

Abstract
FSH is produced by the pituitary gonadotrope to regulate gametogenesis. Production of the β-subunit of FSH is the rate-limiting step in FSH synthesis, and a number of peptide and steroid hormones within the reproductive axis have been found to regulate transcription of the FSH β-subunit gene. Although both activin and glucocorticoids are notable regulators of FSHβ by themselves, we find that cotreatment results in a synergistic interaction on the mouse FSHβ promoter at the level of the gonadotrope using transient transfection of a reporter gene into the LβT2 immortalized gonadotrope-derived cell line. This synergistic interaction is specific to FSHβ, because only additive effects of these two hormones are observed on LH β-subunit, GnRH receptor, and mouse mammary tumor virus gene expression. Components of both activin and glucocorticoid signaling are found to be necessary for synergy, and there are specific cis elements on the mouse FSHβ promoter that contribute to the synergistic response as well. We also identify novel activin-responsive regions in the mouse FSHβ promoter and find that the −120 site can bind Smad2/3 in vitro. In addition, the glucocorticoid receptor and Smad3 are sufficient to confer a striking synergy with glucocorticoids on the mouse FSHβ promoter. Our studies provide the first evidence of a synergistic interaction between activin and glucocorticoids within the gonadotrope cell and demonstrate that this synergy can occur directly at the level of the mouse FSHβ promoter.
FSH is produced solely in the gonadotrope cells of the anterior pituitary and is critical for follicular development (1). Female mice lacking FSH are infertile due to a block in folliculogenesis at the preantral stage (2). FSH levels are coordinately regulated by several hormones including GnRH, activin, and steroid hormones (1). During the estrous cycle, a surge in GnRH during the afternoon of proestrus triggers a surge in both of the gonadotropin hormones, FSH and LH, resulting in ovulation of the mature follicle in response to the LH surge (3). Several hours later, on the morning of estrus, a secondary FSH surge is observed that is independent of GnRH and does not correspond with a surge in LH (4, 5). At the transcriptional level, the secondary FSH surge is preceded by an increase in mRNA levels of the unique FSH β-subunit (6), the synthesis of which is the rate-limiting step in the production of FSH (7, 8). Interestingly, it is the secondary FSH surge that appears to be essential for follicular recruitment, whereas it is unclear what role the primary FSH surge plays (9). Therefore, understanding the mechanisms governing the secondary FSH surge is important in understanding the overall regulation of FSH and its role in fertility.
Glucocorticoids have a well-documented ability to regulate FSH (10), and endogenous glucocorticoids have been reported to be important for maintaining the secondary FSH surge (11). Although traditionally known for their roles in maintaining cardiovascular, metabolic, immunological, and developmental functions, glucocorticoids are also critical for reproductive functions such as lactation (12–14). Glucocorticoids have been shown to positively regulate FSHβ expression both in whole-animal studies (15, 16) as well as in primary pituitary cultures (17–19). Furthermore, our laboratory has recently demonstrated that glucocorticoids can directly regulate FSHβ transcription at the level of the gonadotrope cell using the LβT2 immortalized cell line (20). Together, these data suggest a connection between adrenal steroids and proper regulation of the reproductive axis.
Activin is another potent regulator of FSH, and its disruption results in decreased FSH production and subfertility (21, 22). Activin is produced within the anterior pituitary as well as the gonads and acts in an endocrine, paracrine, and autocrine manner (23). Activin action is antagonized by inhibin and follistatin, both of which fluctuate throughout the estrous cycle. This results in varying levels of bioavailable activin during the cycle (24, 25). Physiologically, normal glucocorticoid levels peak at the same time that follistatin and inhibin decrease, resulting in simultaneously high, active levels of both activin and glucocorticoids (26, 27). An interaction between activin and glucocorticoids in the regulation of FSHβ was previously reported in studies using primary rodent pituitary cultures (18). The mechanism of interaction is currently unknown, although hormonal cooperation in the gonadotrope may be common because activin has been shown recently to synergize with both GnRH and androgens in the regulation of FSHβ gene expression (28, 29).
The immortalized LβT2 gonadotrope cell line has proved invaluable in studying the molecular mechanisms of FSHβ regulation by activin and steroids (20, 28–32). In this study, we investigate the molecular mechanisms by which activin and glucocorticoids synergistically regulate FSHβ expression. We demonstrate that the gonadotrope cell is sufficient to observe synergy between activin and glucocorticoids. Furthermore, we show that the synergy occurs directly on the FSHβ promoter and demonstrate that components of both activin and glucocorticoid signaling pathways are necessary for the synergy. Additionally, we identify novel activin-responsive regions in the FSHβ promoter and find that the −120 site can bind Smad2/3 in vitro. This study furthers our understanding of how both activin and glucocorticoids can regulate FSHβ expression and underscores the profound effects of activin on FSHβ regulation.
Materials and Methods
Hormones
Dexamethasone and corticosterone were obtained from Sigma-Aldrich (St. Louis, MO). Activin A was obtained from Calbiochem (La Jolla, CA) and follistatin from R&D Systems (Minneapolis, MN). Vehicle for all experiments was ethanol unless otherwise noted.
Reporter and expression plasmids
The reporter plasmids −1000FSHluc and −1200GnRHRluc were previously described in Refs. 20 and 33, respectively; the −1800LHluc was kindly provided by Mark Lawson and MMTVluc is described in Ref. 29. The following site-specific mutations in the −1000FSHluc reporter plasmid have been previously described: 120m and 153m in Bailey et al. (32) and 139m, 197m, and 381m in Thackray et al. (20). The remaining site-specific mutations in the −1000FSHluc reporter plasmid were generated using the QuikChange mutagenesis kit (Stratagene, La Jolla, CA) according to the manufacturer's protocol with the following oligonucleotides: 267m (5′-AGATCAGAAAGAAATAGTagctACTCTAGAGTCACATTTAATTTAC-3′) and 106m (5′-GTCTAAACAAaGATctCCTTTCAGCAGGCTTTATGTTGGTATTGG-3′). The 5× reporter contains mutations in the −267, −153, −139, −120, and −106 regions of the FSHβ promoter and was constructed using the 153m reporter and then making the subsequent −267 mutation using the 267m oligonucleotide described. The remaining mutations in the −139/−120/−106 sites were made using the oligonucleotide (5′-TGCTTTGcCGAGGCTTcATgTCCCTGTCCGTagAAACAAaGATctCCT-3′). The expression vector for Smad7 was kindly provided by Rik Derynck, and Smad3 was provided by Theresa Guise. The wild-type GR plasmid was provided by Keith Yamamoto. The GRdim4 mutant is described in Thackray et al. (20). The mutant GRDBDmut contains a mutation in its DNA-binding domain, C476W/R479Q, and is described in Ref. 34. This mutation was generated using the QuikChange mutagenesis kit and the oligonucleotide (5′-CTGCCCAGCATGgCGCTATCaGAAATGTCTTCAGGCTGGAATG-3′).
Cell culture and transient transfections
All of the cell culture and transient transfection experiments were performed with the LβT2 cell line (30, 35). LβT2 cells were maintained in 10-cm-diameter dishes in DMEM (Cellgro; Mediatech, Herndon, VA) supplemented with 10% fetal bovine serum (Omega Scientific, Inc., Tarzana, CA) at 37 C with 5% CO2. One day before transfection, 3 × 105 cells per well were plated into 12-well plates. Transient transfection was performed using Fugene 6 reagent (Roche Molecular Biochemicals, Indianapolis, IN) following the manufacturer's instructions. Each well was transfected with 0.4 μg reporter plasmid. Additionally, the cells were transfected with 0.2 μg glucocorticoid receptor (GR) (unless otherwise noted) to observe a robust hormone induction (20). When applicable, 0.2 μg of the indicated expression vector or its empty vector control was also transfected. As an internal control for transfection efficiency, the LβT2 cells were transfected with 0.2 μg of a reporter plasmid containing β-galactosidase (β-gal) driven by the herpes virus thymidine kinase promoter (TKβ-gal). The cells were switched to serum-free DMEM supplemented with 0.1% BSA, 5 mg/liter transferrin, and 50 nm sodium selenite at 6 h after transfection. The following day, the cells were treated with ethanol (vehicle control) or hormone for 24 h. The cells were washed once with 1× PBS and then lysed with 0.1 m K-phosphate buffer (pH 7.8) containing 0.2% Triton X-100. After lysing, the cells were assayed for luciferase activity using a buffer containing 100 mm Tris-HCl (pH 7.8), 15 mm MgSO4, 10 mm ATP, and 65 μm luciferin. β-Galactosidase activity was measured using the Galacto-light assay (Tropix, Bedford, MA) according to the manufacturer's protocol. Both luciferase and β-galactosidase activity were measured using an EG&G Berthold microplate luminometer (PerkinElmer Corp., Norwalk, CT).
EMSA
Nuclear extracts were prepared from LβT2 cells as previously described (36). Probes were end-labeled using T4 polynucleotide kinase (New England Biolabs, Beverly, MA) and were column purified using Micro Bio-Spin chromatography columns (Bio-Rad Laboratories, Hercules, CA). Binding reactions were carried out using 4 μg nuclear protein and 4 fmol 32P-labeled oligonucleotide at 4 C for 30 min in a DNA-binding buffer containing 5 mm dithiothreitol, 0.025 μg/μl poly-dIdC, 1 mm phenylmethylsulfonyl fluoride, 0.25 mg/ml BSA, and binding buffer, 50 mm HEPES (pH 7.8), 250 mm KCl, 5 mm EDTA, 25 mm spermidine, 30% glycerol, and 10% Ficoll. After 30 min, the DNA-binding reactions were run on a 5% polyacrylamide gel (30:1 acrylamide: bisacrylamide) in 0.5× TAE buffer. A 500-fold excess of the relevant oligonucleotide was used for competition. Antibodies specific for phosphorylated Smad2/3 (Santa Cruz Biotechnology, Santa Cruz, CA) and the nonspecific rabbit IgG (Santa Cruz Biotechnology) were used to detect specific protein binding. Oligonucleotides used in EMSA include 120 WT, 5′-CTCCCTGTCCGTCTAAACAA-3′; 153, 5′-CTGTGGCATTTAGACTGCTTTGG-3′; 153m, 5′-CTGTGGCATTTCTACTGCTTTGG-3′; 139, 5′-AGACTGCTTTGGCGAGGCTTGATCTCCCTGTCCGT-3′; 139m, 5′-AGACTGCTTTGCCGAGCTTCATGTCCCTGTCCGT-3′; 106, 5′-AAACAATGATTCCCTTTCAGCAGGCTTT-3′; and 106m, 5′-AAACAAGGATCTCCTTTCAGCAGGCTTT-3′.
Statistical analyses
Transient transfections were performed in triplicate, and each experiment was repeated at least three times. The data were normalized for transfection efficiency by expressing luciferase activity relative to β-galactosidase activity and relative to the empty pGL3 plasmid to control for hormone effects on the vector DNA. The data were analyzed by the Student's t test for independent samples, by one-way ANOVA followed by post hoc comparisons with the Tukey-Kramer honest significant difference test, or by two-way ANOVA using the statistical package JMP 5.0 (SAS, Cary, NC). Significant differences were designated as P < 0.05. The results are presented as fold induction relative to the vehicle control.
Results
Activin and glucocorticoids specifically activate the FSHβ promoter in a synergistic manner
Numerous hormones regulate gonadotropin production at multiple levels of the reproductive axis. An important focus in the field is to understand how these hormones interact with each other to properly regulate fertility. Both activin and glucocorticoids can regulate FSH levels, and there is evidence that these two hormones interact (18). Although primary gonadotrope cells have been shown to express the GR (37), it is unknown whether this interaction occurs at the level of the gonadotrope cell and by what mechanism. To test whether synergy between activin and glucocorticoids occurs on the FSHβ promoter at the level of the gonadotrope, 1000 bp of the mouse FSHβ promoter fused to a luciferase reporter gene (−1000FSHluc) was transiently transfected into the immortalized gonadotrope-derived cell line, LβT2. Cells were treated for 24 h with vehicle (0.1% ethanol), 10 ng/ml activin, and/or 100 nm dexamethasone, a synthetic glucocorticoid analog, and changes in luciferase activity were monitored. Under these conditions, activin induces the FSHβ promoter 3.7-fold, but there was no significant induction by dexamethasone alone (Fig. 1A). However, cotreatment of both activin and dexamethasone resulted in a 5.4-fold induction, and the interaction between these two hormones was determined to be synergistic by two-way ANOVA (38).
Fig. 1.
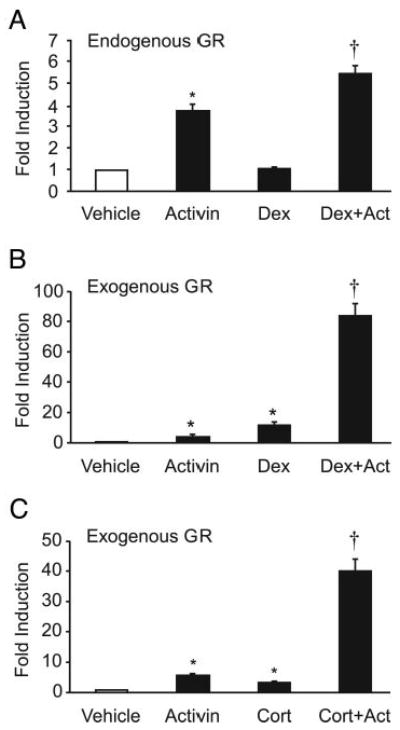
Activin and glucocorticoids synergistically activate FSHβ gene expression in immortalized gonadotropes. A, The −1000FSHβluc reporter gene and TKβgal were transiently transfected into LβT2 cells. After overnight starvation in serum-free media, the cells were treated for 24 h with vehicle (0.1% ethanol), 10 ng/ml activin, and/or 100 nM dexamethasone (Dex). B and C, Exogenous GR (200 ng) was transfected along with FSHβluc reporter gene and TKβgal and treated with vehicle, activin, and/or 100 nm dexamethasone or corticosterone (Cort) as indicated. The results represent the mean ± sem of at least three experiments performed in triplicate and are presented as fold induction relative to the vehicle control. *, Hormone treatment significantly different from the vehicle-treated control, using one-way ANOVA followed by Tukey's post hoc test; †, synergy between activin and dexamethasone by two-way ANOVA.
Because LβT2 cells express a low level of endogenous GR (20) and glucocorticoids and activin can synergize on the FSHβ promoter as seen in Fig. 1A, exogenous GR was transfected into the LβT2 cells to amplify the response. In the presence of exogenous GR, activin induced −1000FSHluc by 4.5-fold, and dexamethasone induced expression by 12-fold, whereas cotreatment with activin and dexamethasone resulted in an 84-fold activation (Fig. 1B). This striking interaction between activin and dexamethasone on the FSHβ promoter is synergistic as determined by two-way ANOVA (38). Synergy is also seen using activin and corticosterone, the endogenous active glucocorticoid metabolite in rodents. Activin alone gave a 5.8-fold induction, corticosterone alone a 3.4-fold induction, and activin plus corticosterone a 41-fold induction (Fig. 1C). These results indicate that activin and glucocorticoids are able to synergistically interact to induce expression of the FSHβ promoter at the level of the gonadotrope cell and that this can occur with endogenous GR present in the LβT2 cells, although a much more dramatic response is seen upon addition of exogenous GR.
To ascertain whether glucocorticoid and activin treatment results in a general stimulatory effect on glucocorticoid-responsive genes, we tested whether there was an interaction between activin and dexamethasone on the mouse mammary tumor virus (MMTV) promoter. The MMTV promoter is highly responsive to glucocorticoids, and its ability to be regulated by GR is well characterized (39, 40). The MMTV promoter fused to a luciferase promoter was transiently transfected in LβT2 cells along with GR and treated with activin, dexamethasone, or both (Fig. 2A). Activin significantly repressed MMTV induction to 0.75-fold of the vehicle control, dexamethasone induced MMTV 97-fold, and treatment with both activin and dexamethasone induced MMTV 60-fold. The interaction between activin and dexamethasone was not synergistic as analyzed by two-way ANOVA, indicating that although both the FSHβ and MMTV promoters are positively regulated by glucocorticoids, the synergy is specific to FSHβ.
Fig. 2.
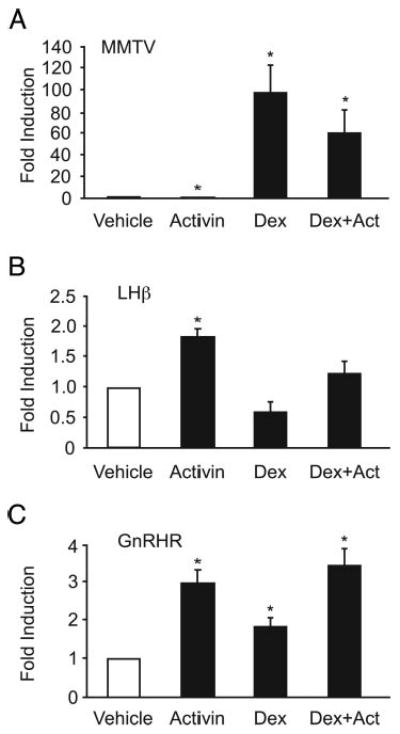
Activin and glucocorticoids do not synergistically activate MMTV, LHβ, or GnRHR gene expression. A–C, The MMTVluc reporter gene (A), the −1800LHβluc reporter gene (B), or the −1200Gn-RHRluc reporter gene (C) were transiently transfected into LβT2 cells along with 200 ng GR and TKβgal. After overnight starvation in serum-free media, the cells were treated for 24 h with vehicle (0.1% ethanol), 10 ng/ml activin, and/or with 100 nM dexamethasone (Dex) as indicated. *, Hormone treatment significantly different from the vehicle-treated control, using one-way ANOVA followed by Tukey's post hoc test. None of the cotreatments were synergistic by two-way ANOVA.
To further address the question of whether the effect seen on FSHβ expression is specific to this promoter or whether it is a result of a global effect on LβT2 cells, we tested the effects of cotreatment of activin and dexamethasone on other genes expressed in the gonadotrope cells, specifically LHβ and GnRH receptor (GnRHR). The proximal promoters of both of these genes have shown to be responsive to activin and glucocorticoids individually (20, 41–43), although co-treatment with both hormones was not previously reported. The 1800 bp of the rat LHβ promoter fused to a luciferase reporter (−1800LHβluc) was transiently transfected into LβT2 cells along with cotransfected GR. Upon treatment for 24 h, activin induced LHβ expression 1.8-fold, whereas dexamethasone repressed LHβ by 40% (Fig. 2B), consistent with previous reports (20, 41). Cotreatment with both hormones essentially canceled out their individual effects on LHβ and resulted in a net fold activation of 1.2. This additive effect was confirmed by two-way ANOVA. A second gonadotrope-expressed gene, GnRHR, was also tested. The 1200 bp of the mouse regulatory region of GnRHR fused to a luciferase reporter (−1200GnRHRluc) was transiently transfected into LβT2 cells along with GR (Fig. 2C). Activin treatment induced GnRHR by 3-fold, whereas dexamethasone resulted in a 1.8-fold activation. Although both of these hormones increased GnRHR expression, cotreatment was not synergistic with only a 3.4-fold activation. These experiments demonstrate that the synergy seen with cotreatment of activin and dexamethasone is specific to FSHβ gene expression.
Activin signaling is important for synergy between activin and glucocorticoids on FSHβ expression
Activin signals via its receptor using the Smad signaling pathway. Upon ligand binding, the activated receptor phosphorylates the receptor-regulated Smads, Smad2 and/or Smad3. Smad2/3 are then released into the cytoplasm, interact with the comediator Smad, Smad4, and translocate to the nucleus (44) where they activate transcription of activin-responsive genes. Activin signaling can be antagonized either through blocking its interaction with its receptor extracellularly using the activin-binding protein follistatin or by inhibiting Smad signaling through overexpression of the inhibitory Smad, Smad7, which binds the receptor intracellularly to block its interaction with Smad2/3 (45). These antagonists of activin action were used to demonstrate that classical activin signaling is necessary for generating a synergistic response on FSHβ. LβT2 cells were transfected with −1000FSHβluc and GR and then treated for 24 h with vehicle (ethanol), activin, or dexamethasone or cotreated with both. Additionally, cells were treated with either the vehicle for follistatin (PBS) and luciferase activity was normalized to the PBS-treated vehicle control (Fig. 3A, black bars) or cells were treated with follistatin (100 ng/ml) and the luciferase response was normalized to the follistatin-treated vehicle control (Fig. 3A, striped bars). Inclusion of follistatin significantly reduced activin induction, an effect that has been previously described (30). Induction with dexamethasone alone was reduced from 25-fold to 7-fold, and the synergy between activin and dexamethasone was reduced from 103-fold to 21-fold. Although one might expect follistatin to have no effect on dexamethasone induction of FSHβ, the LβT2 cells produce endogenous activin that is likely contributing to the dexamethasone induction of FSHβ. The addition of follistatin appears to block the effects of this endogenous activin, resulting in a reduction of dexamethasone induction of FSHβ to 7-fold. These results indicate that the ability of activin to bind its receptor is necessary for maintaining a full synergistic response.
Fig. 3.
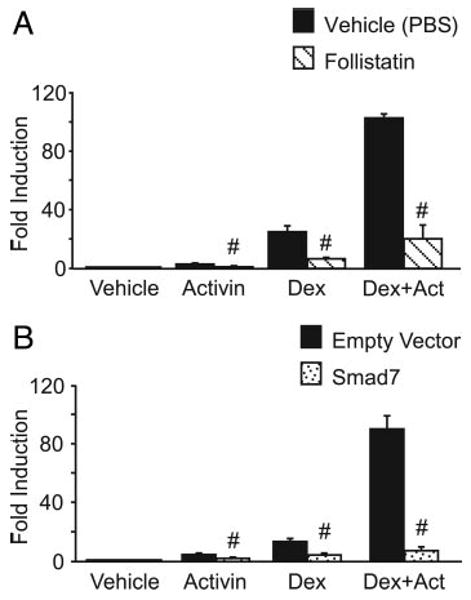
Disruption of activin signaling significantly reduces synergy on FSHβ. The −1000FSHβluc reporter gene and TKβgal were transiently transfected into LβT2 cells along with 200 ng GR. After overnight starvation in serum-free media, the cells were treated for 24 h with 10 ng/ml activin and/or 100 nM dexamethasone (Dex). A, Cells were treated with vehicle (0.1% ethanol), activin, and/or dexamethasone plus vehicle control for follistatin (PBS) and were normalized to the PBS-treated vehicle control (black bars), or cells were treated with vehicle (0.1% ethanol), activin, and/or dexamethasone plus 100 ng/ml follistatin and normalized to the follistatin-treated vehicle control (striped bars). #, Significant reduction with addition of follistatin in comparison with cells without follistatin. B, Cells were treated with vehicle (0.1% ethanol), activin, and/or dexamethasone and were transfected with the empty vector for Smad7 and normalized to the empty vector-transfected vehicle control (black bars), or cells were treated with vehicle (0.1% ethanol), activin, and/or dexamethasone and transfected with 200 ng Smad7 and normalized to the Smad7-transfected vehicle control (dotted bars). #, Significant reduction with overexpression of Smad7 in comparison with the empty vector by Student's t test.
To examine the specific contributions of the activin receptor signaling cascade to the synergy between activin and glucocorticoids, we overexpressed the inhibitory Smad, Smad7, along with −1000FSHβluc and GR in LβT2 cells. Cells were then treated for 24 h with vehicle, activin, and/or dexamethasone as indicated. Black bars represent cells transfected with the empty vector for Smad7, and the luciferase response was normalized to the empty-vector, vehicle-treated control (Fig. 3B). Dotted bars represent cells transfected with Smad7, and the luciferase response was normalized to the Smad7 vehicle-treated control. As can be seen in Fig. 3B, overexpression of Smad7 caused activin induction to be reduced from 4-fold to 1.5-fold, as has been shown previously (46). Dexamethasone induction was reduced from 13-fold to 4-fold, and the synergy between activin and dexamethasone was reduced from 90-fold to 7-fold. These results further confirm that activin signaling is necessary for maintaining a full synergistic response. As with follistatin, the reduction of dexamethasone induction of FSHβ by Smad7 likely represents the inhibition of signaling by endogenous activin produced by these cells.
Multiple sites in the FSHβ promoter contribute to activin induction and the synergy between activin and glucocorticoids
Smads can bind DNA directly through Smad-binding elements (SBEs), such as the palindromic sequence GTCTAGAC or the half sites GTCT or AGAC (47). Several such elements in the mouse FSHβ promoter have been previously implicated in activin signaling including the −267, −153, and −120 sites (28, 31, 32). As can be seen in Fig. 4A, all three of these sites are putative SBEs, either representing a full consensus site (−267) or half-sites (−153 and −120). In addition, we have identified two other elements at −139 and −106 that also play a role in activin induction; however, these sites do not contain putative SBEs. The −139 site was originally identified as a putative steroid hormone response element (HRE) (20, 29, 48), whereas the −106 corresponds to the −120 activator protein (AP)-1 site identified in the ovine FSHβ promoter (49). Transcription factors known to bind these specific elements are indicated in Fig. 4A, and although the −267 has been shown to bind Smad2, Smad3, and Smad4 (28, 31, 50), it is less clear what role Smads play at the other cis elements. We sought to test the relative effects of all five of these cis elements on activin induction to compare and contrast the roles they play in generating a synergistic response with glucocorticoids. Specific mutations were made in the context of the −1000FSHβluc reporter (underlined bases, Fig. 4A) and designated 267m, 153m, 139m, 120m, and 106m. The 5× reporter plasmid contains all five mutations. These mutated plasmids were transiently transfected into LβT2 cells, and relative luciferase activity was assayed after treatment with activin for 24 h. None of these mutations significantly affected basal expression of FSHβ (data not shown). However, all five of these cis mutations significantly reduced activin induction, although only the 5× combination of mutations completely abolished it (Fig. 4B). Interestingly, although the −267 element has been shown to be critical for Smad signaling (28, 31), its mutation reduced the activin response by only 40%. Activin induction was reduced 50% by the −153 and −139 mutations, whereas the −120 mutation produced the most significant reduction of any single mutation with a 70% repression of activin response. These data indicate that multiple cis elements play roles in activin induction.
Fig. 4.
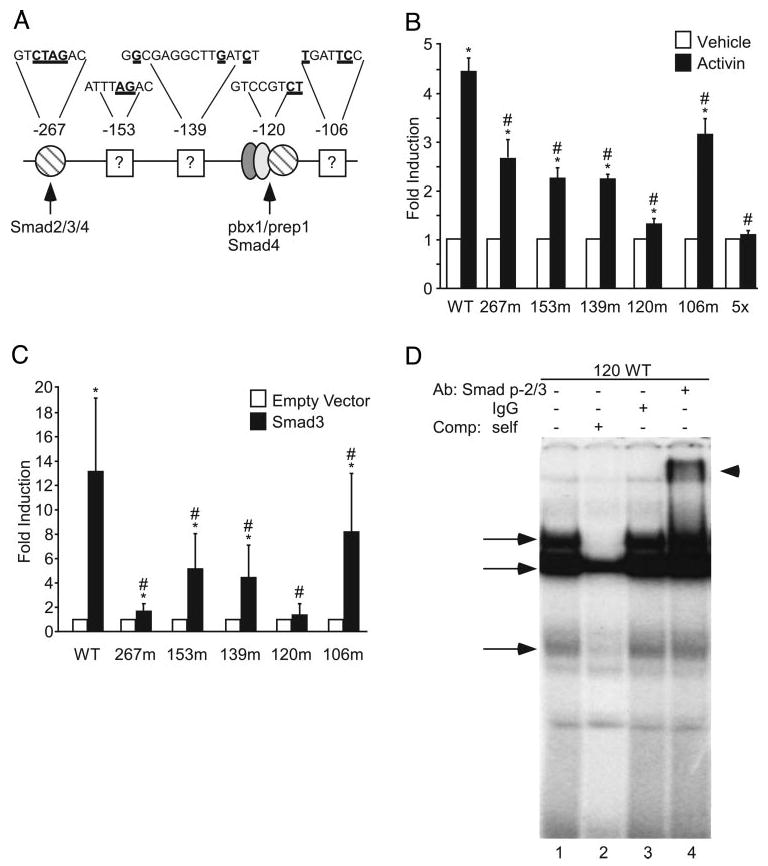
Multiple sites on the FSHβ promoter contribute to activin induction. A, Sites on the FSHβ promoter that contribute to activin responsiveness of the FSHβ gene are denoted. Numbers above the promoter indicate the location of each regulatory element relative to the transcriptional start site. Nucleotides in bold and underlined indicate bases mutated in subsequent transfection experiments. Proteins binding each site are indicated or, if unknown, are denoted with a question mark. B, The wild-type −1000FSHβluc reporter gene or one of six mutants was transiently transfected into LβT2 cells with TKβgal. After overnight starvation in serum-free media, the cells were treated for 24 h with either vehicle (white bars) or 10 ng/ml activin (black bars). *, Hormone treatment significantly different from the vehicle-treated control of the same reporter gene, using Student's t test; #, induction of the mutant reporter gene is significantly different from the induction of the wild-type reporter gene, using one-way ANOVA followed by Tukey's post hoc test. C, The wild-type −1000FSHβluc reporter gene or one of five mutated reporters (with TKβgal) was transiently transfected into LβT2 cells along with either empty vector (white bars) or 200 ng Smad3 (black bars). *, Smad3 induction significantly different from the empty vector control of the same reporter gene, using Student's t test; #, induction of the mutant reporter gene is significantly different from the induction of the wild-type reporter gene, using one-way ANOVA followed by Tukey's post hoc test. D, Nuclear extracts from LβT2 cells were incubated with the −120 probe and tested for complex formation in EMSA. Arrows denote specific complexes that are competed with the addition of 500× unlabeled competitor (lane 2). Inclusion of a nonspecific IgG antibody does not disrupt any complexes (lane 3); however, addition of an antibody specific for phosphorylated Smad2/3 (lane 4) results in a supershift (arrowhead).
It is well established that activin signals through the Smad family members Smad2 and/or 3 (45), and it has been shown that overexpression of Smad3 alone induces FSHβ expression (28, 31, 46, 51), whereas Smad3 knockout mice have reduced FSHβ expression (41). We hypothesized that if Smad3 were acting through these cis elements important for activin induction, then mutations at these sites would also inhibit the ability of Smad3 to activate FSHβ expression. To test this, we overexpressed either Smad3 or its empty vector, along with −1000FSHβluc or one of the five mutated reporter plasmids. All five of these mutations significantly reduced Smad3 induction of FSHβ; however, the −267 and −120 mutations had the most dramatic effects (Fig. 4C). Induction of the 120m reporter was completely abolished, and induction of the 267m reporter was reduced 87%. Induction of the 106m reporter was significantly reduced; however, this mutation had the smallest effect with only a 38% reduction. The −153 and −139 mutations had similar effects with reductions of 61 and 67%, respectively. It appears from these data that the −267 and −120 sites are the most critical elements for Smad3 signaling on FSHβ. In the case of the −267 site, this is in accordance with previous reports (28, 31).
Because all of these sites are important in activin induction and are critical for, or facilitate, Smad3 induction of the FSHβ promoter, we sought to determine whether Smads could bind these sites. EMSA has demonstrated that Smad4 can bind the −267 site (28, 31), and we confirmed this by EMSA as well (data not shown). We have previously demonstrated that Smad4 could form a higher-order complex with Pbx1 and Prep1 on the corresponding −120 putative SBE in the ovine FSHβ promoter. This complex, however, was relatively unstable and could only be visualized when large amounts (20 μg) of LβT2 nuclear extract were used in EMSA (32). To test whether Smad2/3 could bind the −120 putative SBE, an oligonucleotide encompassing −126 to −105 of the FSHβ promoter was end-labeled with 32P and incubated with LβT2 nuclear extracts. Three specific complexes were detected (Fig. 4D, arrows) that were reduced by self-competition (lane 2). Inclusion of a nonspecific control IgG antibody did not disrupt any of these bands (lane 3); however, inclusion of an antibody specific for phosphorylated Smad2/3 resulted in a supershift (lane 4, arrowhead). As an additional control, the antibody for phosphorylated Smad2/3 was incubated with probe alone; however, no corresponding nonspecific band is seen in the absence of extract (data not shown). EMSA was also performed with the −153, −139, and −106 sites; however, although specific complexes formed at each of these sites, complex disruption or supershift was not detected with antibodies to Smads (data not shown) (32). These data indicate that the −120, like the −267 site, is a functional SBE. However, it is still unclear what proteins are interacting at the −153, −139, and −106 activin-responsive regions.
To further characterize the −153, −139, and −106 sites, we used EMSA to determine whether endogenous LβT2 nuclear proteins bind to these elements. Specific protein complexes formed at each of these sites as indicated by competition with excess unlabeled wild-type probe. Mutant oligonucleotides were used that encompassed the same region as the wild-type probe but contained mutations corresponding to the mutations made in the −153, −139, and −106 sites in Fig. 4. Excess unlabeled mutant oligonucleotides were no longer able to compete some complexes at each of these sites (Fig. 5, arrows). This indicates that these sites are necessary for binding of specific LβT2 nuclear proteins as well as being functionally important in activin induction of FSHβ (Fig. 4). There were also bands that were competed by both the wild-type and mutant oligonucleotides. This suggests that there are other binding sites within these regions; however, the functional significance of these sites is unclear. No change in complex formation upon hormone treatment, either with activin alone or with cotreatment of activin plus dexamethasone, was seen at any of these sites. This suggests that these proteins are basal transcription factors or that autocrine activin secreted by the LβT2 cell is sufficient to cause their activation. However, mutations of these sites do not affect basal expression of the FSHβ gene (data not shown).
Fig. 5.
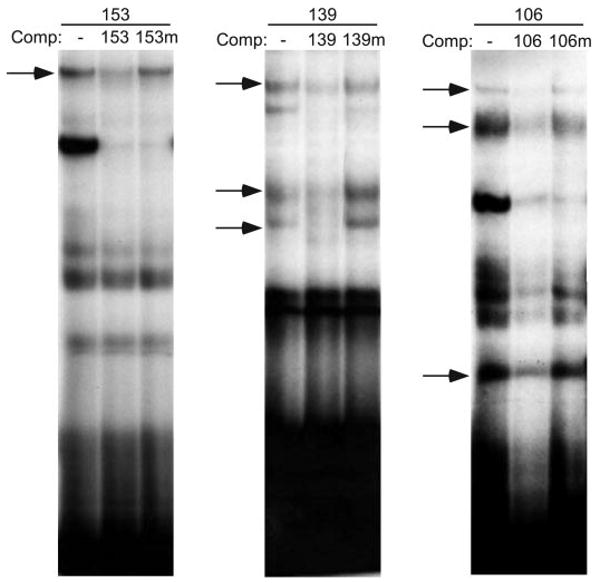
Specific protein complexes from LβT2 nuclear extracts bind the −153, −139, and −106 activin-responsive regions. Nuclear extracts prepared from LβT2 cells were incubated with the 153, 139, or 106 probes and tested for specific complex formation in EMSA. The 500× cold competitors (Comp) were added to test the specificity of complex formation as indicated: 153 probe with no competition (–), self-competition (153), or mutant competition (153m); 139 probe with no competition (–), self-competition (139), or mutant competition (139m); and 106 probe with no competition (–), self-competition (106), or mutant competition (106m). Complexes competed by self-competition, but not by mutant, are indicated with arrows.
As it became evident that multiple elements within the mouse FSHβ promoter contribute to activin induction, it was important to determine whether any of these sites play a role in glucocorticoid induction or in the synergy between activin and glucocorticoids. To test this, wild-type −1000FSHβluc and each of the five activin-responsive element mutants, 267m, 153m, 139m, 120m, or 106m, were transfected along with GR into LβT2 cells and treated with vehicle and dexamethasone (Fig. 6A) or vehicle and cotreatment with both activin and dexamethasone (Fig. 6B). Only the −139, which was originally identified as an HRE, significantly decreased the induction of FSHβ by glucocorticoids alone (Fig. 6A); however, most of the mutations did significantly reduce synergy upon cotreatment of activin and glucocorticoids. A 45% reduction was seen on 267m, a 27% reduction on 153m, a 62% reduction with 139m, and a 60% reduction with 120m, but no reduction was seen with 106m (Fig. 6B). As with activin alone, all five mutations were needed to completely inhibit activin signaling and reduce the synergistic interaction between activin and dexamethasone to the level of dexamethasone alone.
Fig. 6.
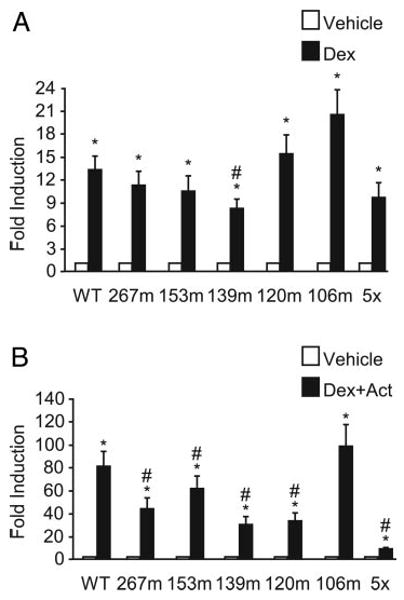
Cis mutations affecting activin responsiveness also reduce synergy. The wild-type −1000FSHβluc reporter gene or one of six mutated reporters was transiently transfected into LβT2 cells along with TKβgal and GR. After overnight starvation in serum-free media, the cells were treated for 24 h with vehicle (white bars) or with either 100 nm dexamethasone (Dex, black bars) (A) or 10 ng/ml activin (Act) and 100 nm dexamethasone (Dex, black bars) (B). *, Hormone treatment significantly different from the vehicle-treated control of the same reporter gene by Student's t test; #, induction of the mutant reporter gene is significantly reduced from the induction of the wild-type reporter gene, using one-way ANOVA followed by Tukey's post hoc test.
To better understand what is occurring at the level of the FSHβ promoter, we performed both EMSA and chromatin immunoprecipitation (ChIP) to visualize changes at the promoter. Specifically, we used EMSA to test whether differences in complex formation could be seen at any of the glucocorticoid or activin-responsive sites upon cotreatment of the hormones, but no differences were observed under any conditions tested (data not shown). We next used ChIP to analyze changes in promoter recruitment of GR, Smad3, or a cofactor used by both, cAMP response element binding protein-binding protein; however, no changes were seen upon hormone stimulation. We also investigated whether increased activation of the FSHβ promoter resulted from increased histone acetylation. Again, no differences were seen upon cotreatment using analysis of the endogenous promoter FSHβ promoter by ChIP with antibodies specific for acetylated H3 and H4 (data not shown). It remains possible that these types of changes are not occurring on the FSHβ promoter or that the conditions and techniques used were not sufficiently sensitive to visualize those changes.
GR binding to DNA is necessary for the synergistic induction of FSHβ gene expression by activin and glucocorticoids
To determine whether direct DNA binding by GR plays a critical role in the transactivation of FSHβ by steroid hormones, we transfected LβT2 cells with two different GR mutants that are incapable of binding DNA. The first mutant, GRdim4, has four point mutations in the dimerization domain. These mutations prevent homodimerization of GR and thus direct DNA binding to glucocorticoid response elements (GREs) but do not disrupt indirect DNA binding through interactions between GR and other transcription factors, such as AP-1, that occur through the DNA-binding domain (DBD) (34, 52). The second GR mutant, GRDBDmut, contains a mutation in the DBD, C476W/R479Q, which also prevents direct DNA binding by the mutant (34). This mutant should not disrupt any potential GR-Smad3 interactions, however, because GR and Smad3 do not interact through the DBD of GR (53). Figure 7A shows LβT2 cells transfected with −1000FSHβluc and wild-type GR (black bars), GRdim4 (dotted bars), or GR DBD mutant (striped bars). As expected, neither GR mutant affected activin induction; however, both completely abolished dexamethasone induction and reduced the induction by both activin and dexamethasone from 130-fold with wild-type GR to 30-fold with GRdim4 and 9-fold with the GR DBD mutant. These data support the hypothesis that direct DNA binding of GR is critical for maintaining a full synergistic response.
Fig. 7.
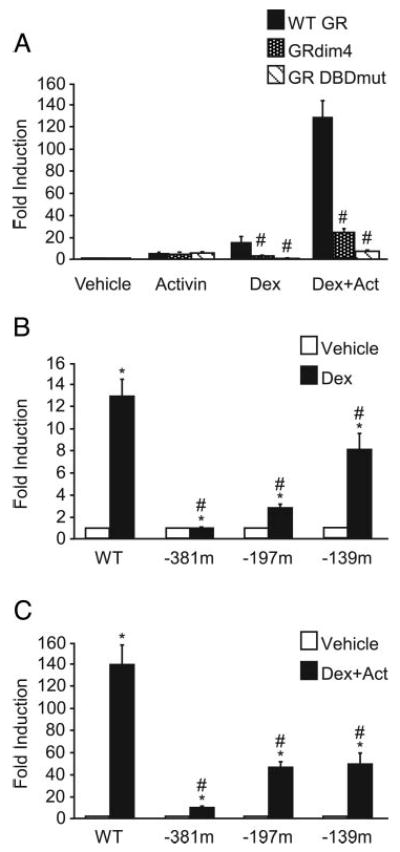
Disruption of GR binding to DNA significantly reduces the ability of activin and glucocorticoids to synergistically activate FSHβ gene expression. A, The −1000FSHβluc reporter gene with TKβgal was transiently transfected into LβT2 cells along with either 200 ng wild-type GR and normalized to the vehicle control transfected with wild-type GR (WT, black bars) or with mutant GR, either GRdim4 (dotted bars) or GR DBDmut (striped bars) and normalized to the vehicle control transfected with the corresponding mutant GR. After overnight starvation in serum-free media, the cells were treated for 24 h with 10 ng/ml activin and/or with 100 nm dexamethasone (Dex) as indicated. #, Induction by the mutant GR is significantly different from induction with wild-type GR by one-way ANOVA followed by Tukey's post hoc test. B and C, The wild-type −1000FSHβluc reporter gene or one of three cis-mutated reporters, GR, and TKβgal were transiently transfected into LβT2 cells. After overnight starvation in serum-free media, the cells were treated for 24 h with either vehicle (white bars) or 100 nm dexamethasone (Dex, black bars) (B) or 10 ng/ml activin (Act) plus 100 nm dexamethasone (Dex, black bars) (C). *, Hormone treatment significantly different from the vehicle-treated control of the same reporter gene by Student's t test; #, induction of the mutant reporter gene is significantly different from the induction of the wild-type reporter gene, using one-way ANOVA followed by Tukey's post hoc test.
We have previously identified three putative GREs in the mouse FSHβ promoter at −381, −197, and −139 relative to the transcriptional start site and have shown GR to bind the −381 GRE in vitro by EMSA (20). These specific sites were mutated in the context of the −1000FSHβluc promoter and are designated 381m, 197m, and 139m, respectively. When LβT2 cells were transfected with GR and either wild-type FSHβluc or one of the three mutant reporters and then treated with dexamethasone for 24 h, the 381m mutation completely abolished the induction by dexamethasone, whereas the 197m reduced induction by 78% and the 139m by 37% (Fig. 7B). These results are in accordance with our earlier results (20).
Next, we sought to determine whether these cis elements in the FSHβ promoter known to be critical for dexamethasone induction are also important for the synergy between activin and dexamethasone. Again, LβT2 cells were transfected with GR and either wild-type −1000FSHβluc or the 381m, 197m, or 139m mutant reporter plasmids and treated for 24 h with vehicle or both activin and dexamethasone. The 381m had the most dramatic effect with a 92% reduction from 138-fold induction with wild-type to 9-fold induction with 381m (Fig. 7C). The 197m and 139m had similar effects, with the 197m reducing induction to 46-fold, a 67% reduction, and 139m reducing the induction to 49-fold, a 65% reduction. None of these mutations reduced basal expression, and only the 139m reduced activin induction of FSHβ (data not shown; and Fig. 4). From these data, it is clear that if a mutation is made that prevents DNA binding by GR, either through a mutation in the DBD of GR or a cis mutation in the mouse FSHβ regulatory region, then synergy is dramatically reduced.
Smad3 and GR are sufficient to induce synergy
Synergy between activin and glucocorticoids is dependent on activin signaling as well as direct DNA binding of GR. Because activin signaling plays such a crucial role in maintaining the synergistic response and because Smad3 plays an important role in activin signaling, we tested whether Smad3 itself was sufficient to confer synergy in the absence of activin. As can be seen in Fig. 8, LβT2 cells were transiently transfected with −1000FSHβluc and GR along with Smad3 or its empty vector. The cells were then treated for 24 h with either vehicle or dexamethasone. Individually, Smad3 and dexamethasone both induced FSHβ 10-fold; however, when cells were transfected with Smad3 and treated with dexamethasone, FSHβ was induced 274-fold. Clearly, Smad3 in conjunction with activated GR is sufficient to induce synergy.
Fig. 8.
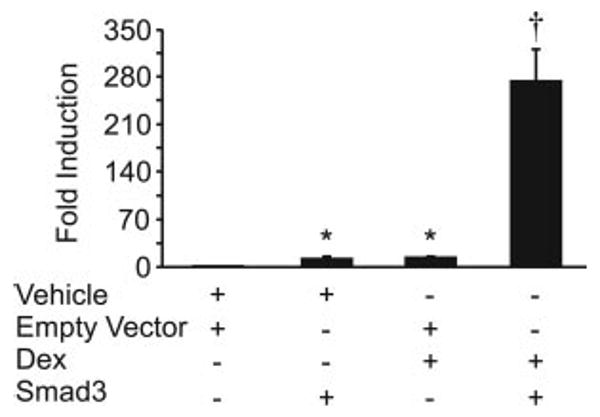
Smad3 and GR are sufficient to induce synergy. The wild-type −1000FSHβluc reporter gene, GR, and TKβgal were transiently transfected into LβT2 cells along with either empty vector or Smad3 as indicated. After overnight starvation in serum-free media, the cells were treated for 24 h with either vehicle or 100 nm dexamethasone (Dex). *, Hormone treatment significantly different from the vehicle-treated control, using one-way ANOVA followed by Tukey's post hoc test; †, synergy between activin and dexamethasone by two-way ANOVA.
Discussion
Tight regulation of FSH levels during the estrous cycle is critical for reproductive fitness. Before the primary ovulatory surge, both LH and FSH are regulated by GnRH, but the regulation of the secondary FSH surge is not known. Several theories have been proposed to explain how the secondary FSH surge is regulated, such as a change in GnRH pulse frequency to favor FSHβ transcription (7, 54), a higher level of bioavailable activin due to the drop in inhibin and follistatin concentration that would selectively activate FSHβ synthesis (24, 25), or a rise in steroid hormone levels such as progestins and androgens that induce FSHβ expression but inhibit LHβ (10, 20). Mounting evidence suggests that glucocorticoids can also have a positive impact on FSHβ gene expression (15–19). The present study adds to this body of evidence by demonstrating that glucocorticoids can synergize with activin to induce FSHβ gene expression.
As mentioned previously, normal levels of glucocorticoids are critical for numerous physiological functions, including reproduction (12–14). However, glucocorticoid levels are also known to rise dramatically during a time of stress, and thus glucocorticoids are also considered stress hormones. It has been postulated that during the time of stress, induction of FSHβ is a protective mechanism (17) needed to maintain the health of the follicle for the following cycle while at the same time preventing ovulation during the stress. Because the primary goal of this study was to address the mechanism of FSHβ regulation, more studies are needed to determine the effects of glucocorticoids on gonadotropin synthesis during normal reproductive function vs. during a stress response.
Both primary gonadotrope cells and the LβT2 cells express GR (20, 37). We demonstrate that synergy between activin and glucocorticoids can occur specifically at the level of the FSHβ promoter using LβT2 cells (Fig. 1A). A similar effect on FSHβ expression upon cotreatment of activin and dexamethasone was previously seen using primary rat pituitary cells (18). Studies in whole animals and primary pituitary culture have established that glucocorticoids regulate FSHβ expression (15, 16), and it has been postulated that glucocorticoids may play a role in the FSHβ secondary surge (11). FSHβ mRNA normally fluctuates within a very tight range of around 4-fold (6). The secondary surge, although necessary for follicular development, is even lower than the primary surge. Therefore, small changes in FSHβ transcript could significantly affect FSH synthesis during the estrous cycle. Even a modest synergistic activation of FSHβ transcription by glucocorticoids and activin is a potential mechanism for quickly changing levels of FSHβ transcript. Furthermore, the magnitude of synergistic induction is highly dependent on the amount of GR, as can be see when exogenous GR is added (Fig. 1B). Whether GR levels fluctuate in the primary gonadotrope during development or during the estrous cycle remains to be investigated. This report focuses on the molecular mechanisms required for the synergistic induction of FSHβ by glucocorticoids and activin.
Synergy on FSHβ is not likely due to overexpression of GR alone or to a global effect on LβT2 cells because no synergistic interaction was seen upon cotreatment with activin and dexamethasone on the MMTV, LHβ, or GnRHR promoters (Fig. 2). Therefore, it is unlikely that the synergy on FSHβ results from an induction of GR or components of activin signaling or a down-regulation of follistatin or inhibin, because these would also affect LHβ and GnRHR regulation. Furthermore, glucocorticoids and activin were previously shown to interact and induce FSHβ in primary rat pituitary cultures, whereas cotreatment with both activin and dexamethasone had no effect on the expression of the activin/inhibin subunit βB, caused only a small repression on activin/inhibin subunit βA, and actually significantly induced follistatin expression (18). This provides additional evidence that changes in the activin-inhibin-follistatin system alone cannot explain the synergistic interaction of activin and dexamethasone.
Not surprisingly, both activin and glucocorticoid signaling must be intact to generate a full synergistic response. If activin signaling is antagonized either through treatment with follistatin or overexpression of Smad7, the synergistic response is dramatically reduced (Fig. 3). Both follistatin and Smad7 also reduced the dexamethasone induction of FSHβ. This is most likely because LβT2 cells express endogenous activin (30), which is probably amplifying the dexamethasone induction. However, even in the presence of follistatin or Smad7, dexamethasone significantly induced FSHβ expression. Functional GR is also necessary to maintain synergy because use of a mutant GR results in a significant reduction in synergy (Fig. 7A).
Because it is unlikely that the synergy between activin and glucocorticoids is produced indirectly via modulation of other genes in the gonadotrope cell and based on the data that both activin and glucocorticoid signaling are necessary in maintaining this synergistic response, we hypothesize that synergy occurs via direct regulation of the promoter by signaling mechanisms instigated by each of these hormones. Our data indicate that multiple sites on the FSHβ promoter are necessary for a full synergistic response providing evidence that events directly at the level of the promoter are important in generating the synergistic response between activin and glucocorticoids.
Specifically, we show that multiple sites on the FSHβ promoter appear to be necessary for activin induction. Three of the five sites, the −267, −153, and −120, were originally characterized because they are putative SBEs (Fig. 4A). We also tested other putative SBEs on the mouse FSHβ promoter at −375 and −355; however, mutations at these sites had no effect on activin induction (data not shown), indicating that the sequence GTCT or AGAC by itself is not enough to confer activin responsiveness. Additionally, two novel activin-responsive regions characterized in this study, the −139 and −106, are not putative SBEs. The −139 site was originally characterized as a putative HRE (20, 29, 48), and the −106 corresponds to the −120 AP-1 site identified in the ovine FSHβ promoter (49). All five of these sites are important for maintaining a full activin response (Fig. 4B), and mutations at these sites reduce Smad3 induction of FSHβ as well (Fig. 4C). Mutations at the −120 and −267 elements had the most dramatic effects on Smad3 signaling, and both of these sites can bind Smads in vitro (28, 31) (Fig. 4D). Shi et al. (55) indicate that there is likely no difference in affinity for Smad binding between the half-sites and full sites, which could explain why the mutation in the half-site at −120 has as dramatic an effect on Smad3 signaling as the mutation in the full site at −267 (Fig. 4C). This may also explain why the mutation at the −120 element had a greater effect on activin responsiveness of the FSHβ promoter (Fig. 4B). Although the −153, −139, and −106 sites also disrupt the ability of Smad3 to activate FSHβ, 40–60% of wild-type induction is retained, and it appears that other base pairs or proteins bound to these regions are able to compensate for the mutation. EMSA analysis of these regions shows that specific LβT2 protein complexes can bind (Fig. 5); however, no supershift indicating Smad binding was detected under our conditions (data not shown). It will be of future interest to examine the LβT2 nuclear proteins binding to the −153, −139, and −106 regions and to investigate how these sites and their associated proteins are contributing to activin induction.
As with activin alone, mutations at these sites also inhibit the synergy between activin and glucocorticoids (Fig. 6B), although there is no effect on glucocorticoid induction, except, as expected, with the mutation at −139 (Fig. 6A). Interestingly, other than mutation of the −139 site, which reduces both glucocorticoid and activin signaling, a mutation in the −120 site produced the greatest reduction in synergy (Fig. 6B). Because this site is critical for Smad3 signaling (Fig. 4C) and can function as an SBE in vitro (Fig. 4D), this may reflect a critical role of Smad3 in generating a synergistic response. In support of this, our data indicate that Smad3 alone is sufficient to generate a synergistic response with glucocorticoids and GR (Fig. 8). For both activin signaling by itself and with synergy, multiple mutations in activin-responsive regions were required to abolish activin signaling (5× mutant, Fig. 4A) and inhibit the synergy between glucocorticoids and activin to the level of glucocorticoids alone (5× mutant, Fig. 6B), indicating that activin signals through multiple response elements.
Glucocorticoid induction and synergy with activin also require direct action at specific cis elements within the FSHβ promoter. Three putative GREs were previously identified in the FSHβ promoter at −381, −197, and −139, and GR was shown to bind the −381 GRE in vitro (20). All three of these sites inhibit glucocorticoid induction, and the −381 completely abolishes induction (Fig. 7B). Mutations in these sites also reduce synergy, with the −381 mutation having the most dramatic effect (Fig. 7C). Inhibiting direct DNA binding of GR through two different mutations of GR, either by inhibiting dimerization with the GRdim4 mutant or mutation of the DBD domain with the GR DBDmut, also significantly reduced synergy (Fig. 7A). The necessity of the −381 element and the data provided by the two GR mutants indicate the importance of direct DNA binding of GR in generating a synergistic response with activin.
The synergistic interaction between activin and glucocorticoids is interesting on multiple levels. Mechanistically, it is fascinating that two hormones could interact to induce the promoter so dramatically. Clearly, synergy is dependent on both the amount of GR (comparing the effect with endogenous vs. exogenous GR in Fig. 1) as well as the amount of activated Smad3 (compare synergy with activin in Fig. 1A with synergy with overexpressed Smad3 in Fig. 8). GR and Smad3 have been found to interact physically in other systems, although this interaction is not dependent on the GR DBD (53, 56), unlike the interaction between GR and other transcription factors such as AP-1 (34). Although it is possible that cotreatment with both hormones allows Smad3 and GR to interact and possibly stabilize one another on the promoter, it is unlikely that synergy is dependent solely on tethering of GR by Smads to the promoter because a mutation in the GR DBD, which still allows their interaction, dramatically reduces synergy (Fig. 7A). As discussed in the Results, no changes were seen at the level of the promoter in response to both activin and glucocorticoids, although these results may reflect an inadequacy in the techniques for visualizing these changes. Future research will be directed toward discovering additional cofactors involved in this synergistic interaction.
At a physiological level, this synergistic interaction raises important questions as well. During the estrous cycle, GnRH peaks during the afternoon of proestrus, and this is immediately followed by a peak in both FSH and LH. However, several hours later, during the early morning of estrus, there is a secondary rise in FSH that is independent of GnRH and is not accompanied by a peak in LH (4, 5). This differential regulation of FSH and LH is also mirrored at the transcriptional level of their respective β-subunits (6, 57). It is possible that the ability of glucocorticoids to synergize with activin is one mechanism by which the secondary FSH surge is generated, independent of GnRH or LH. Glucocorticoid levels, like progesterone, peak during the afternoon of proestrus (26, 27), and reducing glucocorticoid levels through adrenalectomy reduces the secondary FSH peak (11), indicating that glucocorticoids may be necessary for normal regulation of the reproductive axis. Interestingly, in the pituitary, activin/inhibin subunit and the activin receptor, ActIIR, mRNA levels are relatively constant during the estrous cycle (58), although follistatin and inhibin levels fluctuate (24, 25, 58). Therefore, it is tempting to postulate that activin may function to maintain FSH at basal levels, whereas other hormones interacting with activin (either antagonizing its function like inhibin and follistatin or synergistically interacting with it like GnRH and glucocorticoids) provide a means to rapidly and specifically regulate production of FSH. Additional in vivo studies will be required to understand to what extent glucocorticoids play a role in regulating FSH. In particular, it would be helpful to study a gonadotrope-specific knockout of GR to address what role that this receptor plays in regulating FSH and LH expression both in times of stress as well as in the normal functioning of the reproductive axis. Multiple studies have demonstrated a complex array of hormones that have been implicated in the regulation of the FSH secondary surge, including activin, inhibin, follistatin, GnRH, progesterone, and glucocorticoids. It is unlikely that any one hormone is responsible for generating the secondary surge; rather, this endocrine milieu functions together to regulate FSH synthesis.
In summary, this study has demonstrated the ability of activin and glucocorticoids to synergistically activate mouse FSHβ expression in the LβT2 immortalized gonadotrope cell line. This interaction is specific for FSHβ and occurs directly at the level of the promoter. Both activin and glucocorticoid signaling components are required for synergy to occur, and multiple cis elements within the mouse FSHβ promoter are important for generating this synergistic response. Furthermore, Smads play an essential role in the synergy between activin and glucocorticoids, and Smad3 and GR are sufficient to induce this synergistic response with glucocorticoids. This study highlights an interesting and potentially important physiological mechanism for regulation of FSHβ gene expression and emphasizes the importance of future studies investigating the physiological role of glucocorticoids in the regulation of the reproductive axis.
Acknowledgments
We thank Janice Sue Bailey and other members of the Mellon lab for helpful discussions and comments. We are grateful to Malcolm Low for providing the mouse FSHβ genomic clone and Keith Yamamoto for the pSG5-rGR plasmid. We thank Mark Lawson and Marit Kreidel for the 1.8LHβluc plasmid, Rik Derynck for the Smad7 expression plasmid, and Theresa Guise for the Smad3 expression plasmid. We also thank the UCSD Cancer Center DNA Sequencing Shared Resource for dideoxynucleotide sequencing.
This work was supported by National Institute of Child Health and Human Development, National Institutes of Health (NIH), through a cooperative agreement (U54 HD012303) as part of the Specialized Cooperative Centers Program in Reproduction Research (P.L.M.). This work was also supported by NIH Grant R37 HD020377 (to P.L.M.). S.M.M. was partially supported by NIH T32 DK007541. V.G.T. was supported by NIH F32 DK065437 and NIH T32 HD007203. D.C. was partially supported by NIH NRSA F32 HD41301 and the Lalor Foundation
Abbreviations
- AP-1
Activator protein 1
- ChIP
chromatin immunoprecipitation
- DBD
DNA-binding domain
- β-gal
β-galactosidase
- Gn-RHR
gonadotropin-releasing hormone receptor
- GR
glucocorticoid receptor
- GRE
glucocorticoid response element
- HRE
hormone response element
- MMTV
mouse mammary tumor virus
- SBE
Smad-binding element
- TK
thymidine kinase.
Footnotes
The authors have nothing to disclose.
References
- 1.Burns KH, Matzuk MM. Genetic models for the study of gonadotropin actions. Endocrinology. 2002;143:2823–2835. doi: 10.1210/endo.143.8.8928. [DOI] [PubMed] [Google Scholar]
- 2.Kumar TR, Wang Y, Lu N, Matzuk MM. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet. 1997;15:201–204. doi: 10.1038/ng0297-201. [DOI] [PubMed] [Google Scholar]
- 3.Jones RE. Human reproductive biology. 2nd. San Diego: Academic Press; 1997. [Google Scholar]
- 4.Daane TA, Parlow AF. Periovulatory patterns of rat serum follicle stimulating hormone and luteinizing hormone during the normal estrous cycle as revealed by radioimmunoassays: effects of pentobarbital. Endocrinology. 1971;88:653–663. doi: 10.1210/endo-88-3-653. [DOI] [PubMed] [Google Scholar]
- 5.Gay VL, Midgley AR, Jr, Niswender GD. Patterns of gonadotropin secretion associated with ovulation. Fed Proc. 1970;29:1880–1887. [PubMed] [Google Scholar]
- 6.Ortolano GA, Haisenleder DJ, Dalkin AC, Iliff-Sizemore SA, Landefeld TD, Maurer RA, Marshall JC. Follicle-stimulating hormone β-subunit messenger ribonucleic acid concentrations during the rat estrous cycle. Endocrinology. 1988;123:2946–2948. doi: 10.1210/endo-123-6-2946. [DOI] [PubMed] [Google Scholar]
- 7.Kaiser UB, Jakubowiak A, Steinberger A, Chin WW. Differential effects of gonadotropin-releasing hormone (GnRH) pulse frequency on gonadotropin subunit and GnRH receptor messenger ribonucleic acid levels in vitro. Endocrinology. 1997;138:1224–1231. doi: 10.1210/endo.138.3.4968. [DOI] [PubMed] [Google Scholar]
- 8.Papavasiliou SS, Zmeili S, Khoury S, Landefeld TD, Chin WW, Marshall JC. Gonadotropin-releasing hormone differentially regulates expression of the genes for luteinizing hormone α and β subunits in male rats. Proc Natl Acad Sci USA. 1986;83:4026–4029. doi: 10.1073/pnas.83.11.4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hoak DC, Schwartz NB. Blockade of recruitment of ovarian follicles by suppression of the secondary surge of follicle-stimulating hormone with porcine follicular field. Proc Natl Acad Sci USA. 1980;77:4953–4956. doi: 10.1073/pnas.77.8.4953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burger LL, Haisenleder DJ, Dalkin AC, Marshall JC. Regulation of gonadotropin subunit gene transcription. J Mol Endocrinol. 2004;33:559–584. doi: 10.1677/jme.1.01600. [DOI] [PubMed] [Google Scholar]
- 11.Tebar M, Bellido C, Sanchez-Criado JE. Luteinizing hormone (LH) and corticosterone in proestrous afternoon restore the follicle-stimulating hormone secretion at early estrus in adrenalectomized LH-releasing hormone antagonist-treated rats. Biol Reprod. 1995;52:63–67. doi: 10.1095/biolreprod52.1.63. [DOI] [PubMed] [Google Scholar]
- 12.Bamberger CM, Schulte HM, Chrousos GP. Molecular determinants of glucocorticoid receptor function and tissue sensitivity to glucocorticoids. Endocr Rev. 1996;17:245–261. doi: 10.1210/edrv-17-3-245. [DOI] [PubMed] [Google Scholar]
- 13.Murphy VE, Smith R, Giles WB, Clifton VL. Endocrine regulation of human fetal growth: the role of the mother, placenta, and fetus. Endocr Rev. 2006;27:141–169. doi: 10.1210/er.2005-0011. [DOI] [PubMed] [Google Scholar]
- 14.Marti A, Lazar H, Ritter P, Jaggi R. Transcription factor activities and gene expression during mouse mammary gland involution. J Mammary Gland Biol Neoplasia. 1999;4:145–152. doi: 10.1023/a:1018721107061. [DOI] [PubMed] [Google Scholar]
- 15.Ringstrom SJ, McAndrews JM, Rahal JO, Schwartz NB. Cortisol in vivo increases FSH-β mRNA selectively in pituitaries of male rats. Endocrinology. 1991;129:2793–2795. doi: 10.1210/endo-129-5-2793. [DOI] [PubMed] [Google Scholar]
- 16.McAndrews JM, Ringstrom SJ, Dahl KD, Schwartz NB. Corticosterone in vivo increases pituitary follicle-stimulating hormone (FSH)-β messenger ribonucleic acid content and serum FSH bioactivity selectively in female rats. Endocrinology. 1994;134:158–163. doi: 10.1210/endo.134.1.8275929. [DOI] [PubMed] [Google Scholar]
- 17.Kilen SM, Szabo M, Strasser GA, McAndrews JM, Ringstrom SJ, Schwartz NB. Corticosterone selectively increases follicle-stimulating hormone β-subunit messenger ribonucleic acid in primary anterior pituitary cell culture without affecting its half-life. Endocrinology. 1996;137:3802–3807. doi: 10.1210/endo.137.9.8756550. [DOI] [PubMed] [Google Scholar]
- 18.Leal AM, Blount AL, Donaldson CJ, Bilezikjian LM, Vale WW. Regulation of follicle-stimulating hormone secretion by the interactions of activin-A, dexamethasone and testosterone in anterior pituitary cell cultures of male rats. Neuroendocrinology. 2003;77:298–304. doi: 10.1159/000070896. [DOI] [PubMed] [Google Scholar]
- 19.Bohnsack BL, Szabo M, Kilen SM, Tam DH, Schwartz NB. Follistatin suppresses steroid-enhanced follicle-stimulating hormone release in vitro in rats. Biol Reprod. 2000;62:636–641. doi: 10.1095/biolreprod62.3.636. [DOI] [PubMed] [Google Scholar]
- 20.Thackray VG, McGillivray SM, Mellon PL. Androgens, progestins and glucocorticoids induce follicle-stimulating hormone β-subunit gene expression at the level of the gonadotrope. Mol Endocrinol. 2006;20:2062–2079. doi: 10.1210/me.2005-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matzuk MM, Kumar TR, Bradley A. Different phenotypes for mice deficient in either activins or activin receptor type II. Nature. 1995;374:356–560. doi: 10.1038/374356a0. [DOI] [PubMed] [Google Scholar]
- 22.Vassalli A, Matzuk MM, Gardner HA, Lee KF, Jaenisch R. Activin/inhibin β B subunit gene disruption leads to defects in eyelid development and female reproduction. Genes Dev. 1994;8:414–427. doi: 10.1101/gad.8.4.414. [DOI] [PubMed] [Google Scholar]
- 23.Corrigan AZ, Bilezikjian LM, Carroll RS, Bald LN, Schmelzer CH, Fendly BM, Mason AJ, Chin WW, Schwall RH, Vale W. Evidence for an autocrine role of activin B within rat anterior pituitary cultures. Endocrinology. 1991;128:1682–1684. doi: 10.1210/endo-128-3-1682. [DOI] [PubMed] [Google Scholar]
- 24.Woodruff TK, Besecke LM, Groome N, Draper LB, Schwartz NB, Weiss J. Inhibin A and inhibin B are inversely correlated to follicle-stimulating hormone, yet are discordant during the follicular phase of the rat estrous cycle, and inhibin A is expressed in a sexually dimorphic manner. Endocrinology. 1996;137:5463–5467. doi: 10.1210/endo.137.12.8940372. [DOI] [PubMed] [Google Scholar]
- 25.Besecke LM, Guendner MJ, Sluss PA, Polak AG, Woodruff TK, Jameson JL, Bauer-Dantoin AC, Weiss J. Pituitary follistatin regulates activin-mediated production of follicle-stimulating hormone during the rat estrous cycle. Endocrinology. 1997;138:2841–2848. doi: 10.1210/endo.138.7.5279. [DOI] [PubMed] [Google Scholar]
- 26.Buckingham JC, Dohler KD, Wilson CA. Activity of the pituitary-adrenocortical system and thyroid gland during the oestrous cycle of the rat. J Endocrinol. 1978;78:359–366. doi: 10.1677/joe.0.0780359. [DOI] [PubMed] [Google Scholar]
- 27.Nichols DJ, Chevins PF. Plasma corticosterone fluctuations during the oestrous cycle of the house mouse. Experientia. 1981;37:319–320. doi: 10.1007/BF01991678. [DOI] [PubMed] [Google Scholar]
- 28.Gregory SJ, Lacza CT, Detz AA, Xu S, Petrillo LA, Kaiser UB. Synergy between activin A and gonadotropin-releasing hormone in transcriptional activation of the rat follicle-stimulating hormone-beta gene. Mol Endocrinol. 2005;19:237–254. doi: 10.1210/me.2003-0473. [DOI] [PubMed] [Google Scholar]
- 29.Spady TJ, Shayya R, Thackray VG, Ehrensberger L, Bailey JS, Mellon PL. Androgen regulates FSHβ gene expression in an activin-dependent manner in immortalized gonadotropes. Mol Endocrinol. 2004;18:925–940. doi: 10.1210/me.2003-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pernasetti F, Vasilyev VV, Rosenberg SB, Bailey JS, Huang HJ, Miller WL, Mellon PL. Cell-specific transcriptional regulation of FSHβ by activin and GnRH in the LβT2 pituitary gonadotrope cell model. Endocrinology. 2001;142:2284–2295. doi: 10.1210/endo.142.6.8185. [DOI] [PubMed] [Google Scholar]
- 31.Suszko MI, Lo DJ, Suh H, Camper SA, Woodruff TK. Regulation of the rat follicle-stimulating hormone β-subunit promoter by activin. Mol Endocrinol. 2003;17:318–332. doi: 10.1210/me.2002-0081. [DOI] [PubMed] [Google Scholar]
- 32.Bailey JS, Rave-Harel N, Coss D, McGillivray SM, Mellon PL. Activin regulation of the follicle-stimulating hormone β-subunit gene involves Smads and the TALE homeodomain proteins Pbx1 and Prep1. Mol Endocrinol. 2004;18:1158–1170. doi: 10.1210/me.2003-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGillivray SM, Bailey JS, Ramezani R, Kirkwood BJ, Mellon PL. Mouse GnRH receptor gene expression is mediated by the LHX3 homeodomain protein. Endocrinology. 2005;146:2180–2185. doi: 10.1210/en.2004-1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Heck S, Kullmann M, Gast A, Ponta H, Rahmsdorf HJ, Herrlich P, Cato AC. A distinct modulating domain in glucocorticoid receptor monomers in the repression of activity of the transcription factor AP-1. EMBO J. 1994;13:4087–4095. doi: 10.1002/j.1460-2075.1994.tb06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alarid ET, Windle JJ, Whyte DB, Mellon PL. Immortalization of pituitary cells at discrete stages of development by directed oncogenesis in transgenic mice. Development. 1996;122:3319–3329. doi: 10.1242/dev.122.10.3319. [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg SB, Mellon PL. An Otx-related homeodomain protein binds an LHβ promoter element important for activation during gonadotrope maturation. Mol Endocrinol. 2002;16:1280–1298. doi: 10.1210/mend.16.6.0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kononen J, Honkaniemi J, Gustafsson JA, Pelto-Huikko M. Glucocorticoid receptor colocalization with pituitary hormones in the rat pituitary gland. Mol Cell Endocrinol. 1993;93:97–103. doi: 10.1016/0303-7207(93)90144-9. [DOI] [PubMed] [Google Scholar]
- 38.Slinker BK. The statistics of synergism. J Mol Cell Cardiol. 1998;30:723–731. doi: 10.1006/jmcc.1998.0655. [DOI] [PubMed] [Google Scholar]
- 39.Ham J, Thomson A, Needham M, Webb P, Parker M. Characterization of response elements for androgens, glucocorticoids and progestins in mouse mammary tumour virus. Nucleic Acids Res. 1988;16:5263–5276. doi: 10.1093/nar/16.12.5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beato M. Gene regulation by steroid hormones. Cell. 1989;56:335–344. doi: 10.1016/0092-8674(89)90237-7. [DOI] [PubMed] [Google Scholar]
- 41.Coss D, Thackray VG, Deng CX, Mellon PL. Activin regulates luteinizing hormone β-subunit gene expression through smad-binding and homeobox elements. Mol Endocrinol. 2005;19:2610–2623. doi: 10.1210/me.2005-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maya-Nunez G, Conn PM. Transcriptional regulation of the GnRH receptor gene by glucocorticoids. Mol Cell Endocrinol. 2003;200:89–98. doi: 10.1016/s0303-7207(02)00419-7. [DOI] [PubMed] [Google Scholar]
- 43.Norwitz ER, Xu S, Jeong KH, Bedecarrats GY, Winebrenner LD, Chin WW, Kaiser UB. Activin A augments GnRH-mediated transcriptional activation of the mouse GnRH receptor gene. Endocrinology. 2002;143:985–997. doi: 10.1210/endo.143.3.8663. [DOI] [PubMed] [Google Scholar]
- 44.Lebrun JJ, Takabe K, Chen Y, Vale W. Roles of pathway-specific and inhibitory Smads in activin receptor signaling. Mol Endocrinol. 1999;13:15–23. doi: 10.1210/mend.13.1.0218. [DOI] [PubMed] [Google Scholar]
- 45.Ethier JF, Findlay JK. Roles of activin and its signal transduction mechanisms in reproductive tissues. Reproduction. 2001;121:667–675. doi: 10.1530/rep.0.1210667. [DOI] [PubMed] [Google Scholar]
- 46.Bernard DJ. Both SMAD2 and SMAD3 mediate activin-stimulated expression of the follicle-stimulating hormone β-subunit in mouse gonadotrope cells. Mol Endocrinol. 2004;18:606–623. doi: 10.1210/me.2003-0264. [DOI] [PubMed] [Google Scholar]
- 47.Zawel L, Dai JL, Buckhaults P, Shou S, Kinzler KW, Vogelstein B, Karn SE. Human Smad3 and Smad 4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- 48.Webster JC, Pedersen NR, Edwards DP, Beck CA, Miller WL. The 5′-flanking region of the ovine follicle-stimulating hormone-beta gene contains six progesterone response elements: three proximal elements are sufficient to increase transcription in the presence of progesterone. Endocrinology. 1995;136:1049–1058. doi: 10.1210/endo.136.3.7867558. [DOI] [PubMed] [Google Scholar]
- 49.Strahl BD, Huang HJ, Pedersen NR, Wu JC, Ghosh BR, Miller WL. Two proximal activating protein-1-binding sites are sufficient to stimulate transcription of the ovine follicle-stimulating hormone-beta gene. Endocrinology. 1997;138:2621–2631. doi: 10.1210/endo.138.6.5205. [DOI] [PubMed] [Google Scholar]
- 50.Lamba P, Santos MM, Philips DP, Bernard DJ. Acute regulation of murine follicle-stimulating hormone β-subunit transcription by activin A. J Mol Endocrinol. 2006;36:201–220. doi: 10.1677/jme.1.01961. [DOI] [PubMed] [Google Scholar]
- 51.Suszko MI, Balkin DM, Chen Y, Woodruff TK. Smad3 mediates activin-induced transcription of follicle-stimulating hormone β-subunit gene. Mol Endocrinol. 2005;19:1849–1858. doi: 10.1210/me.2004-0475. [DOI] [PubMed] [Google Scholar]
- 52.Rogers SL, Nabozny G, McFarland M, Pantages-Torok L, Archer J, Kalkbrenner F, Zuvela-Jelaska L, Haynes N, Jiang H. Four mutations in the GR dimerization domain result in perinatal lethal mice. Keystone Symposia, Nuclear Receptors: Steroid Sisters; Keystone, CO. 2004. Abstract 316. [Google Scholar]
- 53.Li G, Wang S, Gelehrter TD. Identification of glucocorticoid receptor domains involved in transrepression of TGFβ action. J Biol Chem. 2003;278:41779–41788. doi: 10.1074/jbc.M305350200. [DOI] [PubMed] [Google Scholar]
- 54.Haisenleder D, Dalkin A, Ortolano G, Marshall J, Shupnik M. A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991;128:509–517. doi: 10.1210/endo-128-1-509. [DOI] [PubMed] [Google Scholar]
- 55.Shi Y, Wang YF, Jayaraman L, Yang H, Massague J, Pavletich NP. Crystal structure of a Smad MH1 domain bound to DNA: insights on DNA binding in TGF-β signaling. Cell. 1998;94:585–594. doi: 10.1016/s0092-8674(00)81600-1. [DOI] [PubMed] [Google Scholar]
- 56.Song CZ, Tian X, Gelehrter TD. Glucocorticoid receptor inhibits transforming growth factor-β signaling by directly targeting the transcriptional activation function of Smad3. Proc Natl Acad Sci USA. 1999;96:11776–11781. doi: 10.1073/pnas.96.21.11776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zmeili SM, Papavasiliou SS, Thorner MO, Evans WS, Marshall JC, Landefeld TD. Alpha and luteinizing hormone β-subunit messenger ribonucleic acids during the rat estrous cycle. Endocrinology. 1986;119:1867–1869. doi: 10.1210/endo-119-4-1867. [DOI] [PubMed] [Google Scholar]
- 58.Halvorson LM, Weiss J, Bauer-Dantoin AC, Jameson JL. Dynamic regulation of pituitary follistatin messenger ribonucleic acids during the rat estrous cycle. Endocrinology. 1994;134:1247–1253. doi: 10.1210/endo.134.3.8119165. [DOI] [PubMed] [Google Scholar]