

Abstract
Background and purpose:
Inhibitors of fatty acid amide hydrolase (FAAH), the enzyme responsible for the metabolism of the endogenous cannabinoid (CB) receptor ligand anandamide (AEA), are effective in a number of animal models of pain. Here, we investigated a series of isoflavones with respect to their abilities to inhibit FAAH.
Experimental approach:
In vitro assays of FAAH activity and affinity for CB receptors were used to characterize key compounds. In vivo assays used were biochemical responses to formalin in anaesthetized mice and the ‘tetrad’ test for central CB receptor activation.
Key results:
Of the compounds tested, biochanin A was adjudged to be the most promising. Biochanin A inhibited the hydrolysis of 0.5 µM AEA by mouse, rat and human FAAH with IC50 values of 1.8, 1.4 and 2.4 µM respectively. The compound did not interact to any major extent with CB1 or CB2 receptors, nor with FAAH-2. In anaesthetized mice, URB597 (30 µg i.pl.) and biochanin A (100 µg i.pl.) both inhibited the spinal phosphorylation of extracellular signal-regulated kinase produced by the intraplantar injection of formalin. The effects of both compounds were significantly reduced by the CB1 receptor antagonist/inverse agonist AM251 (30 µg i.pl.). Biochanin A (15 mg·kg−1 i.v.) did not increase brain AEA concentrations, but produced a modest potentiation of the effects of 10 mg·kg−1 i.v. AEA in the tetrad test.
Conclusions and implications:
It is concluded that biochanin A, in addition to its other biochemical properties, inhibits FAAH both in vitro and peripherally in vivo.
This article is part of a themed issue on Cannabinoids. To view the editorial for this themed issue visit http://dx.doi.org/10.1111/j.1476-5381.2010.00831.x
Keywords: fatty acid amide hydrolase, pain, cannabinoid, isoflavones, biochanin A, anandamide
Introduction
The endocannabinoid system, comprising the cannabinoid CB1 and CB2 receptors, their endogenous ligands anandamide (arachidonoylethanolamide, AEA) and 2-arachidonoylglycerol (2-AG) and the endocannabinoid synthetic and metabolic enzymes, is involved in the regulation of a variety of physiological processes including cognition, pain perception and appetite (see Pacher et al., 2006). Δ9-Tetrahydrocannabinol, the main psychoactive ingredient of cannabis, has long been known to produce pain relief (see Pertwee, 2001), but the psychotropic properties of the compound place a limitation upon the doses (and thereby the level of benefit) that can be used.
AEA is metabolized by the enzyme fatty acid amide hydrolase (FAAH), and selective inhibitors of the enzyme such as 3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate (URB597) and 1-oxo-1[5-(2-pyridyl)-2-yl]-7-phenylheptane (OL-135) produce potentially useful effects in animal models of inflammatory, visceral, cancer and in some cases neuropathic pain (see e.g. Chang et al., 2006; Jayamanne et al., 2006;Khasabova et al., 2008; Naidu et al., 2009). These compounds do not produce the behavioural changes seen as a result of a general activation of central CB1 receptors (in the classical ‘tetrad’ of catalepsy, inhibition of locomotion, reduction of body temperature and thermal analgesia, only the latter is affected), and are thus an attractive target for drug development (reviews, see Seierstad and Breitenbucher, 2008; Fowler et al., 2009).
In 2004, McFarland et al. reported that the soy isoflavone genistein could interfere with the cellular uptake of AEA. Subsequent studies showed that this property was shared by daidzein and was due to the ability of the compounds to inhibit FAAH (Thors et al., 2007a,b;). The Ki values for the inhibition of the rat enzyme were 2.8 and 1.7 µM, respectively (Thors et al., 2007a,b;). In Asian countries, where soy consumption is higher than in Western countries, serum concentrations of genistein and daidzein can reach as high as 2–4 µM (Morton et al., 2002), suggesting that the concentrations required for inhibition of FAAH are reachable in vivo, at least in theory. In the present study, we have investigated a series of analogues of genistein and daidzein in order to shed light on the structural requirements for FAAH inhibition, and to determine whether more potent analogues could be found. Among the analogues tested, biochanin A, a naturally occurring compound found in red clover, was shown to be a more potent inhibitor of FAAH than genistein in vitro, and to produce biochemical effects upon a spinal cord pain signalling pathway consistent with FAAH inhibition in vivo.
Methods
Expression of human FAAH and FAAH-2 in COS7 cells (La Jolla)
The cDNA sequence for human FAAH was subcloned into the pFLAG-CMV vector, and the constructs were transfected into COS7 cells using the FuGENE6 transfection reagent as described previously (Wei et al., 2006). The cells were harvested by scraping 48 h post-transfection, dounce homogenized in phosphate-buffered saline (PBS) and sonicated. The resulting lysate was centrifuged at 100 000×g for 45 min at 4°C to provide the cytosolic fraction in the supernatant and the membrane fraction as a pellet. Total protein concentration of the membrane fractions was determined by a colorimetric protein assay kit (Bio-Rad), and the expression of FAAH was confirmed both by labelling membrane fractions with FP-rhodamine, and by the ability of the membranes to hydrolyse [14C]oleamide, in contrast to membranes from mock-transfected cells (data not shown).
FAAH activity measurements (Umeå)
For experiments with FAAH, rat liver homogenates, mouse brain homogenates and membranes from COS7 cells transfected with the human enzyme were used. Frozen (−80°C) livers from adult C57BL/6 mice and frozen brains (minus cerebella) from adult Wistar or Sprague-Dawley rats were thawed and homogenized in 20 mM HEPES, 1 mM MgCl2, pH 7. The homogenates were centrifuged at ∼35 000×g for 20 min at 4°C. After resuspension in buffer followed by recentrifugation and a second resuspension in buffer, the pellets were incubated at 37°C for 15 min. This incubation was undertaken in order to hydrolyse all endogenous FAAH substrates. The homogenates were then centrifuged as above, recentrifuged and resuspended in 50 mM Tris–HCl buffer, pH 7.4, containing 1 mM EDTA and 3 mM MgCl2. The homogenates were then frozen at −80°C in aliquots until used for assay. FAAH was assayed in the homogenates and in the COS7 cell membranes by the method of Boldrup et al. (2004) using 0.5 µM (unless otherwise stated) [3H]AEA labelled in the ethanolamine part of the molecule. Blank values were obtained by the use of buffer rather than homogenate. In the experiments comparing effects of biochanin A upon FAAH and FAAH-2, the same assay was used but with 16 nM [3H]oleoylethanolamide ([3H]OEA) as substrate and with an incubation phase at room temperature. The choice of OEA rather than AEA for FAAH-2 was motivated by the relative rates of hydrolysis: OEA is metabolized four times faster than AEA by FAAH-2, whereas for FAAH the rate of hydrolysis of OEA is about a third of that for AEA (Wei et al., 2006). When 0.5 µM [3H]AEA was used as substrate, assay conditions for rat brain and mouse liver were chosen so that <10% of added substrate was metabolized. For the human FAAH samples, <5% of the [3H]AEA was metabolized in all cases. For 16 nM [3H]OEA, a limited supply of an expensive ligand meant that optimization was not possible, and the amount of substrate utilized was higher (34 ± 1 and 0.5 ± 0.1% for FAAH and its corresponding mock-transfected, respectively; 40 ± 2 and 21 ± 0.4 for FAAH-2 and its corresponding mock-transfected respectively). However, when we ran experiments with human FAAH and 0.5 µM [3H]AEA with assay conditions giving these higher utilization rates, the activity was still inhibited by biochanin A, genistein, formononetin and daidzein in the low micromolar range (IC50 values of 6.0, 8.4, 12 and 30 µM, respectively; data not shown).
URB597-sensitive accumulation of tritium in RBL2H3 cell membranes following incubation of cells in suspension with [3H]AEA (Umeå)
RBL2H3 cells were cultured in minimum essential medium with Earl's salts, 15% fetal bovine serum and 100 U·mL−1 penicillin + 100 µg·mL−1 streptomycin (‘medium’). After centrifugation and resuspension in medium in Eppendorf tubes (2 × 105 cells per tube; 5 × 105 cells·mL−1), the cells were pre-incubated with either the test compounds or URB597 for 10 min at 37°C. [3H]AEA (assay concentration 100 nM; substrate labelled in the arachidonoyl part of the molecule) in medium was added and the cells were incubated for a further 10 min (final assay volume 200 µL). After incubation, the cells were sedimented using a microcentrifuge (1 min, 1000×g), then washed twice with ice cold medium, and the tritium retained by the cell membranes was determined by rapid filtration and washing with deionized water as described by Thors et al. (2008). The URB597-sensitive accumulation of tritium was defined as the radioactivity seen for the test compounds or vehicle control minus the corresponding radioactivity for the cells treated with 100 nM URB597.
Radioligand binding experiments (Aberdeen)
The ability of the isoflavones to interact with CB1 and CB2 receptors was assessed using mouse brain membranes as a source of the former, and membranes from CHO cells transfected with the human CB2 receptor as a source of the latter. A standard filtration assay was used (for details, see Ross et al., 1999; Thomas et al., 2005), and the concentration of radioligand ([3H]CP 55940) was 0.7 nM. Non-specific binding was determined in the presence of 1 µM of unlabelled CP 55940.
Formalin-induced phosphorylation of extracellular signal-regulated kinase (ERK) (St Louis, Umeå)
The method was based upon that of Karim et al. (2001). Male adult C57BL/6 mice (body weight ∼22 g) were anaesthetized with an i.p. injection of 0.075 mL av 100 mg·mL−1 pentobarbital. Upon anaesthesia, the animals were injected with test compounds (URB597, biochanin A ± AM251) or vehicle (3% Tween-80 in physiological saline) into the right rear paw (injection volume 20 µL). This vehicle was chosen because it has been used by others investigating the effects of i.pl. URB597 in pain models (Jhaveri et al., 2006;Sagar et al., 2008), thereby allowing comparison of active doses. Ten minutes later, the animals were injected with formalin (10 µL of a 3% solution dissolved in saline) into the same paw. Five minutes (unless otherwise stated) after the formalin injection, the mice were perfused transcardially with warm saline (37°C, 0.9% NaCl), followed by 300 mL of ice-cold 4% paraformaldehyde solution. A 5 min time was used because the pERK response to formalin was fully developed at this time-point (see Results). Spinal cords were dissected and placed in Falcon tubes containing ice-cold 4% paraformaldehyde solution. After 4 h at 4°C, the samples were transferred to Falcon tubes containing 30% sucrose in 0.1 M phosphate buffer and kept at 4°C, the cryoprotection solution being exchanged until the spinal cord samples were saturated. The lumbar enlargement was localized and dissected out, and a nick was made on the contralateral side to allow identification under the microscope. The sample was placed in a cryomold and affixed on dry ice using OCT compound (Tissue-Tek, Miles Inc., Elkhart, IN, USA). Coronal sections (30 µM) were cut using a freezing sliding cryostat, and the sections were transferred to 24-well culture plates containing 0.1 M phosphate buffer. After washing with phosphate buffer (twice) and PBS (once) at room temperature, the sections were incubated for 30 min at room temperature with peroxidase suppression solution (10% MeOH, 0.3% H2O2 in 0.1 M PBS). After three washes with PBS at room temperature, 1% normal goat serum in 0.1 M PBS contaning 0.02% (v/v, final concentration) Triton X-100 (1% NGST) was added. The primary pERK R6 polyclonal antibody (9101S, Cell Signaling Technology, Danvers, MA, USA) (1:500, in NGST) was added, and the sections were incubated for 72 h at 4°C before being washed three times at RT with 1% NGST. The secondary antibody (biotinylated anti-rabbit IgG, BA-1000, Vector Laboratories, Burlingame, CA, USA, diluted 1:100 in 1% NGST) was incubated with the slices for 2 h at room temperature, the plates being covered with aluminium foil. The samples were washed three times with 1% NGST prior to addition of ExtrAvidin peroxidise (1:1000, Sigma, St Louis, MO, USA) in 1% NGST and incubation for 1 h at room temperature. After two washes with PBS and one wash with phosphate buffer at room temperature, the samples were stained using the DAB kit (Vector Laboratories). The sections were mounted on slides, dehydrated, dried overnight and viewed under the microscope. Four sections from each animal were analysed under the microscope by an investigator who was blind to the treatment given to the animal in question, and the mean score from the four sections was calculated.
Behavioural effects of biochanin A (Richmond)
ICR mice (Harlan Laboratories, Indianapolis, IN, USA) were used for the behavioural tests measuring spontaneous activity (over a 10 min testing period), rectal temperature, ring immobility (over a 5 min testing period) and nociceptive threshold (tail flick tests) (for details, see Pertwee, 1972; Martin et al., 1991; Burston et al., 2008). AEA and biochanin A were dissolved in a vehicle consisting of ethanol, Emulphor-620 (Sanofi-Aventis, Bridgewater, NJ, USA) and physiological saline in a ratio of 1:1:18 v/v, and administered i.v. to the animals via the tail vein (injection volume 10 µL·g−1 body weight). The degree of antinociception is expressed as percentage of maximum possible effect (%MPE), defined as [(test – control time)/(10 – control time)]× 100.
Quantification of AEA levels in the brain (Richmond)
The mice above treated with vehicle rather than AEA were killed by decapitation 1 h after the injection of the isoflavones. Brains were collected and immersed in liquid nitrogen to ensure rapid cooling. Following extraction of lipids with methanol/chloroform (Hardison et al., 2006), AEA levels were determined by liquid chromatography/mass spectrometry analysis (Kingsley and Marnett, 2003).
Ethical considerations
The experiments reported in this paper were approved by the appropriate regional Animal Care and Use Committees.
Statistical analyses
The pI50 and IC50 values, linear regressions and confidence limits were determined using the GraphPad Prism computer programme (GraphPad Software Inc., San Diego, CA, USA). The pI50, and hence IC50, values were calculated using the built-in programme ‘sigmoidal dose–response (variable slope)’ from the data expressed as % of control using top (i.e. uninhibited) values of 100% and bottom (residual activity) values that were either set to zero or allowed to float. The two curves were compared using Akaike's informative criteria. In all but one case, statistical analysis suggested that the simple 0% residual inhibition model fitted the curve more appropriately. Kmapp and Vmaxapp values were determined using the Direct Linear plot and the Enzyme Kinetics v1.4 computer programme for the Macintosh (Trinity Software, Campton, NH, USA).
Compounds
Biochanin A (5,7-dihydroxy-4′-methoxyisoflavone), formononetin (7-hydroxy-4′-methoxyisoflavone), genistein (4′,5,7-trihydroxyisoflavone), genistein dimethyl ether (4′,7-dimethoxy-5-hydroxyisoflavone), genistin (genistein-7-O-β-d-glucopyranoside), prunetin (4′,5-dihydroxy-7-methoxyisoflavone), puerarin (8-(β-d-glucopyranosyl-daidzein)), 3′,4′,7-trihydroxyisoflavone, 4′,6,7-trihydroxyisoflavone, ExtrAvidin peroxidase and the pFLAG-CMV6 vector were obtained from the Sigma Chemical Co. Daidzein (4′,7-dihydroxyisoflavone) was obtained from Sigma and from the Cayman Chemical Co. (Ann Arbor, MI, USA). The source of daidzein was from Sigma unless otherwise indicated. Equol (4′,7-dihydroxyisoflavane) and 7-hydroxyisoflavone were obtained from Apin Chemicals Ltd (Abingdon, UK) and Sequoia Research Products Ltd (Pangbourne, UK) respectively. GW9662 (2-chloro-5-nitrobenzanilide) was obtained from the Cayman Chemical Co. AEA [ethanolamine-1-3H] (specific activity 2.22 TBq·mmol−1, for the FAAH experiments), AEA [arachidonoyl 5,6,8,9,11,12,14,15-3H] (specific activity 7.4 TBq·mmol−1, for the membrane tritium accumulation experiments) and oleoylethanolamide [ethanolamine-1-3H] (specific activity 0.74 TBq·mmol−1) were purchased from American Radiolabeled Chemicals, Inc (St Louis, MO, USA). CP 55940, [side chain-2,3,4-3H(N)]- (specific activity 3.7–6.66 TBq)·mmol−1 was obtained from PerkinElmer Life and Analytical Sciences, Inc (Boston, MA, USA). URB597 and non-radioactive AEA were purchased from the Cayman Chemical Company. Non-radioactive CP 55940 was obtained from Tocris Bioscience (Ellisville, MO, USA). The pERK R6 polyclonal antibody (9101S; phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody) was obtained from Cell Signaling Technology, Inc. Biotinylated anti-rabbit IgG (BA-1000) was obtained from Vector Laboratories. FuGENE6 transfection reagent was obtained from Roche Applied Science (Indianapolis, IN, USA). Rat RBL2H3 basophilic leukaemia cells (passage range 21–27) were obtained from the American Type Culture Collection (Manassas, VA, USA). The culture medium, sera and supplements were obtained from Invitrogen (Stockholm, Sweden).
Nomenclature
The drug/molecular target nomenclature used here conforms to BJP's Guide to Receptors and Channels (Alexander et al., 2008).
Results
Structure–activity relationships for the interaction of isoflavones with FAAH
The effects of 11 isoflavone compounds upon the hydrolysis of 0.5 µM [3H]AEA by rat brain homogenates are shown in Table 1. Examples of the inhibition curves from which the pI50 values were derived are shown in Figure 1. A number of conclusions can be drawn from the data with respect to the chromone part of the molecule:
Table 1.
Inhibition of FAAH by a series of isoflavones related to daidzein and genistein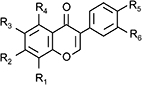
| Name | R1 | R2 | R3 | R4 | R5 | R6 | pI50 | IC50 (µM) |
|---|---|---|---|---|---|---|---|---|
| 7-Hydroxyisoflavone | H | OH | H | H | H | H | ||
| EtOH | 5.50 ± 0.02 | 3.1 | ||||||
| DMSO | 5.38 ± 0.02 | 4.2 | ||||||
| Daidzeina | H | OH | H | H | OH | H | ||
| Sigma | 5.60 ± 0.02 | 2.5 | ||||||
| Cayman | 5.42 ± 0.02 | 3.8 | ||||||
| Puerarin | β-d-gluco-pyranosyl | OH | H | H | OH | H | <4b | >100b |
| Formononetin | H | OH | H | H | OCH3 | H | 6.25 ± 0.02c | 0.57 |
| Genistein | H | OH | H | OH | OH | H | ||
| EtOH | 5.51 ± 0.05 | 3.1 | ||||||
| DMSO | 5.66 ± 0.05 | 2.2 | ||||||
| 3′,4′,7-Trihydroxyisoflavone | H | OH | H | H | OH | OH | 5.61 ± 0.05 | 2.5 |
| 4′,6,7-Trihydroxyisoflavone | H | OH | OH | H | OH | H | 4.42 ± 0.02 | 38 |
| Biochanin A | H | OH | H | OH | OCH3 | H | ||
| EtOH | 5.85 ± 0.02 | 1.4 | ||||||
| DMSO | 6.03 ± 0.06 | 0.93 | ||||||
| Prunetin | H | OCH3 | H | OH | OH | H | 4.99 ± 0.03 | 10 |
| Genistein 4′,7-dimethyl ether | H | OCH3 | H | OH | OCH3 | H | <4b | >100b |
| Genistin | H | Glucoside-O linkage | H | OH | OH | H | <4b | >100b |
‘Sigma’ and ‘Cayman’ refer to the commercial sources of daidzein.
<8% Inhibition at highest concentration tested (100 µM).
Prism analysis suggested that the maximal inhibition was slightly smaller (97%), but the difference between statistically demonstrable and pharmacologically relevant is such that we have assumed a maximal inhibition here.
For comparative purposes, the selective FAAH inhibitor URB597 inhibits the hydrolysis of 0.5 µM AEA at an assay pH of 7.0 with pI50 values of 7.76 ± 0.07 and 8.20 ± 0.07 following pre-incubation phases of 0 and 60 min, respectively (Holt et al., 2005).
Figure 1.
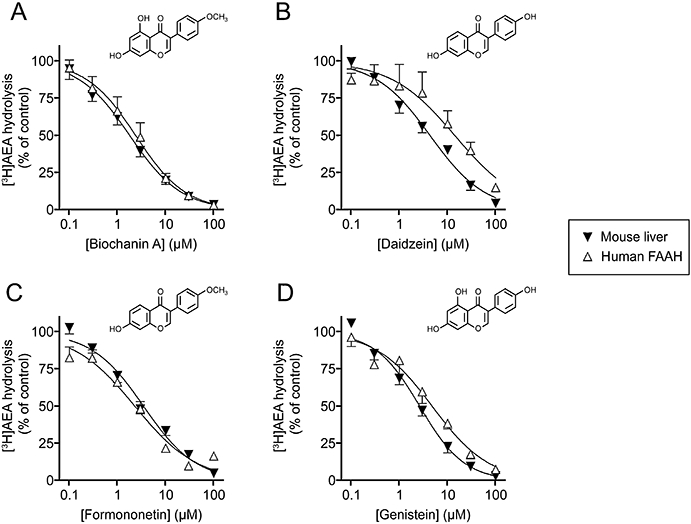
Inhibition of the FAAH-catalysed hydrolysis of 0.5 µM [3H]AEA in mouse liver homogenates and human recombinant FAAH-expressing COS7 cell membrane homogenates by (A) biochanin, (B) daidzein, (C) formononetin and (D) genistein. Shown are means and SEM. n= 9 (using three homogenates) and n= 4–5 for mouse liver and human FAAH, respectively.
Addition of a bulky group at either R1 or R2 results in a loss of actvity towards FAAH (puerarin vs. daidzein; genistin vs. genistein)
Methylation of the hydroxy group at R2 reduces activity (prunetin vs. genistein; genistein 4′, 7-dimethyl ether vs. biochanin A)
Inclusion of a hydroxyl group at R3 reduces activity (4′,6,7-trihydroxyflavone vs. daidzein)
The absence or presence of a hydroxy group at position R4 has little (formononetin vs. biochanin A) or no (daidzein vs. genistein) influence on the activity of the molecule towards FAAH
These data suggest that the main requirement for activity towards FAAH of the molecules is a free hydroxy group at position R2, and a free hydrogen substituent at position R3 of the chromone part of the molecule.
With respect to the phenyl substituent, the situation is less clear:
Addition of a hydroxy group at position R6 does not improve activity (3′,4′,7-trihydroxyflavone vs. daidzein)
Replacement of the hydroxy group at position R5 with a hydrogen group has little effect upon the activity (7-hydroxyisoflavone vs. daidzein)
Replacement of the hydroxy group at position R5 with a methoxy group increases activity when the molecule has a free hydroxy group at position R2 (formononetin vs. daidzein; biochanin A vs. genistein), but reduces it if the molecule has a methoxy group at position R2 (genistein 4′,7-dimethyl ether vs. prunetin).
In order to determine whether the inhibition of FAAH by biochanin A, daidzein, formononetin and genistein was species dependent, their effects upon the FAAH-catalysed hydrolysis of AEA in homogenates of mouse liver and in membranes from COS7 cells transfected with human FAAH were investigated (Figure 1). The pI50 and corresponding IC50 values for the compounds are summarized in Table 2, together with the data for rat brain (and in the case of daidzein, rat liver, for comparative purposes). For biochanin A and genistein, there was a reasonably consistent inhibitory potency across the species, while for daidzein (mouse = rat > human) and formononetin (rat > mouse = human) there were slight species differences (rank orders given for each compound in brackets). Biochanin A was shown further to act as a mixed-type inhibitor of the rat enzyme (i.e. to affect both Kmapp and Vmaxapp), with Kislope and Kiintercept values of 1.1 and 8.2 µM, respectively (Figure 2).
Table 2.
Comparison of the inhibition of mouse, rat and human FAAH by biochanin A, daidzein, formononetin and genistein
| Compound |
pI50 Value (IC50 value, µM): |
||
|---|---|---|---|
| Mouse liver homogenates | Rat brain (b) or liver (l) homogenates | Human FAAH-transfected COS7 cell membranes | |
| Biochanin A | 5.74 ± 0.05 (1.8) | 5.85 ± 0.02 (1.4) (b) | 5.62 ± 0.07 (2.4) |
| 6.03 ± 0.06a (0.93) (b) | |||
| Daidzein | 5.36 ± 0.05 (4.4) | 5.60 ± 0.02a (2.5) (b) | 4.85 ± 0.14 (14) |
| 5.36 ± 0.01a (4.4) (l) | |||
| Formononetin | 5.46 ± 0.05 (3.5) | 6.25 ± 0.02a (0.57) (b) | 5.64 ± 0.07 (2.3) |
| Genistein | 5.58 ± 0.04 (2.7) | 5.51 ± 0.05 (3.1) (b) | 5.32 ± 0.07 (4.8) |
| 5.66 ± 0.05a (2.2) (b) | |||
For the mouse brain and human FAAH samples, the compounds were dissolved in ethanol. Rat brain samples were dissolved in either ethanol or
DMSO. The values for rat brain are the same as those given in Table 1, and are inluded here for comparative purposes.
Figure 2.
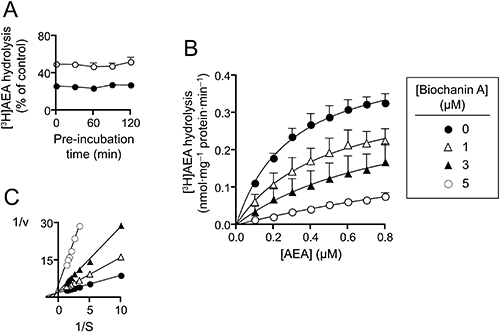
(A) Lack of effect of a pre-incubation phase upon the inhibition of FAAH by 1 µM (unfilled symbols) and 3 µM (filled symbols) biochanin A. Rat brain homogenates were incubated with biochanin A for the times shown prior to addition of 0.5 µM [3H]AEA and incubation for a further 10 min. Shown are means and SEM, n= 3. (B) Mode of inhibition of the FAAH-catalysed hydrolysis of [3H]AEA in rat brain homogenates by biochanin A. Shown are means and SEM, n= 3. (C) Double reciprocal plot of the mean data to illustrate the mixed-type nature of the inhibition. The Kmapp values calculated from the mean data by direct linear plots (Eisenthal and Cornish-Bowden, 1974) were 0.30, 0.56 and 0.86 µM for 0, 1 and 3 µM biochanin A respectively. The corresponding Vmaxapp values were 0.45, 0.41 and 0.33 nmol·(mg protein)−1·min−1 respectively. At 5 µM biochanin A, the Kmapp >>the highest AEA concentration used, and was thus not considered sufficiently reliable. The Kislope and Kiintercept values given in the text were in consequence calculated from the Kmapp and Vmaxapp values for 0, 1 and 3 µM biochanin A.
Effect of biochanin A upon FAAH-2 activity
FAAH-2 is a second N-acylethanolamine-hydrolyzing enzyme sharing a ∼20% sequence identity with FAAH, and which is found in primates, but not in rats or mice (Wei et al., 2006). The effect of biochanin A towards the activity of FAAH-2 was investigated using 16 nM [3H]oleoylethanolamine as substrate. The FAAH-transfected membranes were also tested for comparative purposes (Figure 3). As expected, FAAH was inhibited by biochanin A with a pI50 value of 6.21 ± 0.02, corresponding to an IC50 value of 0.62 µM. In these experiments, the contribution to the total activity by the cells per se (i.e. the mock transfectants) was very low. The catalytic activity of the membranes from FAAH-2-transfected cells, however, was also low. Hence, at the higher protein concentration needed (compared to the situation for FAAH), there was a significant hydrolysis of [3H]OEA in the mock-transfected cell membranes. This basal hydrolysis was sensitive to biochanin A and so presumably represents the low level of FAAH native to the cells. The difference between the mock-transfected and the FAAH-transfected cells, used as an estimate of FAAH-2 activity, was not inhibited by biochanin A. Thus, this compound is selective for FAAH versus FAAH-2.
Figure 3.
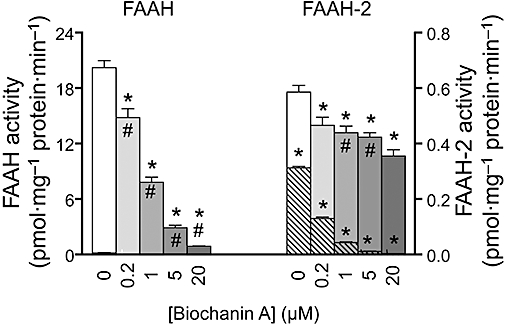
Inhibition by biochanin A of the hydrolysis of 16 nM [3H]oleoylethanolamide by membranes of COS7 cells transfected with either FAAH or FAAH-2. Values are means and SEM, n= 3, following an 18 h incubation of the membranes (0.05 and 2 µg per assay for FAAH and FAAH-2, respectively) with biochanin A and substrate at room temperature. Enclosed within the columns are the corresponding values for membranes from mock-transfected cells, used at the same protein concentrations. *Significantly different from the corresponding values in the abence of biochanin A, Dunnett's post hoc test following significant one-way anova for repeated measures. #Indicates significance for the net activities (i.e. calculated as the activity for the enzyme-transfected cells minus the corresponding activity for the mock-transfected cells).
Inhibition of cellular processing of AEA in intact cells by genistein, daidzein, biochanin A and equol
We have previously shown that neither genistein nor daidzein affects the uptake of AEA into cells either lacking FAAH, or after inhibition of the enzyme with URB597, whereas the FAAH-driven uptake into cells is inhibited by these compounds (Thors et al., 2007b). Here, we incubated rat RBL2H3 basophilic leukaemia cells in suspension with [3H]AEA labelled in the arachidonate part of the molecule, and measured the retention of tritium label in the membranes following incubation. Following FAAH-catalysed hydrolysis in these cells, the arachidonic acid is recycled to the cell plasma membrane (McFarland et al., 2004). The tritium accumulation for the cells in the absence (i.e. FAAH intact) and presence of 0.1 µM URB597 (a concentration producing a complete inhibition of FAAH) was 0.50 ± 0.03 and 0.14 ± 0.02 pmol per well respectively. The corresponding value for wells alone (no URB597) was 0.07 ± 0.01 pmol per well. A compound with FAAH inhibitory properties (or a compound preventing the recycling of the arachidonic acid produced by hydrolysis of AEA to the membrane) should behave like URB597 in this assay. This can easily be quantitated by measuring the retention of tritium by the membranes in the absence or presence of the test compound, and comparing it with the value seen with URB597. For example, if the values produced: (i) in the absence of compound; (ii) in the presence of a given concentration of test compound; and (iii) in the presence of 0.1 µM URB597 are 0.50, 0.32 and 0.14 pmol per well, respectively, then it can be seen that reduction of the tritium retention produced by the test compound, at the concentration used, is half of that seen with URB597. In other words, inhibition of the URB597-sensitive tritium labelling is a measure of the ability of the compounds to inhibit FAAH and/or the recycling of the arachidonate to the membrane in the intact cells (Thors et al., 2008; Thors and Fowler, 2009). Biochanin A, daidzein and genistein produced significant inhibition of the URB597-sensitive tritium retention at high nanomolar–low micromolar concentrations (Figure 4). The daidzein metabolite equol (structure shown in Figure 4), which was found to inhibit rat brain FAAH with a pI50 value of 5.26 ± 0.02 (EC50 value 5.5 µM) (data not shown), was also effective in the intact cells (Figure 4).
Figure 4.
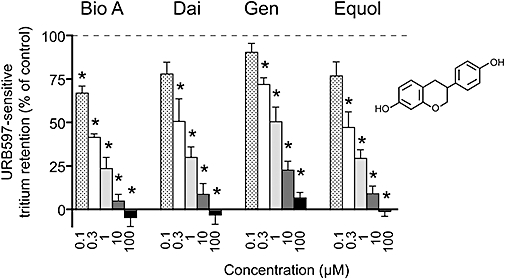
Inhibition by biochanin A (Bio A), daidzein (Dai), genistein (Gen) and equol of the URB597 (0.1 µM)-sensitive retention of tritium in RBL2H3 rat basophilic cell membranes following pre-incubation and incubation of the intact cells with 0.1 µM [3H]AEA (labelled in the arachidonate part of the molecule) for 15 and 5 min, respectively, prior to the filtration and washing phase of the assay (see Materials and Methods). Shown are means and SEM, n= 3–6. *Indicates where both 95% confidence levels were below 100%. The pI50 values (with IC50 values in brackets) were: biochanin A, 6.65 ± 0.07 (0.22 µM); daidzein, 6.43 ± 0.10 (0.37 µM); genistein, 5.88 ± 0.09 (1.3 µM); and equol, 6.47 ± 0.08 (3.4 µM). The structure of equol is shown on the right to aid the reader.
Interaction of biochanin A, daidzein, formononetin and genistein with CB receptors
The ability of the four isoflavones to inhibit the binding of the CB receptor agonist ligand [3H]CP 55940 to brain CB1 receptors and recombinant CB2 receptors is shown in Figure 5. Although the compounds could produce significant reductions in the specific binding of [3H]CP 55940 to both receptors, these effects were modest. Thus, the maximum inhibition found was 27 ± 7 and 33 ± 5% for brain CB1 receptors and recombinant CB2 receptors, respectively, produced in both cases with 30 µM biochanin A.
Figure 5.
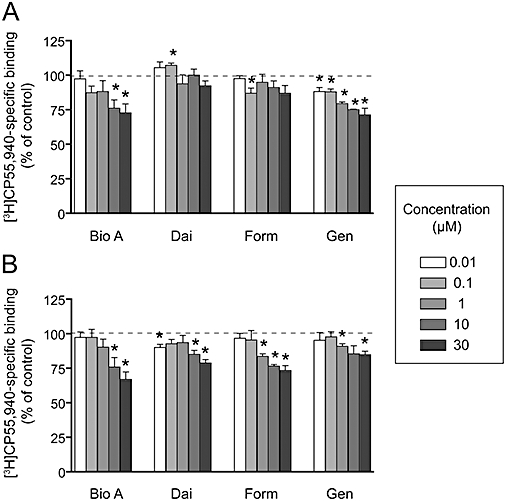
Effects of biochanin A (Bio A), daidzein (Dai), formononetin (Form) and genistein (Gen) upon the specific binding of 0.7 nM [3H]CP 55940 to A, mouse brain membranes (CB1 receptors) and B, membranes from hCB2-CHO cells. Data are expressed as a percentage of the specific binding in the presence of vehicle alone (0.01% DMSO). Shown are means and SEM, n= 4–8. *Indicates that both 95% confidence levels were either above (for 0.1 µM daidzein) or below 100% (where 100% means no effect of the test compound on [3H]CP 55940 specific binding).
Effects of i.pl. URB597 and biochanin A upon formalin-induced phosphorylation of ERK in the spinal cord
The i.pl. administration of formalin into the hindpaw of pentobarbital-anaesthetized mice produced a rapid phosphorylation of ERK in L4-S1 lumbar spinal cord ipsi-, but not contralateral to the injection (Figure 6A–C), consistent with data in awake animals (Karim et al., 2001). This phosphorylation was reduced by the i.pl. administration of 100, but not 30 or 10, µg of the selective FAAH inhibitor URB597. The effect of 100 µg URB597 was significantly inhibited by concomitant administration of the CB1 receptor antagonist/inverse agonist AM251 (30 µg). (Figure 6D). Biochanin A was tested at doses of 30, 100 and 300 µg. The highest dose also reduced formalin-induced ERK phosphorylation in a manner antagonized by AM251 (Figure 6E). Thus, biochanin A behaved like URB597 after local administration to the paw.
Figure 6.
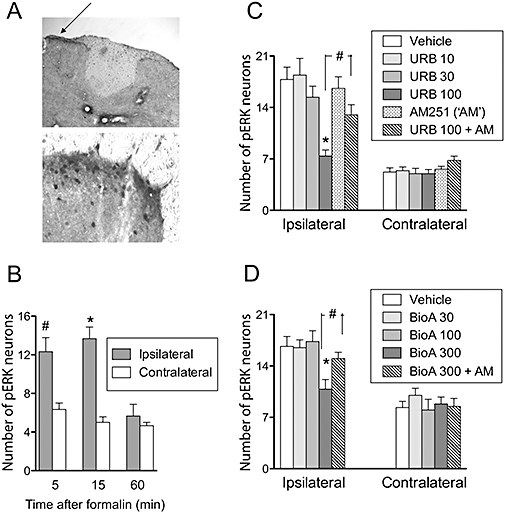
The i.pl. administration of formalin to pentobarbital-anaesthetized mice produces phosphorylation of ERK ipsi-, but not contralaterally, and in a manner reduced by the FAAH inhibitor URB597 and by biochanin A. (A) Top: magnification 10×, the pERK is indicated with the arrow; bottom: a higher magnification (40×). No immunoreactivity was seen for samples incubated with the secondary, but not the primary, antibody. (B) Time-course of the pERK response to formalin. Means and SEM, n= 3. *P < 0.05, #P= 0.06, two-tailed paired t-test versus the corresponding contralateral value. (C,D) Number of pERK-positive neurons produced by the formalin in the mice after i.pl. administration of URB597 (‘URB’, C) or Biochanin A (‘BioA’, D) per se or together with the CB1 receptor antagonist/inverse agonist AM251 (30 µg/paw). Doses are given as µg/paw, and the data are means and SEM, n= 5 (C) or n= 6 (D). Significance levels (*P < 0.01; #P < 0.05) are versus vehicle or for the comparison indicated and were determined by post hoc tests (Dunnetts and Neumann–Keul's multiple comparison, respectively) following significant one-way anova (P= 0.0006 and P= 0.0071 for the ipsilateral paws for C and D respectively). The anova values for the contralateral paws did not reach significance (P≥ 0.24).
Behavioural effects of i.v. biochanin A alone or in combination with AEA
The compounds were given either together with different doses of AEA. As expected, AEA produced dose-dependent effects upon all four components of the tetrad test (locomotion, nociception, body temperature and ring immobility). Biochanin A (15 mg·kg−1 i.v.) was without effects on its own, but significantly potentiated the effects of AEA (10 mg·kg−1 i.v.) (Figure 7). The AEA levels in the brains of the biochanin A-treated animals were not affected (Figure 8). Levels of other N-acylethanolamines were not measured. Daidzein (10 mg·kg−1 i.p.) was also tested. In this case, however, there was a higher level of behavioural response to AEA alone, and no significant potentiation could be seen, although the combination of AEA + daidzein produced results similar to those seen with biochanin + AEA (data not shown). The experiments were undertaken on different occasions (chronologically, daidzein was the first, and the results with this compound led to the choice of a different route of administration for biochanin A).
Figure 7.
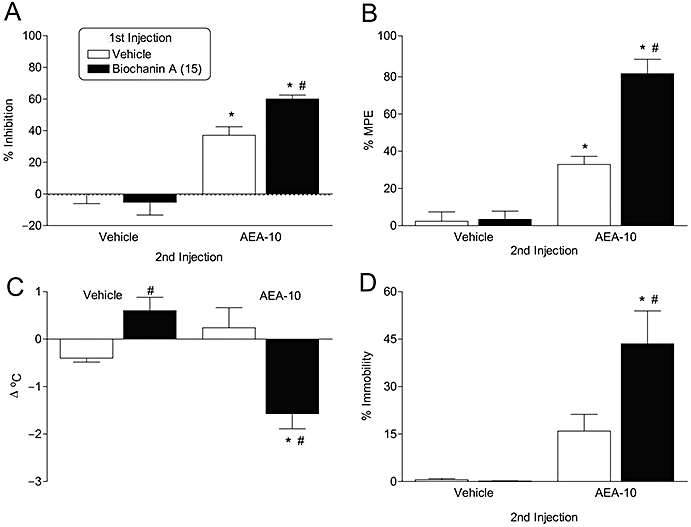
(A) Locomotor activity, (B) antinociception, (C) body temperature and (D) ring immobility of ICR mice following i.v. treatment with either vehicle or biochanin A (15 mg kg−1) (shown in the figure as ‘1st injection’) followed by i.v. administration of either vehicle or AEA (10 mg kg−1) (shown in the figure as ‘2nd injection’). Shown are means and SEM, n= 6. *Indicates significant difference from respective vehicle injection (P < 0.05). # indicates significant effect of biochanin A alone (V/V vs. B/V) or in combination with AEA (V/AEA vs. B/AEA) [P < 0.05].
Figure 8.
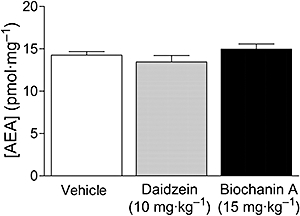
AEA levels in the brains of mice following either vehicle, biochanin A (15 mg·kg−1 i.v.) or daidzein (10 mg·kg−1 i.p.) treatment. The animals were first investigated behaviourally following a second vehicle injection (see Figure 7 for details) and killed 60 min after the isoflavone administration. Shown are means ± SEM, n= 6 each for daidzein and biochanin A and 12 for vehicle. There were no significant differences between the groups (P > 0.05, one-way anova).
Discussion
In the present study, the ability of a series of isoflavones related to genistein and daidzein has been investigated with respect to their effects upon FAAH. Of the compounds tested, the isoflavone biochanin A was chosen for further study. Biochanin A was a mixed-type inhibitor of the rat FAAH, and showed similar low micromolar potencies towards rat, mouse and recombinant human FAAH. In intact RBL2H3 cells, 1 µM biochanin A produced a large (∼75%) decrease in the URB597-sensitive accumulation of tritium label following incubation of the cells with [3H]AEA (labelled in the arachidonoyl portion of the molecule), consistent with inhibition of FAAH in these cells. In contrast, biochanin A had modest effects upon the binding of [3H]CP 55940 to CB1 and CB2 receptors.
In the formalin model of persistent pain, the behavioural responses (such as flinching, licking and biting of the paw) to the i.pl. injection of formalin are relatively rapid and follow two phases: an initial phase within seconds of the injection due to direct stimulation of nociceptors followed, after a short lag, by a second phase, which is the result of a central nociceptive sensitization. The second phase of the formalin test is sensitive to FAAH inhibitors (Lichtman et al., 2004a; Maione et al., 2007; Sit et al., 2007) and to genetic ablation of the enzyme (Lichtman et al., 2004b). There is good evidence that the expression of pERK in L4-S1 lumbar spinal cord is a useful marker of the activation of central nociceptive sensitization, such as is seen after the administration of formalin to the hindpaw (review, see Gao and Ji, 2009). Thus, pERK expression is induced by noxious, but not by innocuous, stimuli to the hindpaws of anaesthetized rats (Ji et al., 1999), and in formalin-treated mice and rats, the expression pattern follows the same time-course as the second phase of nocifensive behaviour (Ji et al., 1999; Karim et al., 2001). Importantly, the second phase of nocifensive behaviour in both species is blocked by PD98059, a MEK kinase inhibitor that prevents the phosphorylation of ERK (Ji et al., 1999; Karim et al., 2001). In spinal cord slices, the CB1 receptor agonist arachidonoyl-2′-chlorethylamide reduces the pERK expression produced by capsaicin (Kawasaki et al., 2006). The latter finding would suggest that this pathway may be responsive to an indirect activation of CB1 receptors secondary to FAAH inhibition. We found that the formalin-induced phosphorylation of spinal ERK ipsi-, but not contralaterally to the injection, was indeed inhibited by URB597 in the mouse strain used (C57BL/6, the strain used by Karim et al., 2001). The effective dose of URB597 is in the same range as found previously to produce effects in other pain models, albeit in the rat rather than the mouse (Jhaveri et al., 2006; Desroches et al., 2008; Sagar et al., 2008). AM251 given concomitantly produced a large reduction of the effect of URB597, indicating that the response to URB597 is mediated at least in part by CB1 receptors. Biochanin A produced the same pattern of AM251-sensitive inhibition of spinal ERK phosphorylation as seen with URB597, consistent with a local FAAH inhibition in vivo. The small difference in the relative potencies of biochanin A and URB597 (as compared with the large difference in vitro, where URB597 inhibits FAAH in the low nanomolar range; Kathuria et al., 2003) may reflect the reduced local pH of the inflamed tissue (Häbler, 1929), because URB597 inhibits FAAH in intact cells in a pH-sensitive manner (Paylor et al., 2006).
While the present data show that in anaesthetized C57BL/6 mice, biochanin A and URB597 produced the same effects upon spinal cord ERK phosphorylation in response to formalin, and that both are antagonized by the CB1 receptor inverse agonist AM251, the data do not rule out the involvement of other pathways. Although a detailed treatment is outside the scope of the present investigation, several potential pathways can be considered, given both the ability of AEA to interact with other receptors (such as CB2, PPARγ and TRPV1), and that the inhibition of FAAH will produce changes in the levels of other endogenous substrates such as palmitoylethanolamide, which have their own cellular targets, such as PPARα (for reviews, see Ross, 2003; O'Sullivan, 2007). Preliminary data with the PPARγ antagonist GW9662 suggest that this PPAR isoform does not contribute to any obvious extent in the effects of biochanin A in the pERK model (L. Thors, unpubl. data), but further studies are needed to determine the extent to which these other pathways can contribute to the effects of biochanin A on ERK phosphorylation in response to formalin.
When given i.v. to ICR mice at a dose of 15 mg·kg−1, biochanin A was without effect on brain levels of AEA, but produced a potentiation of the behavioural effects of AEA in the tetrad test. Given that i.v. administered AEA will be subject to both peripheral and central metabolism by FAAH, the data are consistent with the hypothesis that the behavioural effects in the ICR mice are due to a reduced peripheral FAAH activity. Certainly, the centrally permeable (albeit non-selective) FAAH inhibitor phenylmethylsulphonyl fluoride produces a very robust potentiation of AEA in the tetrad test in ICR mice (Compton and Martin, 1997), compared to the more modest effects of biochanin A seen here. With respect to pharmacokinetic studies of isoflavones, biochanin A is converted to genistein followed by conjugation in vivo (Setchell et al., 2001; Moon et al., 2006), and although genistein can be detected in the rat brain after administration of 30 mg·kg−1 i.v. (but not after 10 mg·kg−1 i.v.), the brain-to-blood-distribution ratio is very low (0.04 ± 0.01) (Tsai, 2005). Yueh and Chu (1977) reported that 15 min after i.v. administration of 40 mg·kg−1 of daidzein to rats, plasma concentrations were ∼30 µg·mL−1 (corresponding to ∼120 µM). The liver, lung and kidney concentrations of daidzein were ∼0.1 µmol·(mg wet weight)−1, while the brain concentration was about fivefold lower (Yueh and Chu, 1977).
Given that biochanin A is a naturally occurring compound, an obvious issue is dietary isoflavone consumption. Although the study was not primarily aimed at considering the effects of normal consumption of isoflavones upon FAAH activity, discussion of this is unavoidable given the suggested (and questioned) health benefits of dietary isoflavones in atherosclerosis, and breast and prostate cancer (Sirtori et al., 2005; Messina and Wood, 2008). In the case of genistein and daidzein, inhibition of FAAH was deemed possible, at least in individuals with a high soy intake (Morton et al., 2002), on the basis of the potencies towards the rodent enzyme (Thors et al., 2007a,b;). Daidzein is less potent towards the recombinant human FAAH (Table 1), but the suggestion still holds for genistein. Biochanin A and formononetin are not present in soy products, but are found in red clover extracts, which are available in health food stores for their purported effects in menopause (review see Booth et al., 2006). In a study where volunteers were given 80 mg of red clover isoflavone extracts (a double-strength tablet) per day for 2 weeks, maximum plasma concentrations of biochanin A and formononetin were 48 (range 18–80) and 11 (range <5–35) ng·mL−1, respectively, corresponding to ∼170 and ∼40 nM, respectively (Howes et al., 2002). The peak plasma genistein and daidzein concentrations were 114 and 63 ng·mL−1, respectively, corresponding to ∼420 and ∼250 nM, respectively (Howes et al., 2002). Thus, in contrast to the situation for individuals with a high soy intake, it is rather unlikely that sufficiently high isoflavone concentrations are achieved for inhibition of FAAH to occur after ingestion of red clover extracts.
In conclusion, the present study has demonstrated that biochanin A, a naturally occuring compound with limited central penetration, inhibits FAAH in a mixed-type manner, does not affect FAAH-2 activity and has only modest effects upon CB receptors. In vivo, the compound can inhibit a biochemical correlate of peripheral pain sensitization, spinal cord ERK phosphorylation following i.pl. formalin treatment to anaesthetized mice. FAAH inhibitors are currently under drug development as potential analgesic (and antidepressant) agents, and the leading compounds are in early clinical development (review, see Seierstad and Breitenbucher, 2008). Although these compounds do not produce ‘cannabinoid-like’ behaviours in vivo, other behavioural effects of central FAAH inhibition may be overlooked, and there is evidence that polymorphisms of FAAH are associated with certain traits, such as drug-seeking behaviour (Flanagan et al., 2006). In this respect, both genetic ablation of FAAH and the administration of URB597 affect ethanol preference and consumption (Basavarajappa et al., 2006; Blednov et al., 2007). FAAH−/− mice also show an increased susceptibililty to chemically induced seizures compared to wild-type animals (Clement et al., 2003). Given that i.pl. administration of AEA and URB597 produced effects in several pain models (Calignano et al., 2001; Sokal et al., 2003; Guindon et al., 2006; Jhaveri et al., 2006; Desroches et al., 2008; Khasabova et al., 2008; Sagar et al., 2008), a case can be made for the identification of peripherally restricted FAAH inhibitors as novel analgesic agents. The data presented here, together with previously published pharmacokinetic studies, suggest that biochanin A acts primarily as a peripheral inhibitor of FAAH. This raises the possibility that the compound may be useful as a template for the design of more potent, peripherally restricted compounds. Of course, the isoflavones are a class of compounds with a variety of biochemical and pharmacological effects. Such properties include actions upon oestrogen receptors, tyrosine kinase, peroxisome proliferator-activated receptor isoforms, DNA topoisomerase II, NFκB activation and transforming growth factor β1 signalling, among others (reviews, see Kurzer and Xu, 1997; Kim et al., 1998; Banerjee et al., 2008; see also Lam et al., 2004; Shen et al., 2006), and thus the isoflavones are certainly a long way from being FAAH-selective compounds. However, a screening programme could incorporate screens to reduce unwanted actions (such as effects upon oestrogen receptors) to tailor more selective FAAH inhibitors, or to design compounds with actions upon this enzyme and other potentially useful targets, such as, for example, cyclooxygenase-2 (see Fowler et al., 2009, for discussion).
Acknowledgments
The authors would like to thank Britt Jacobsson, Eva Hallin and Lesley Stevenson for excellent technical assistance. This work was supported by grants from the JC Kempe Memorial Foundation (to L.T.), the Swedish Research Council (grant no. 12158, medicine; to C.J.F.), the Research Funds of the Medical Faculty, Umeå University (to C.J.F.), the National Institute on Drug Abuse (grant nos. DA-03672 and DA-09789 to J.L.W., B.F.C., R.G.P. and R.A.R.).
Glossary
Abbreviations:
- AEA
anandamide
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- CB
cannabinoid
- CP 55940
(–)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)-phenyl]-trans-4-(3-hydroxy-propyl)-cyclohexanol
- ERK
extracellular signal-regulated kinase
- FAAH
fatty acid amide hydrolase
- GW9662
2-chloro-5-nitrobenzanilide
- NGST
normal goat serum in 0.1 M phosphate-buffered saline contaning Triton X-100
- OEA
oleoylethanolamide
- OL-135
1-oxo-1-[5-(2-pyridyl)-2-yl]-7-phenylheptane
- PD98059
2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one
- pERK
phosphorylated ERK
- PPAR
peroxisome proliferator-activated receptor
- URB597
cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester
Conflicts of interest
The authors state no conflicts of interests with respect to the work presented here.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 3rd edition (2008 revision) Br J Pharmacol. 2008;153(Suppl 2):S1–S209. doi: 10.1038/sj.bjp.0707746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee S, Li Y, Wang Z, Sarkar FH. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008;269:226–242. doi: 10.1016/j.canlet.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basavarajappa BS, Yalamanchili R, Cravatt BF, Cooper TB, Hungund BL. Increased ethanol consumption and preference and decreased ethanol sensitivity in female FAAH knockout mice. Neuropharmacology. 2006;5:834–844. doi: 10.1016/j.neuropharm.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Cravatt BF, Boehm SL, II, Walker D, Harris RA. Role of endocannabinoids in alcohol consumption and intoxication: studies of mice lacking fatty acid amide hydrolase. Neuropsychopharmacology. 2007;32:1570–1582. doi: 10.1038/sj.npp.1301274. [DOI] [PubMed] [Google Scholar]
- Boldrup L, Wilson SJ, Barbier AJ, Fowler CJ. A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J Biochem Biophys Methods. 2004;60:171–177. doi: 10.1016/j.jbbm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Booth NL, Piersen CE, Banuvar S, Geller SE, Shulman LP, Farnsworth NR. Clinical studies of red clover (Trifolium pratense) dietary supplements in menopause: a literature review. Menopause. 2006;13:251–264. doi: 10.1097/01.gme.0000198297.40269.f7. [DOI] [PubMed] [Google Scholar]
- Burston JJ, Sim-Selley LJ, Harloe JP, Mahadevan A, Razdan RK, Selley DE, et al. N-arachidonyl maleimide potentiates the pharmacological and biochemical effects of the endocannabinoid 2-arachidonylglycerol through inhibition of monoacylglycerol lipase. J Pharmacol Exp Ther. 2008;327:546–553. doi: 10.1124/jpet.108.141382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Piomelli D. Antinociceptive activity of the endogenous fatty acid amide, palmitylethanolamide. Eur J Pharmacol. 2001;419:191–198. doi: 10.1016/s0014-2999(01)00988-8. [DOI] [PubMed] [Google Scholar]
- Chang L, Luo L, Palmer JA, Sutton S, Wilson SJ, Barbier AJ, et al. Inhibition of fatty acid amide hydrolase produces analgesia by multiple mechanisms. Br J Pharmacol. 2006;148:102–113. doi: 10.1038/sj.bjp.0706699. Corrigendum published in Br J Pharmacol 148: 114 (2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement AB, Hawkins EG, Lichtman AH, Cravatt BF. Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Compton DR, Martin BR. The effect of the enzyme inhibitor phenylmethylsulfonyl fluoride on the pharmacological effect of anandamide in the mouse model of cannabimimetic activity. J Pharmacol Exp Ther. 1997;283:1138–1143. [PubMed] [Google Scholar]
- Desroches J, Guindon J, Lambert C, Beaulieu P. Modulation of the anti-nociceptive effects of 2-arachidonoyl glycerol by peripherally administered FAAH and MGL inhibitors in a neuropathic pain model. Br J Pharmacol. 2008;155:913–924. doi: 10.1038/bjp.2008.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenthal R, Cornish-Bowden A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem J. 1974;139:715–720. doi: 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JM, Gerber AL, Cadet JL, Beutler E, Sipe JC. The fatty acid amide hydrolase 385 A/A (P129T) variant: haplotype analysis of an ancient missense mutation and validation of risk for drug addiction. Hum Genet. 2006;120:581–588. doi: 10.1007/s00439-006-0250-x. [DOI] [PubMed] [Google Scholar]
- Fowler CJ, Naidu PS, Lichtman A, Onnis V. The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or transient receptor potential vanilloid type 1. Br J Pharmacol. 2009;156:412–419. doi: 10.1111/j.1476-5381.2008.00029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y-J, Ji R-R. c-Fos and pERK, which is a better marker for neuronal activation and central sensitization after noxious stimulation and tissue injury? Open Pain J. 2009;2:11–17. doi: 10.2174/1876386300902010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon J, De Léan A, Beaulieu P. Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain. 2006;121:85–93. doi: 10.1016/j.pain.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Häbler C. Über den K- und Ca-gehalt von eiter und exsudaten und seine beziehungen zum entzündungsschmerz. Klin Wochenschr. 1929;8:1569–1572. [Google Scholar]
- Hardison S, Weintraub ST, Giuffrida A. Quantification of endocannabinoids in rat biological samples by GC/MS: technical and theoretical considerations. Prostaglandins Other Lipid Mediat. 2006;81:106–112. doi: 10.1016/j.prostaglandins.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146:467–476. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes J, Waring M, Huang L, Howes LG. Long-term pharmacokinetics of an extract of isoflavones from red clover (Trifolium pratense) J Altern Complement Med. 2002;8:135–142. doi: 10.1089/107555302317371424. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jhaveri MD, Richardson D, Kendall DA, Barrett DA, Chapman V. Analgesic effects of fatty acid amide hydrolase inhibition in a rat model of neuropathic pain. J Neurosci. 2006;26:13318–13327. doi: 10.1523/JNEUROSCI.3326-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji R-R, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Karim F, Wang CC, Gereau RW., 4th Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–78. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Ji R-R. Different effects of opioid and cannabinoid receptor agonists on C-fiber-induced extracellular signal-regulated kinase activation in dorsal horn neurons in normal and spinal nerve-ligated rats. J Pharmacol Exp Ther. 2006;316:601–607. doi: 10.1124/jpet.105.093583. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Khasabov SG, Harding-Rose C, Coicou LG, Seybold BA, Lindberg AE, et al. A decrease in anandamide signaling contributes to the maintenance of cutaneous mechanical hyperalgesia in a model of bone cancer pain. J Neurosci. 2008;28:11141–11152. doi: 10.1523/JNEUROSCI.2847-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Peterson TG, Barnes S. Mechanisms of action of the soy isoflavone genistein: emerging role for its effects via transforming growth factor beta signaling pathways. Am J Clin Nutr. 1998;68:1418S–1425S. doi: 10.1093/ajcn/68.6.1418S. [DOI] [PubMed] [Google Scholar]
- Kingsley PJ, Marnett LJ. Analysis of endocannabinoids by Ag+ coordination tandem mass spectrometry. Anal Biochem. 2003;314:8–15. doi: 10.1016/s0003-2697(02)00643-7. [DOI] [PubMed] [Google Scholar]
- Kurzer MS, Xu X. Dietary phytoestrogens. Annu Rev Nutr. 1997;17:353–381. doi: 10.1146/annurev.nutr.17.1.353. [DOI] [PubMed] [Google Scholar]
- Lam AN, Demasi M, James MJ, Husband AJ, Walker C. Effect of red clover isoflavones on cox-2 activity in murine and human monocyte/macrophage cells. Nutr Cancer. 2004;49:89–93. doi: 10.1207/s15327914nc4901_12. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Leung D, Shelton C, Saghatelian A, Hardouin C, Boger D, et al. Reversible inhibitors of fatty acid amide hydrolase that promote analgesia: evidence for an unprecedented combination of potency and selectivity. J Pharmacol Exp Ther. 2004a;311:441–448. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- Lichtman AH, Shelton CC, Advani T, Cravatt BF. Mice lacking fatty acid amide hydrolase exhibit a cannabinoid receptor-mediated phenotypic hypoalgesia. Pain. 2004b;109:319–327. doi: 10.1016/j.pain.2004.01.022. [DOI] [PubMed] [Google Scholar]
- McFarland MJ, Porter AC, Rakhshan FR, Rawat DS, Gibbs RA, Barker EL. A role for caveolae/lipid rafts in the uptake and recycling of the endogenous cannabinoid anandamide. J Biol Chem. 2004;279:41991–41997. doi: 10.1074/jbc.M407250200. [DOI] [PubMed] [Google Scholar]
- Maione S, De Petrocellis L, de Novellis V, Moriello AS, Petrosino S, Palazzo E, et al. Analgesic actions of N-arachidonoyl-serotonin, a fatty acid amide hydrolase inhibitor with antagonistic activity at vanilloid TRPV1 receptors. Br J Pharmacol. 2007;150:766–781. doi: 10.1038/sj.bjp.0707145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin BR, Compton DR, Thomas BF, Prescott WR, Little PJ, Razdan RK, et al. Behavioral, biochemical, and molecular modeling evaluations of cannabinoid analogs. Pharmacol Biochem Behav. 1991;40:471–478. doi: 10.1016/0091-3057(91)90349-7. [DOI] [PubMed] [Google Scholar]
- Messina MJ, Wood CE. Soy isoflavones, estrogen therapy, and breast cancer risk: analysis and commentary. Nutr J. 2008;7:17. doi: 10.1186/1475-2891-7-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon YJ, Sagawa K, Frederick K, Zhang S, Morris ME. Pharmacokinetics and bioavailability of the isoflavone biochanin A in rats. AAPS J. 2006;8:E433–E442. doi: 10.1208/aapsj080351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton MS, Arisaka O, Miyake N, Morgan LD, Evans BA. Phytoestrogen concentrations in serum from Japanese men and women over forty years of age. J Nutr. 2002;132:3168–3171. doi: 10.1093/jn/131.10.3168. [DOI] [PubMed] [Google Scholar]
- Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paylor B, Holt S, Fowler CJ. The potency of the fatty acid amide hydrolase inhibitor URB597 is dependent upon the assay pH. Pharmacol Res. 2006;54:481–485. doi: 10.1016/j.phrs.2006.07.006. Corrigendum published in Pharmacol Res 55: 80 (2007. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The ring test: a quantitative method for assessing the ‘cataleptic’ effect of Cannabis in mice. Br J Pharmacol. 1972;46:753–763. doi: 10.1111/j.1476-5381.1972.tb06900.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Cannabinoid receptors and pain. Prog Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- Ross RA. Anandamide and vanilloid TRPV1 receptors. Br J Pharmacol. 2003;40:790–801. doi: 10.1038/sj.bjp.0705467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Stevenson LA, Murphy VL, Templeton F, Makriyannis A, et al. Agonist-inverse agonist characterization at CB1 and CB2 cannabinoid receptors of L759633, L759656 and AM630. Br J Pharmacol. 1999;126:665–672. doi: 10.1038/sj.bjp.0702351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar DR, Kendall DA, Chapman V. Inhibition of fatty acid amide hydrolase produces PPAR-α-mediated analgesia in a rat model of inflammatory pain. Br J Pharmacol. 2008;155:1297–1306. doi: 10.1038/bjp.2008.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seierstad M, Breitenbucher JG. Discovery and development of fatty acid amide hydrolase (FAAH) inhibitors. J Med Chem. 2008;51:7327–7343. doi: 10.1021/jm800311k. [DOI] [PubMed] [Google Scholar]
- Setchell KDR, Brown NM, Desai P, Zimmer-Nechemias L, Wolfe BE, Brashear WT, et al. Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J Nutr. 2001;131:1362S–1375S. doi: 10.1093/jn/131.4.1362S. [DOI] [PubMed] [Google Scholar]
- Shen P, Liu MH, Ng TY, Chan YH, Yong EL. Differential effects of isoflavones, from Astragalus membranaceus and Pueraria thomsonii, on the activation of PPARα, PPARγ, and adipocyte differentiation in vitro. J Nutr. 2006;136:899–905. doi: 10.1093/jn/136.4.899. [DOI] [PubMed] [Google Scholar]
- Sirtori CR, Arnoldi A, Johnson SK. Phytoestrogens: end of a tale? Ann Med. 2005;37:423–438. doi: 10.1080/07853890510044586. [DOI] [PubMed] [Google Scholar]
- Sit SY, Conway C, Bertekap R, Xie K, Bourin C, Burris K, et al. Novel inhibitors of fatty acid amide hydrolase. Bioorg Med Chem Lett. 2007;17:3287–3291. doi: 10.1016/j.bmcl.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Sokal DM, Elmes SJ, Kendall DA, Chapman V. Intraplantar injection of anandamide inhibits mechanically-evoked responses of spinal neurones via activation of CB2 receptors in anaesthetised rats. Neuropharmacology. 2003;45:404–411. doi: 10.1016/s0028-3908(03)00195-3. [DOI] [PubMed] [Google Scholar]
- Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, et al. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146:917–926. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thors L, Fowler CJ. Effect of nitric oxide donors on membrane tritium accumulation of endocannabinoids and related endogenous lipids. Eur J Pharmacol. 2009;621:10–18. doi: 10.1016/j.ejphar.2009.08.028. [DOI] [PubMed] [Google Scholar]
- Thors L, Alajakku K, Fowler CJ. The ‘specific’ tyrosine kinase inhibitor genistein inhibits the enzymic hydrolysis of anandamide: implications for anandamide uptake. Br J Pharmacol. 2007a;150:951–960. doi: 10.1038/sj.bjp.0707172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thors L, Eriksson J, Fowler CJ. Inhibition of the cellular uptake of anandamide by genistein and its analogue daidzein in cells with different levels of fatty acid amide hydrolase-driven uptake. Br J Pharmacol. 2007b;152:744–750. doi: 10.1038/sj.bjp.0707401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thors L, Belghiti M, Fowler CJ. Inhibition of fatty acid amide hydrolase by kaempferol and related naturally occurring flavonoids. Br J Pharmacol. 2008;155:244–252. doi: 10.1038/bjp.2008.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai TH. Concurrent measurement of unbound genistein in the blood, brain and bile of anesthetized rats using microdialysis and its pharmacokinetic application. J Chromatogr A. 2005;1073:317–322. doi: 10.1016/j.chroma.2004.10.048. [DOI] [PubMed] [Google Scholar]
- Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. A second fatty acid amide hydrolase with variable distribution among placental mammals. J Biol Chem. 2006;281:36569–36578. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- Yueh T-L, Chu H-Y. The metabolic fate of daidzein. Sci Sin. 1977;20:513–521. [PubMed] [Google Scholar]