

Abstract
Single-molecule force spectroscopy is providing unique, and sometimes unexpected, insights into the free-energy landscapes of proteins. Despite the complexity of the free-energy landscapes revealed by mechanical probes, forced unfolding experiments are often analyzed using one-dimensional models that predict a logarithmic dependence of the unfolding force on the pulling velocity. We previously found that the unfolding force of the protein filamin at low pulling speed did not decrease logarithmically with the pulling speed. Here we present results from a large number of unfolding simulations of a coarse-grain model of the protein filamin under a broad range of constant forces. These show that a two-path model is physically plausible and produces a deviation from the behavior predicted by one-dimensional models analogous to that observed experimentally. We also show that the analysis of the distributions of unfolding forces (p[F]) contains crucial and exploitable information, and that a proper description of the unfolding of single-domain proteins needs to account for the intrinsic multidimensionality of the underlying free-energy landscape, especially when the applied perturbation is small.
Introduction
Over the past decade, single-molecule atomic force microscopy studies have revealed the complexity of the folding and unfolding of single-domain proteins. Examples include the observation of parallel unfolding pathways (1, 2), unfolding intermediates (3), and compact collapsed structures along the folding pathway (4). Recently, the mechanical unfolding kinetics of the native to intermediate (N→I) transition of the protein filamin was characterized in a broad range of pulling velocities (1–4000 nm/s), and an unexpected plateau in the modal (i.e., most probable) unfolding force, F∗, was observed at low pulling velocities/loading rates (Fig. 1) (5). The convex curvature in the plot of F∗ as a function of ln[RF∗] represents a sharp departure from the behavior predominantly observed for simple two-state proteins where F∗ is generally assumed to vary linearly with the logarithm of the loading rate, ln[RF∗] (6). The linear relationship between F∗ and ln[RF∗] relies on the validity of the irreversible two-state Bell model (7), which states that the unfolding rate decreases exponentially with the applied force,
where ku(0) is the unfolding rate in the absence of force and xu is the force dependence of the unfolding barrier. However, if xu changes with the force (8, 9, 10, 11) or the refolding reaction is nonnegligible (12), the dependence of F∗ on ln[RF∗] will no longer be linear. A close inspection of the force traces of filamin showed no evidence of refolding or additional unfolding intermediates at low pulling velocities (5). In addition, using a two-state model that accounts for the force-dependence of xu (10) did not result in better agreement with the experimental data (5). Although anomalous curvatures are not uncommon in the mechanical unbinding of receptor-ligand complexes (13, 14, 15, 16), to our knowledge they have never been directly observed for the unfolding of single-domain proteins. One reason could be the highly cooperative unfolding kinetics of small, single-domain proteins. Previous simulation (17, 18, 19) and experimental (20) studies, however, have suggested that such anomalous kinetic signatures can, in principle, be observed for single-domain proteins.
Figure 1.
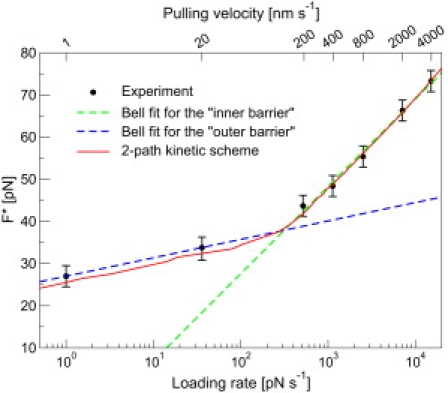
The modal unfolding force, F∗, as a function of the loading rate, RF∗, calculated at each F∗. The pulling velocities at which the F∗ were determined are indicated (top of the graph). The loading rates, RF∗, were calculated by modeling the polymeric spacer as a wormlike chain (see Methods for details). (Error bars) Bin-width used to construct the unfolding force histograms. (Dashed lines) Piecewise fits of the data at 200–4000 nm/s (dark dashed line/red online) and at 1–20 nm/s (light dashed line/green online) to (kBT/xin/out)ln(RF∗) + Cin/out), respectively. The force-dependencies are xF∗in = 4.6 Å and xF∗out = 21.6 Å for the high- and low-velocity regimes, respectively. (Continuous line/red online) Fit to the kinetic scheme shown at the bottom of Fig. 5.
The convex curvature is typically explained by the presence of two sequential barriers along the unfolding pathway of the protein (Fig. 2). At low pulling velocities, unfolding or unbinding occurs over the outer barrier, located far from the native state (Fig. 2 A), while at high pulling velocities, the outer barrier is sufficiently lowered by the force that the inner barrier becomes rate-limiting instead (Fig. 2 B). This sequential barriers (SB) model was first proposed by Merkel et al. (13), who used it to rationalize the mechanical unbinding kinetics of the streptavidin-biotin complex. If the free-energy landscape of the protein can be represented by a single reaction coordinate, the end-to-end extension, the SB model predicts that the plot of F∗ as a function of ln[RF∗] will consist of a series of quasilinear regimes that get increasingly steeper with the loading rate (Fig. 2, A and B). It can be shown that within each quasilinear regime (12, 21)
where xin/out can be thought of as the distance between the native minimum and the inner or outer barrier (Fig. 2), and Cin/out contains information about the heights of inner or outer barriers in the absence of force. Therefore, the steepness of each the quasilinear regime is inversely related to xin/out.
Figure 2.
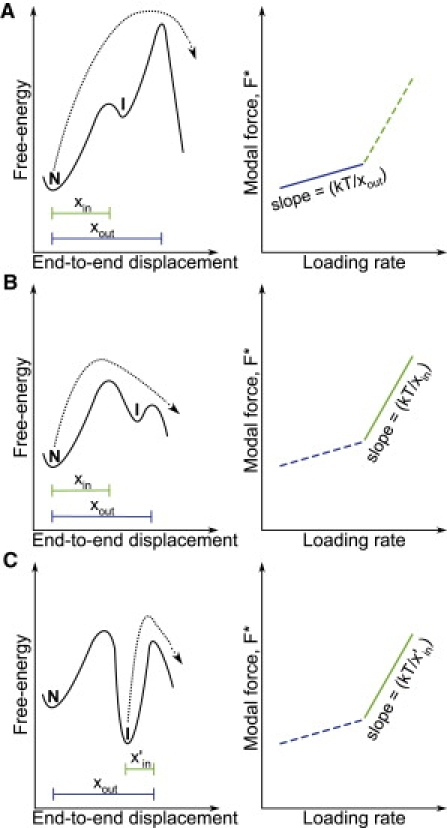
One-dimensional free-energy profiles with two sequential barriers. The reaction coordinate is typically taken to be the end-to-end extension of the protein, allowing the force-dependencies, xin/out, to be interpreted as the difference in extension between the native and transition states. The resulting force spectrum (i.e., the F∗ versus ln[RF∗] plot) is depicted for each free-energy profile (right, solid line). A key assumption is that the force and end-to-end vectors do not change direction throughout the unfolding transition. (A) At low loading rates, the unfolding of the native state (N) is impeded by the outer barrier, which is located furthest away from the native state (i.e., xout Å). (B) At sufficiently high loading rates, the height of the outer barrier becomes low enough such that the inner barrier, located xin Å away from the native state, becomes rate-limiting. This switch in transition barriers shows up as a sharp change in the slope of the force spectrum. (C) If the intermediate is stabilized by the force such that it becomes more stable than the native state, the outer barrier becomes rate-limiting again. In this case, the slope of the force spectrum at high loading rates will be governed by x′in, the distance between the intermediate state (I) and the outer transition state. Therefore, when x′in = xin, the change in the force spectrum at high loading rates could be misattributed to the unfolding of the native state over the inner barrier.
In contrast to the conventional explanation, the convex curvature observed for filamin was explained using a kinetic scheme with two unfolding pathways (see Fig. 5, denoted henceforth as the two-path kinetic scheme) (5). From Fig. 1, it can be seen that the SB model describes the RF∗-dependence of F∗ of filamin quite nicely (dashed lines), which begs the question: why use a more complex model when a more parsimonious one is able to adequately describe the data?
Figure 5.
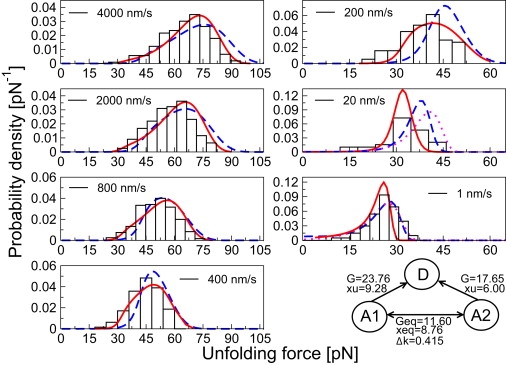
The distribution of unfolding forces at each pulling velocity (black histograms). (Continuous lines/red online) Fits to the kinetic scheme shown in the last panel (Geq and G are in units of kBT; xeq and xu are in units of Å; Δκ is in units of Å/pN). (Dashed lines/blue online) Fits to a kinetic model (denoted as the SB variant in the main text) where the pathway A1→D is absent (Geq = 7.45 kBT, xeq = 1.01 Å, xu = 4.2 Å, G = 15.75 kBT, and Δκ = 0.303 Å/pN). The dotted line (magenta online) at 1 nm/s is a fit to the unfolding force histogram using the rate equations of Bell (7) (k[0] = 2 × 10−4 s−1, xpfout = 11 Å). The dotted line at 1 nm/s cannot be seen clearly, because it overlaps perfectly with the dashed line. The dotted line (magenta online) at 20 nm/s is a fit to the unfolding force histogram calculated using the parameters determined at 1 nm/s.
Due to the difficulty in performing single-molecule mechanical unfolding experiments, the unfolding kinetics of a protein is often studied in a narrow range of pulling velocities, and/or with limited sampling within the range of the pulling velocities investigated. In addition, the measured unfolding forces are often noisy (e.g., ΔFrms ∼15–25 pN), which makes the accurate estimation of the distribution of unfolding forces, p(F), difficult. For this reason, the p(F) is less commonly analyzed and an analysis of F∗ is preferred instead. Analyzing only F∗, however, means that a large amount of the information (e.g., the variance and skew) contained in p(F) is neglected. Theoretical analyses of the forced unbinding of receptor-ligand complexes (17, 21) have shown that the variation of F∗ with ln[RF∗] is insufficient to discriminate between various kinetic models. For instance, even if the plot of F∗ as a function of ln[RF∗] can be described by the traditional SB model (13), the force-dependence at high pulling velocities need not correspond to the inner barrier that is encountered when unfolding from the native state; it can be equally consistent with the unfolding of a force stabilized intermediate state over the outer barrier (Fig. 2 C) (17, 21).
In this article, we present results from a large number of unfolding simulations of a coarse-grain model of the protein filamin under a broad range of constant forces. These show that a two-path model is physically plausible and produces a deviation from the behavior predicted by one-dimensional models, analogous to that observed experimentally. We also show that the analysis of the distributions of unfolding forces (p[F]) contains crucial and exploitable information, and that a proper description of the unfolding of single-domain proteins needs to account for the intrinsic multidimensionality of the underlying free-energy landscape, especially when the applied perturbation is small.
Methods
Molecular dynamics simulations
All simulations were performed using a structure-based coarse-grain model (22), where each residue is represented by a Cα atom and only native interactions are attractive. The polymeric properties of the chain are governed by an amino-acid-specific dihedral potential. The topology of the model and interaction parameters were based on the lowest energy NMR structure of filamin (PDB ID: 1KSR). The dynamics of the coarse-grain model were simulated using a Langevin integrator with a friction coefficient of 1 ps−1, a timestep of 15 fs, and holonomic constraints on all bonds. Constant forces ranging from 10 to 60 pN were applied to the N- and C-termini of the coarse-grain model. The simulations were performed at 330 K to ensure that good statistics were obtained at small forces (i.e., 10–20 pN). The unfolding time was defined as the time required for the N-C distance of the coarse-grain model to be >200 Å. The average unfolding time, τu, was computed using at least 1000 simulations at each force.
The τu as a function of force was fitted to the Dudko-Hummer-Szabo (DHS) rate equation (see Eq. 4 below), the Saffman-Delbrück (SB) model, and the two-path kinetic scheme (Fig. 3 B) by nonlinear least-squares. The SB model is
where τin/out(0) is the time needed to unfold over the inner (i.e., in) or outer (i.e., out) barrier at zero force, and xin/out represents the force-dependence of the height of the inner or outer barrier. For the two-path model, the unfolding time for each unfolding pathway (i.e., starting from either A1 or A2) was modeled using the Bell model (7),
where xu is the force-dependence of the unfolding time. The equilibrium between the states A1 and A2 was modeled assuming linear force-dependence of the relevant free-energy, i.e.,
where xeq is the force-dependence of the equilibrium free-energy.
Figure 3.
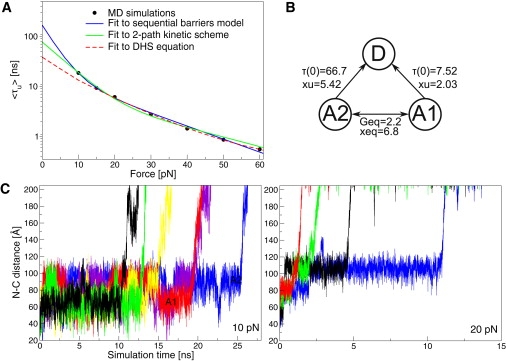
(A) The average unfolding time, τu, of a coarse-grain model of ddFLN4 as a function of the applied force at 330 K. (Dark-dashed line/red online) Fit of τu to the DHS equation, Eq. 4, over the entire force range. The DHS parameters are τDHS(0) = 38.5 ns, G‡ = 6.5 kBT, and xu = 4.92 Å. (Dark continuous line/blue online) Fit to the SB model. The parameters for the inner and outer barriers are τin(0) = 17.7 ns and xin = 2.7 Å; τout(0) = 150.8 ns and xout = 12.9 Å, respectively. (Light continuous line/green online) Fit to the kinetic scheme shown in panel B. At forces <30 pN, a significant convex curvature is observed. Please refer to Methods for details of the fitting procedure. (B) The kinetic scheme used to fit τu shown in panel A. The kinetic scheme was inspired by the rapid equilibrium between A1 (centered ∼70 Å) and A2 (centered ∼95 Å) observed in the time series seen in the time series in panels C and D. The τ(0) are in units of nanoseconds; Geq and G‡ are in units of kBT; and xu and xeq are in units of Å. (C) (Left) The time series of the N-C distance of the coarse-grain filamin model at 10 pN. Unfolding can be initiated either from A1 (red, black, and green online) or from A2 (yellow, blue, and purple online). (Right) The time series of the N-C distance of the coarse-grain filamin model at 20 pN. The fast switching between A1 and A2 is lost, and unfolding proceeds mainly from A2.
Fitting of the unfolding force distributions
There were 66, 34, 91, 107, 231, 149, and 199 unfolding force measurements at pulling velocities of 1–4000 nm/s, respectively. The distribution of unfolding forces, p(F), at each pulling velocity was determined by constructing a histogram of the measured unfolding forces using a bin-width of ∼4–5 pN. The bin-width was chosen according to the Freedman-Diaconis rule, i.e.,
where IQ is the interquartile range. The root-mean-square fluctuation in the unfolding force due to the fluctuations of the cantilever,
was of similar magnitude. A total of 56 data-points (i.e., eight points per pulling velocity) was used for the fitting procedure.
To fit the p(F) to the various kinetic models, we calculated the theoretical force distributions according to the Evans-Ritchie formalism (6),
| (1) |
where F is the force, ku(F) is the unfolding rate function that describes a particular kinetic model, and R(F, L, v) is the loading rate.
The loading rate, R(F, L, v) was calculated by modeling the polymeric spacer as a wormlike chain (23)
| (2) |
where β = 1/kBT; v is the pulling velocity (1–4000 nm/s); κc = 6 pN/nm is the cantilever spring constant; lp = 0.5 nm is the persistence length; and L values represent the contour lengths: 79 nm (200–4000 nm/s) (24), 98 nm (20 nm/s) (5), and 92 nm (1 nm/s) (5).
For the two-path kinetic scheme, ku(F) is given by
| (3) |
where kA1–D(F) and kA2–D(F) are the unfolding rate constants for each of the two pathways, and Keq(F) is the equilibrium constant between states A1 and A2.
The rate constants kA1–D(F) and kA2–D(F) were modeled using the DHS rate equation—specifically the cusp model (10),
| (4) |
where k(0) is the usual Kramer's unfolding rate in the absence of force, and G‡(0) and xu are the zero force barrier height and transition state distance, respectively. We assumed
thus reducing the DHS rate equation to a two-parameter model.
The equilibrium constant Keq(F) between the states A2 and A1 is given by
| (5) |
where
is the difference in the free-energies and extensions of A2 and A1 at zero force, respectively; and Δκ is a parameter that accounts for the force-dependence of the extensions of the states (8, 11).
The best-fit parameters for each kinetic model were determined by fitting the p(F) from all seven pulling velocities simultaneously (unless indicated otherwise). The best-fitting parameters were determined by minimizing the sum of squared deviation from the experimental p(F), and a visual inspection of the resulting fits. Parameter space was explored using a two-step procedure programmed in MATLAB (The MathWorks, Natick, MA):
-
Step 1.
A broad search of parameter space using a genetic algorithm; and
-
Step 2.
A fine optimization of the best-fitting parameters from Step 1 using the Nelder-Mead algorithm.
To make the search of parameter space more efficient, all parameters were constrained to be in the following bounds: 1–30 kBT for free-energies, and 1–30 Å for xu and xeq.
Results and Discussion
Molecular dynamics simulations as a proof of principle
The pitfalls of solely relying on the relationship between F∗ and ln[RF∗] to extract the features of the free-energy landscapes of proteins are illustrated in this section by molecular dynamics simulations of a coarse-grain model of filamin. In Fig. 3 A, the average unfolding time, τu, is shown as a function of the applied force. It is clear that the unfolding kinetics of the coarse-grain model exhibits two distinct regimes: the force-dependence of τu is greater at small forces (10–20 pN) than at larger forces (30–60 pN). This is analogous to the convex curvature observed for filamin experimentally (Fig. 1), because the force-dependence of τu and the dependence of F∗ on RF∗ are related through the parameter
It should be noted that the steep curvature at low forces cannot be accounted for by the DHS equation, i.e., Eq. 4 (Fig. 3 A, dashed line/red online). As will be clear later, the main reason for the convex curvature is the complex multistate unfolding kinetics of the filamin model. To clearly illustrate the underlying physical picture, we will use the Bell model as a simple parameterization of the rate constant in the rest of the simulation analysis.
As shown in Fig. 3 A (dark continuous line/blue online), the simulation results can be reproduced by the SB model. Inspection of the time series of the N-C distance at 10 pN (Fig. 3 C, left) and 20 pN (Fig. 3 C, right), however, reveal that the SB model is not consistent with the unfolding mechanism of the coarse-grain model. The time series at 10 pN shows that before unfolding, the system equilibrates rapidly between two states, A1 and A2, centered at N-C distances of ∼70 Å and 95 Å, respectively. Full unfolding then proceeds from either A1 or A2. At 20 pN, the fast interconversion between A1 and A2 is lost and unfolding occurs predominantly from the more extended state, A2. There are thus two parallel unfolding pathways available to the system in the small force regime (Fig. 3 C and Fig. 4). In contrast, the SB model predicts that the system will only unfold via one pathway in the small force regime (Fig. 2). Taken together, the two sets of time series suggest that the unfolding kinetics of the system can be explained by a three-state kinetic scheme with parallel unfolding pathways (Fig. 3 B). The kinetic scheme is able to reproduce the force-dependence of τu relatively well (Fig. 3 A, light continuous line/green online), but with rate parameters (i.e., τu(0) and xu) that are distinctly different from those emerging from the SB model. It should be noted that without the insights provided by the simulations, the SB model would have been deemed the superior model due to 1), its parsimony—it only requires four parameters compared to six for the two-path kinetic scheme; and 2), its slightly better overall fit to τu.
Figure 4.
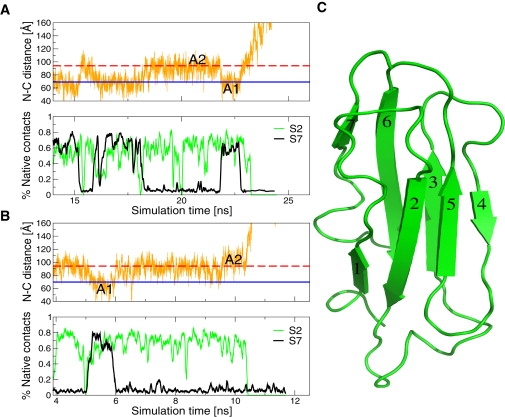
The time series of the fraction of native contacts between pairs of neighboring strands at 10 pN. (Light/green online and dark/black online) Lines represent the nativeness of strands 2 and 7, respectively (see panel C). Strand 1 unfolds immediately upon force application while the other strands (i.e., S3–S6) track the behavior of strand 2 or stay native (∼80%) until full unfolding occurs. (A) A representative trajectory unfolding from state A1, characterized by the cooperative unfolding of strands 2–7. State A1 consists of conformations that have strand 1 distorted. (B) A representative trajectory unfolding from state A2. State A2 consists of conformations that have strands 1 and 7 unfolded. (C) The structure of the lowest energy NMR structure of filamin (PDB ID: 1KSR) with the strands numbered in order of the amino-acid sequence.
Although a coarse-grain model of a protein was used in the simulations, the model is realistic in the sense that it captures key proteinlike features—a defined native structure, and native interactions that vary depending on the type of interactions observed in the experimentally determined structure. Thus, while the simplified model used here may not reproduce all the experimentally characterized properties of a particular protein, it is perfectly valid for looking at the general properties of proteins. In this regard, the simulations show that the curvature observed experimentally for filamin is physically possible for an Immunoglobulin-like domain, and that an alternative kinetic model can account for the convex curvature at low pulling velocities.
Unlike the SB model, there is no requirement in the two-path kinetic scheme for the unfolding pathways to be confined to a single reaction coordinate, or for unfolding to be a sequential process. The proposed kinetic scheme is thus a multidimensional generalization of the SB model that is consistent with the new view (25) of protein folding/unfolding.
Fitting the experimentally determined distribution of unfolding forces
Given a limited number of data-points in typical plots of ln [τu] versus F or F∗ versus ln[RF∗] and the noise inherent in single-molecule measurements, the SB model is almost guaranteed to provide a good fit to any set of data consisting of two quasilinear regimes. The apparent good agreement between the data and the SB model may then lead to a potentially misleading picture of the underlying free-energy landscape. The situation, however, is not entirely dire. Using unfolding force data obtained for filamin in a broad range of pulling velocities (1–4000 nm/s) (5), we show that the distribution of unfolding forces, p(F), contains additional information regarding the underlying free-energy landscape.
As mentioned in the Introduction, the plot of F∗ as a function of ln[RF∗] for filamin revealed a convex curvature at low pulling velocities (Fig. 1) that could be adequately described by the SB model (Fig. 1, dashed lines) as well as a three-state kinetic scheme with two distinct pathways (Fig. 1, continuous line/red online). If based solely on Fig. 1, it is difficult to argue against the SB model because it requires fewer parameters, and fits F∗ slightly better at pulling velocities of 1–20 nm/s. From the fits of F∗ to the SB model, the force-dependence of the outer barrier is xF∗out = 21.6 Å. This means that at pulling velocities of 1–20 nm/s, filamin unfolds from the native state over a barrier characterized by xF∗out = 21.6 Å (Fig. 2). Fitting the p(F) at pulling velocities of 1 nm/s and 20 nm/s however, reveal that the p(F) at these pulling velocities cannot be simultaneously described by the same xpfout, in contradiction to the SB model. The p(F) at 20 nm/s cannot be reproduced by using the parameters, k(0) and xpfout, determined at 1 nm/s (Fig. 5, dotted line/magenta online). Furthermore, the xpfout = 11 Å estimated from fitting the p(F) at 1 nm/s is one-half of that estimated from fitting F∗. In contrast, the two-path kinetic scheme (Fig. 5, last panel) is able to describe the p(F) of all seven pulling velocities simultaneously. The above results show that despite the good fits to F∗, the SB is inconsistent with the p(F), while the two-path kinetic scheme fits both the p(F) and F∗ well.
As a control, we fitted the p(F) to a model where the A1→D pathway in the two-path kinetic scheme is ignored, and the A2→D pathway is modeled as before using the DHS rate equation. The resulting model is a variant of the traditional SB model where unfolding can also be initiated from the intermediate state at high pulling velocities. The fits to the SB variant (Fig. 5, dashed lines/blue online) are clearly not as good as those to the two-path kinetic scheme (Fig. 5, continuous lines/red online), especially at 20–200 nm/s where the fitted distributions are significantly displaced from the p(F). In 250 iterations of the fitting procedure (see Methods), a large proportion (28%) returned, numerically, very similar parameter estimates for the SB variant (Fig. 5). Significantly, further local optimization (i.e., Nelder-Mead minimization) of poorer fits yielded the same parameters, indicating that the parameter values that we obtained for the SB variant are likely to be optimal. Taken together, these results suggest that the additional unfolding pathway is crucial to the good fit of p(F) generated by the two-path model, and not a redundant feature of the kinetic scheme.
Conclusions
Force spectroscopy is providing unique insights into the free-energy landscapes of proteins thanks to the single-molecule feature of the technique and the quantifiable effect of the perturbation applied on the free-energy landscape. Forced unfolding experiments are most often interpreted by fitting the data to two-state models. Such analyses relate the features (e.g., xu) of the underlying free-energy landscape to the dependence of the modal unfolding force (F∗) on the loading rates. Most often an approximately logarithmic dependence of F∗ on the cantilever retraction speed is observed, which is what the Bell model (the most widely used model) predicts. Increasingly, however, mechanical probes are revealing complexity in the free-energy landscapes of proteins (26, 27, 28, 29) that cannot be easily accounted-for by the conventional kinetic models.
Here, by using a combination of atomistic simulations and an analysis of the distributions of unfolding forces (p[F]) for the protein filamin, which was previously shown to exhibit anomalous unfolding kinetics (5), we attempted to illustrate that an analysis solely based on F∗ may give a misleading picture of the free-energy landscape. Specifically, we demonstrated that while kinetic models may fit the modal or average value of the unfolding force/time to good approximation, they may dramatically violate the whole distribution of forces. For filamin, we find that the p(F) can be well described by a model with two unfolding pathways, but not by a model with sequential barriers. At sufficiently low pulling velocities or forces, it is likely that a protein unfolds through both its forced and unforced pathway (11, 18). In this regard, previous work (20, 30, 31) has shown that the forced and unforced pathways are distinct. In addition, extensive simulations of a coarse-grain model of filamin show that the two-path model is physically plausible for a small single-domain protein, and can produce a curvature similar to that observed experimentally.
Recently, it was elegantly demonstrated that anomalous kinetic signatures can also be explained by the smooth deformation of a single unfolding pathway by the applied force (32). In this scenario, the force changes the properties of the ground state and/or transition state resulting in the alteration of the force-dependence of the unfolding pathway. We note that the approach used here, which is analogous to a master-equation-type description of the kinetics, is compatible with the single-path picture. The interpretation of the kinetic equations in terms of the structure of the underlying free-energy landscape, however, is more subtle; e.g., how many coordinates are required to adequately describe the free-energy landscape? Are there two competing, coexisting transition barriers; or is there a single, albeit kinetically distinct, pathway at different forces? We previously showed that the experimental data for ddFLN4 can be fitted to a kinetic model with two pathways with very good agreement (5). However, the currently available experimental data does not allow a distinction between a free-energy landscape with two coexisting pathways and one with a single pathway. To better understand the free-energy landscape of this rather simple and very common protein fold, further experiments as well as all-atom simulations at experimental pulling velocities would be required.
Indeed, the correspondence of any model with reality cannot be guaranteed due to: 1), the use of simplifying assumptions; 2), the impossibility of examining all possible kinetic models; 3), the lack of detailed structural information due to the vast difference in timescales between full-atom molecular-dynamics simulations and mechanical unfolding experiments; and/or 4), the difficulty in performing the relevant experiments.
The conclusions drawn here should not be taken as a criticism of the validity of existing analyses (e.g., the Bell model), which are often justified in the context of the experiments (e.g., comparing between mutants). Instead, the results here illustrate the currently underexploited potential of force spectroscopy techniques, particularly the wealth of information contained in the p(F).
The main problem in the analysis of the p(F) is the need to obtain large amounts of data (e.g., 1000 unfolding events per pulling velocity) in a large range of pulling velocities (stability and force noise). However, with the advent of improved instruments and techniques (33, 34), such problems are likely to be circumvented in the near future. Despite such difficulties, current datasets may already be sufficient to discriminate between kinetic models and/or suggest alternative interpretations, which may serve as the basis of future investigations.
Acknowledgments
We thank Peter Rein ten Wolde for comments on a previous version of the manuscript and Godfrey Beddard, Dave Brockwell, and Neal Crampton for stimulating discussions.
E.P. and Z.T.Y. are supported by the Wellcome Trust and the University of Leeds. M.S. was supported in the framework of the Elite Network of Bavaria by the International Graduate School-NanoBioTechnology (grant No. IDK-NBT). M.R. was supported by an SFB 863 Grant of Deutsche Forschungsgemeinschaft.
Editor: Gerhard Hummer.
Footnotes
Michael Schlierf's present address is B CUBE, Molecular Bioengineering, TU Dresden, Dresden, Germany.
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
References
- 1.Li L., Huang H.H., Fernandez J.M. Mechanical unfolding intermediates observed by single-molecule force spectroscopy in a fibronectin type III module. J. Mol. Biol. 2005;345:817–826. doi: 10.1016/j.jmb.2004.11.021. [DOI] [PubMed] [Google Scholar]
- 2.Peng Q., Li H. Atomic force microscopy reveals parallel mechanical unfolding pathways of T4 lysozyme: evidence for a kinetic partitioning mechanism. Proc. Natl. Acad. Sci. USA. 2008;105:1885–1890. doi: 10.1073/pnas.0706775105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schwaiger I., Kardinal A., Rief M. A mechanical unfolding intermediate in an actin-crosslinking protein. Nat. Struct. Mol. Biol. 2004;11:81–85. doi: 10.1038/nsmb705. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Manyes S., Dougan L., Fernandez J.M. Direct observation of an ensemble of stable collapsed states in the mechanical folding of ubiquitin. Proc. Natl. Acad. Sci. USA. 2009;106:10534–10539. doi: 10.1073/pnas.0901213106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yew Z.T., Schlierf M., Paci E. Direct evidence of the multidimensionality of the free-energy landscapes of proteins revealed by mechanical probes. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2010;81:031923. doi: 10.1103/PhysRevE.81.031923. [DOI] [PubMed] [Google Scholar]
- 6.Evans E., Ritchie K. Dynamic strength of molecular adhesion bonds. Biophys. J. 1997;72:1541–1555. doi: 10.1016/S0006-3495(97)78802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bell G.I. Models for the specific adhesion of cells to cells. Science. 1978;200:618–627. doi: 10.1126/science.347575. [DOI] [PubMed] [Google Scholar]
- 8.Sachs F., Lecar H. Stochastic models for mechanical transduction. Biophys. J. 1991;59:1143–1145. doi: 10.1016/S0006-3495(91)82329-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hummer G., Szabo A. Kinetics from nonequilibrium single-molecule pulling experiments. Biophys. J. 2003;85:5–15. doi: 10.1016/S0006-3495(03)74449-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dudko O.K., Hummer G., Szabo A. Intrinsic rates and activation free energies from single-molecule pulling experiments. Phys. Rev. Lett. 2006;96:108101. doi: 10.1103/PhysRevLett.96.108101. [DOI] [PubMed] [Google Scholar]
- 11.Yew Z.T., Krivov S., Paci E. Free-energy landscapes of proteins in the presence and absence of force. J. Phys. Chem. B. 2008;112:16902–16907. doi: 10.1021/jp807316e. [DOI] [PubMed] [Google Scholar]
- 12.Evans E. Probing the relation between force, lifetime, and chemistry in single molecular bonds. Annu. Rev. Biophys. Biomol. Struct. 2001;30:105–128. doi: 10.1146/annurev.biophys.30.1.105. [DOI] [PubMed] [Google Scholar]
- 13.Merkel R., Nassoy P., Evans E. Energy landscapes of receptor-ligand bonds explored with dynamic force spectroscopy. Nature. 1999;397:50–53. doi: 10.1038/16219. [DOI] [PubMed] [Google Scholar]
- 14.Thomas W.E., Trintchina E., Sokurenko E.V. Bacterial adhesion to target cells enhanced by shear force. Cell. 2002;109:913–923. doi: 10.1016/s0092-8674(02)00796-1. [DOI] [PubMed] [Google Scholar]
- 15.Marshall B.T., Long M., Zhu C. Direct observation of catch bonds involving cell-adhesion molecules. Nature. 2003;423:190–193. doi: 10.1038/nature01605. [DOI] [PubMed] [Google Scholar]
- 16.Evans E., Leung A., Zhu C. Mechanical switching and coupling between two dissociation pathways in a P-selectin adhesion bond. Proc. Natl. Acad. Sci. USA. 2004;101:11281–11286. doi: 10.1073/pnas.0401870101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bartolo D., Derényi I., Ajdari A. Dynamic response of adhesion complexes: beyond the single-path picture. Phys. Rev. E Stat. Nonlin. Soft Matter Phys. 2002;65:051910. doi: 10.1103/PhysRevE.65.051910. [DOI] [PubMed] [Google Scholar]
- 18.West D.K., Olmsted P.D., Paci E. Mechanical unfolding revisited through a simple but realistic model. J. Chem. Phys. 2006;124:154909. doi: 10.1063/1.2185100. [DOI] [PubMed] [Google Scholar]
- 19.Best R.B., Paci E., Dudko O.K. Pulling direction as a reaction coordinate for the mechanical unfolding of single molecules. J. Phys. Chem. B. 2008;112:5968–5976. doi: 10.1021/jp075955j. [DOI] [PubMed] [Google Scholar]
- 20.Williams P.M., Fowler S.B., Clarke J. Hidden complexity in the mechanical properties of titin. Nature. 2003;422:446–449. doi: 10.1038/nature01517. [DOI] [PubMed] [Google Scholar]
- 21.Derényi I., Bartolo D., Ajdari A. Effects of intermediate bound states in dynamic force spectroscopy. Biophys. J. 2004;86:1263–1269. doi: 10.1016/S0006-3495(04)74200-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Karanicolas J., Brooks C.L., 3rd The structural basis for biphasic kinetics in the folding of the WW domain from a formin-binding protein: lessons for protein design? Proc. Natl. Acad. Sci. USA. 2003;100:3954–3959. doi: 10.1073/pnas.0731771100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dudko O.K., Hummer G., Szabo A. Theory, analysis, and interpretation of single-molecule force spectroscopy experiments. Proc. Natl. Acad. Sci. USA. 2008;105:15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schlierf M., Rief M. Single-molecule unfolding force distributions reveal a funnel-shaped energy landscape. Biophys. J. 2006;90:L33–L35. doi: 10.1529/biophysj.105.077982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dill K.A., Chan H.S. From Levinthal to pathways to funnels. Nat. Struct. Biol. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 26.Forman J.R., Yew Z.T., Clarke J. Non-native interactions are critical for mechanical strength in PKD domains. Structure. 2009;17:1582–1590. doi: 10.1016/j.str.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serquera D., Lee W., Itzhaki L.S. Mechanical unfolding of an ankyrin repeat protein. Biophys. J. 2010;98:1294–1301. doi: 10.1016/j.bpj.2009.12.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brujic J., Hermans R.I., Fernandez J.M. Single-molecule force spectroscopy reveals signatures of glassy dynamics in the energy landscape of ubiquitin. Nat. Phys. 2006;2:282–286. [Google Scholar]
- 29.Dougan L., Li J., Fernandez J.M. Single homopolypeptide chains collapse into mechanically rigid conformations. Proc. Natl. Acad. Sci. USA. 2009;106:12605–12610. doi: 10.1073/pnas.0900678106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paci E., Karplus M. Unfolding proteins by external forces and temperature: the importance of topology and energetics. Proc. Natl. Acad. Sci. USA. 2000;97:6521–6526. doi: 10.1073/pnas.100124597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klimov D.K., Thirumalai D. Native topology determines force-induced unfolding pathways in globular proteins. Proc. Natl. Acad. Sci. USA. 2000;97:7254–7259. doi: 10.1073/pnas.97.13.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki Y., Dudko O.K. Single-molecule rupture dynamics on multidimensional landscapes. Phys. Rev. Lett. 2010;104:048101. doi: 10.1103/PhysRevLett.104.048101. [DOI] [PubMed] [Google Scholar]
- 33.Schlierf M., Berkemeier F., Rief M. Direct observation of active protein folding using lock-in force spectroscopy. Biophys. J. 2007;93:3989–3998. doi: 10.1529/biophysj.107.114397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu R., Garcia-Manyes S., Fernandez J.M. Mechanical characterization of protein L in the low-force regime by electromagnetic tweezers/evanescent nanometry. Biophys. J. 2009;96:3810–3821. doi: 10.1016/j.bpj.2009.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]