, and Gly-141, which was proposed to interact with the inhibitor korormicin (11), is marked by a rhomboid.
FIGURE 10.

Partial sequence alignment of NqrB and flavodoxin. Amino acid sequences of flavodoxin from D. vulgaris (P00323) and Methanosarcina acetivorans (Q8TMF9) in the regions of the flavin-binding loops were aligned with the corresponding region of NqrB from C. pneumoniae (Q9Z8B6) and V. cholerae (A6XUU9). Residues of flavodoxin that mediate side-chain contact to riboflavin, including the two aromatic residues of flavodoxin, which stack to the isoalloxazine ring, are marked by arrows. Thr-236 of NqrB, which carries the covalently linked FMN, is indicated by an asterisk. Black bar, proposed transmembrane helix; gray bar, hydrophobic stretch.
The proposed flavin-binding site seems to be in conflict with a previous topology model of NqrB (51), which predicts a transmembrane helix between residues 205 and 225, thereby placing the two loops proposed to participate in binding of flavin on different sides of the membrane. We note that this part of NqrB which shows similarity to flavodoxin has not been subjected to an experimental topology analysis (51). By summing up the results of 11 different topology prediction methods, we constructed a consensus model, defining a TMH where at least two third of the prediction models predicted a helix (supplemental Fig. S2). According to this topology model, the region between residues 206 and 226 of NqrB (V. cholerae numbering, Fig. 10) does not necessarily represent a transmembrane region, but could also represent a hydrophobic stretch mediating contact to the bacterial membrane.
Riboflavin was proposed to participate in the final reduction of membrane-bound quinone to quinol by Na+-NQR (3, 4, 67, 68), and a mutagenesis study identified NqrB as being potentially important for quinone binding (11). An open question concerning the mechanism of Na+-NQR is how one-electron transfer reactions initiated by the 2Fe-2S cluster on NqrF are coupled to the two-electron reduction of quinone to quinol (2, 4, 8, 67). Both riboflavin and the covalently linked FMNs were proposed to exclusively act as one-electron converters in Na+-NQR (4, 67–69). Our finding that both riboflavin and one covalently bound FMN are located in NqrB suggests that these cofactors are in electron-transfer distance and supports the view that NqrB catalyzes the ultimate reduction steps leading to formation of ubiquinol from ubiquinone.
Supplementary Material
Acknowledgments
We thank Birgit Dreier and Christophe Briand, University of Zurich, and Stefan Schauer, Functional Genomics Centre Zurich, for technical assistance.
This work was supported by fellowships from the Research Commission of the University of Zurich and the EMDO Stiftung (to M. S. C.), and by grants from the Swiss National Science Foundation and from Parkinson Schweiz (to J. S.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs S1 and S2 and Table S1.
- Na+-NQR
- sodium ion-translocating NADH:quinone oxidoreductase
- DDM
- n-dodecyl-β-d-maltoside
- Q1
- ubiquinone-1
- TMH
- transmembrane helix
- PDB
- Protein Data Bank.
REFERENCES
- 1.Hayashi M., Nakayama Y., Unemoto T. (2001) Biochim. Biophys. Acta 1505, 37–44 [DOI] [PubMed] [Google Scholar]
- 2.Bogachev A. V., Verkhovsky M. I. (2005) Biochemistry 70, 143–149 [DOI] [PubMed] [Google Scholar]
- 3.Tao M., Casutt M. S., Fritz G., Steuber J. (2008) Biochim. Biophys. Acta 1777, 696–702 [DOI] [PubMed] [Google Scholar]
- 4.Juárez O., Morgan J. E., Barquera B. (2009) J. Biol. Chem. 284, 8963–8972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Juárez O., Nilges M. J., Gillespie P., Cotton J., Barquera B. (2008) J. Biol. Chem. 283, 33162–33167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pfenninger-Li X. D., Albracht S. P. J., van Belzen R., Dimroth P. (1996) Biochemistry 35, 6233–6242 [DOI] [PubMed] [Google Scholar]
- 7.Bogachev A. V., Bertsova Y. V., Barquera B., Verkhovsky M. I. (2001) Biochemistry 40, 7318–7323 [DOI] [PubMed] [Google Scholar]
- 8.Türk K., Puhar A., Neese F., Bill E., Fritz G., Steuber J. (2004) J. Biol. Chem. 279, 21349–21355 [DOI] [PubMed] [Google Scholar]
- 9.Nakayama Y., Yasui M., Sugahara K., Hayashi M., Unemoto T. (2000) FEBS Lett. 474, 165–168 [DOI] [PubMed] [Google Scholar]
- 10.Hayashi M., Nakayama Y., Yasui M., Maeda M., Furuishi K., Unemoto T. (2001) FEBS Lett. 488, 5–8 [DOI] [PubMed] [Google Scholar]
- 11.Hayashi M., Shibata N., Nakayama Y., Yoshikawa K., Unemoto T. (2002) Arch. Biochem. Biophys. 401, 173–177 [DOI] [PubMed] [Google Scholar]
- 12.Taylor R. G., Walker D. C., McInnes R. R. (1993) Nucleic Acids Res. 21, 1677–1678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barquera B., Hellwig P., Zhou W., Morgan J. E., Häse C. C., Gosink K. K., Nilges M., Bruesehoff P. J., Roth A., Lancaster C. R., Gennis R. B. (2002) Biochemistry 41, 3781–3789 [DOI] [PubMed] [Google Scholar]
- 14.Smith P. K., Krohn R. I., Hermanson G. T., Mallia A. K., Gartner F. H., Provenzano M. D., Fujimoto E. K., Goeke N. M., Olson B. J., Klenk D. C. (1985) Anal. Biochem. 150, 76–85 [DOI] [PubMed] [Google Scholar]
- 15.Schägger H., von Jagow G. (1987) Anal. Biochem. 166, 368–379 [DOI] [PubMed] [Google Scholar]
- 16.Roughead Z. K., McCormick D. B. (1990) J. Nutr. 120, 382–388 [DOI] [PubMed] [Google Scholar]
- 17.Brown P. H., Schuck P. (2006) Biophys. J. 90, 4651–4661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuck P., Rossmanith P. (2000) Biopolymers 54, 328–341 [DOI] [PubMed] [Google Scholar]
- 19.Demeler B. (2005) in Modern Analytical Ultracentrifugation: Techniques and Methods (Scott D. J., Harding S. E., Rowe A. J. eds), Royal Society of Chemistry, London, UK [Google Scholar]
- 20.le Maire M., Arnou B., Olesen C., Georgin D., Ebel C., Møller J. V. (2008) Nat. Protoc. 3, 1782–1795 [DOI] [PubMed] [Google Scholar]
- 21.Fleming K. G. (2008) Curr. Protoc. Protein Sci. Unit 7.12, 1–12 [DOI] [PubMed] [Google Scholar]
- 22.Møller J. V., le Maire M. (1993) J. Biol. Chem. 268, 18659–18672 [PubMed] [Google Scholar]
- 23.Folta-Stogniew E., Williams K. R. (1999) J. Biomol. Tech. 10, 51–63 [PMC free article] [PubMed] [Google Scholar]
- 24.Slotboom D. J., Duurkens R. H., Olieman K., Erkens G. B. (2008) Methods 46, 73–82 [DOI] [PubMed] [Google Scholar]
- 25.Dubois M., Gilles K. A., Hamilton J. K., Rebers P. A., Smith F. (1956) Anal. Chem. 28, 350–356 [Google Scholar]
- 26.Mokhonov V., Mokhonova E., Yoshihara E., Masui R., Sakai M., Akama H., Nakae T. (2005) Protein Expr. Purif. 40, 91–100 [DOI] [PubMed] [Google Scholar]
- 27.Sambrook J., Russel D. W. (2001) Molecular Cloning: A Laboratory Manual, 3rd Ed., Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 28.Inoue H., Nojima H., Okayama H. (1990) Gene 96, 23–28 [DOI] [PubMed] [Google Scholar]
- 29.Marcus H., Ketley J. M., Kaper J. B., Holmes R. K. (1990) FEMS Microbiol. Lett. 56, 149–154 [DOI] [PubMed] [Google Scholar]
- 30.Gemperli A. C., Dimroth P., Steuber J. (2002) J. Biol. Chem. 277, 33811–33817 [DOI] [PubMed] [Google Scholar]
- 31.Steuber J., Krebs W., Dimroth P. (1997) Eur. J. Biochem. 249, 770–776 [DOI] [PubMed] [Google Scholar]
- 32.Nakayama Y., Hayashi M., Yoshikawa K., Mochida K., Unemoto T. (1999) Biol. Pharm. Bull. 22, 1064–1067 [DOI] [PubMed] [Google Scholar]
- 33.Burgdorf T., van der Linden E., Bernhard M., Yin Q. Y., Back J. W., Hartog A. F., Muijsers A. O., de Koster C. G., Albracht S. P., Friedrich B. (2005) J. Bacteriol. 187, 3122–3132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.The UniProt Consortium (2009) Nucleic Acids Res. 37, D169–D174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson J. D., Higgins D. G., Gibson T. J. (1994) Nucleic Acids Res. 22, 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hall T. A. (1999) Nucleic Acids Symposium Series41, 95–98 [Google Scholar]
- 37.Henikoff S., Henikoff J. G. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 10915–10919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Waterhouse A. M., Procter J. B., Martin D. M., Clamp M., Barton G. J. (2009) Bioinformatics 25, 1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sonnhammer E. L., von Heijne G., Krogh A. (1998) Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182 [PubMed] [Google Scholar]
- 40.Hofmann K., Stoffel W. (1993) Biol. Chem. Hoppe-Seyler 374, 166. [DOI] [PubMed] [Google Scholar]
- 41.Hirokawa T., Boon-Chieng S., Mitaku S. (1998) Bioinformatics 14, 378–379 [DOI] [PubMed] [Google Scholar]
- 42.Cserzö M., Wallin E., Simon I., von Heijne G., Elofsson A. (1997)). Protein Eng. 10, 673–676 [DOI] [PubMed] [Google Scholar]
- 43.Tusnády G. E., Simon I. (1998) J. Mol. Biol. 283, 489–506 [DOI] [PubMed] [Google Scholar]
- 44.Tusnády G. E., Simon I. (2001) Bioinformatics 17, 849–850 [DOI] [PubMed] [Google Scholar]
- 45.Claros M. G., von Heijne G. (1994) Comput. Appl. Biosci. 10, 685–686 [DOI] [PubMed] [Google Scholar]
- 46.Jones D. T., Taylor W. R., Thornton J. M. (1994) Biochemistry 33, 3038–3049 [DOI] [PubMed] [Google Scholar]
- 47.Juretić D., Zoranić L., Zucić D. (2002) J. Chem. Inf. Comput. Sci. 42, 620–632 [DOI] [PubMed] [Google Scholar]
- 48.Rost B., Casadio R., Fariselli P., Sander C. (1995) Protein Sci. 4, 521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Milpetz F., Argos P., Persson B. (1995) Trends Biochem. Sci. 20, 204–205 [DOI] [PubMed] [Google Scholar]
- 50.Käll L., Krogh A., Sonnhammer E. L. (2005) Bioinformatics 21, i251–i257 [DOI] [PubMed] [Google Scholar]
- 51.Duffy E. B., Barquera B. (2006) J. Bacteriol. 188, 8343–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barquera B., Zhou W., Morgan J. E., Gennis R. B. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 10322–10324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bogachev A. V., Bertsova Y. V., Bloch D. A., Verkhovsky M. I. (2006) Biochemistry 45, 3421–3428 [DOI] [PubMed] [Google Scholar]
- 54.Tao M., Türk K., Diez J., Grütter M. G., Fritz G., Steuber J. (2006) Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 110–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bogachev A. V., Belevich N. P., Bertsova Y. V., Verkhovsky M. I. (2009) J. Biol. Chem. 284, 5533–5538 [DOI] [PubMed] [Google Scholar]
- 56.Barquera B., Häse C. C., Gennis R. B. (2001) FEBS Lett. 492, 45–49 [DOI] [PubMed] [Google Scholar]
- 57.Meier T., Yu J., Raschle T., Henzen F., Dimroth P., Muller D. J. (2005) FEBS J. 272, 5474–5483 [DOI] [PubMed] [Google Scholar]
- 58.Rath A., Glibowicka M., Nadeau V. G., Chen G., Deber C. M. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 1760–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayashi M., Miyoshi T., Sato M., Unemoto T. (1992) Biochim. Biophys. Acta 1099, 145–151 [PubMed] [Google Scholar]
- 60.Nakayama Y., Hayashi M., Unemoto T. (1998) FEBS Lett. 422, 240–242 [DOI] [PubMed] [Google Scholar]
- 61.Hempel J. (2002) in Modern Protein Chemistry: Practical Aspects (Howard G. C., Brown W. E. eds), CRC Press, Boca Raton, FL [Google Scholar]
- 62.Fadeeva M. S., Bertsova Y. V., Verkhovsky M. I., Bogachev A. V. (2008) Biochemistry 73, 123–129 [DOI] [PubMed] [Google Scholar]
- 63.Berman H. M., Westbrook J., Feng Z., Gilliland G., Bhat T. N., Weissig H., Shindyalov I. N., Bourne P. E. (2000) Nucleic Acids Res. 28, 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walsh M. A., McCarthy A., O'Farrell P. A., McArdle P., Cunningham P. D., Mayhew S. G., Higgins T. M. (1998) Eur. J. Biochem. 258, 362–371 [DOI] [PubMed] [Google Scholar]
- 65.Pueyo J. J., Curley G. P., Mayhew S. G. (1996) Biochem. J. 313, 855–861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monaco H. L. (1997) EMBO J. 16, 1475–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bogachev A. V., Bloch D. A., Bertsova Y. V., Verkhovsky M. I. (2009) Biochemistry 48, 6299–6304 [DOI] [PubMed] [Google Scholar]
- 68.Bogachev A. V., Kulik L. V., Bloch D. A., Bertsova Y. V., Fadeeva M. S., Verkhovsky M. I. (2009) Biochemistry 48, 6291–6298 [DOI] [PubMed] [Google Scholar]
- 69.Barquera B., Ramirez-Silva L., Morgan J. E., Nilges M. J. (2006) J. Biol. Chem. 281, 36482–36491 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.