

Abstract
In adult tissues, multi-potent progenitor cells are some of the most primitive members of the developmental hierarchies that maintain homeostasis. That progenitors and their more mature progeny share identical genomes, suggests that fate decisions are directed by interactions with extrinsic soluble factors, ECM, and other cells, as well as physical properties of the ECM. To understand regulation of fate decisions, therefore, would require a means of understanding carefully choreographed combinatorial interactions. Here we used microenvironment protein microarrays to functionally identify combinations of cell-extrinsic mammary gland proteins and ECM molecules that imposed specific cell fates on bipotent human mammary progenitor cells. Micropatterned cell culture surfaces were fabricated to distinguish between the instructive effects of cell–cell versus cell–ECM interactions, as well as constellations of signaling molecules; and these were used in conjunction with physiologically relevant 3 dimensional human breast cultures. Both immortalized and primary human breast progenitors were analyzed. We report on the functional ability of those proteins of the mammary gland that maintain quiescence, maintain the progenitor state, and guide progenitor differentiation towards myoepithelial and luminal lineages.
Introduction
Within a given tissue, cell types with distinct functions are distributed into specific regions, suggesting that microenvironment begets cell function. The fact that tissue-specific functions are often lost in normal cells when they are explanted into culture conditions suggests that the lowest functional unit of a tissue, is a cell associated with a particular microenvironment.1 Adult stem cells are maintained inside a specialized microenvironment called a niche,2 whereas their mature progeny reside in surrounding microenvironments that are distinct from the niche. The ability of adult stem cells to self-maintain, as well as to give rise to differentiated progeny, reflects an ability to respond to the changing demands of their tissue.3 It has been predicted that those demands are communicated to stem cells by the combinatorial application of cell–cell, cell–ECM, and cell–soluble factor interactions as well as by geometric and physical properties of the microenvironment4–6 Understanding how microenvironmental milieus interact with stem and progenitor cells to impose discrete cell fate decisions is of fundamental interest.
The mammary gland is widely used as a model system to study the role of the microenvironment in cancer, morphogenesis, and lactational differentiation,7 but little is known about the components of the extracellular milieu that orchestrate mammary stem and progenitor cell activities. Human mammary gland stem cells reside in nests within terminal ducts, and give rise to the progenitor cells, luminal epithelial, and myoepithelial cells8 that are required to maintain homeostasis in this dynamic, branching, and secretory organ. Using candidate-based cell culture approaches, a couple of developmental pathways have been implicated to regulate aspects of human mammary progenitor cell differentiation and self-renewal.9,10 However, stem and progenitor cells in vivo experience a large number of cell extrinsic interactions simultaneously, the net result of which are discrete cell fate decisions. Here we have utilized highly parallel microenvironment microarrays (MEArrays™), and 3D organotypic culture models to demonstrate that different cell fate decisions in human bipotent mammary progenitor cells are imposed by distinct combinations of microenvironment constituents. Taking a combinatorial approach, relative to candidate-based approach, allowed us to screen unique combinations of multiple known mammary gland microenvironment proteins to identify previously unrecognized activities with respect to stem cell behavior that occur as a result of those combinations. In doing so, we have identified putative functional roles for a number of molecules that are known to be expressed in human mammary gland, but heretofore had not been ascribed a role for mammary stem or progenitor cell regulation.
Insight, innovation, integration.
Stem cells in adult tissues reside within niches and maintain homeostasis within an organ for the life of an organism. To do so they must self-maintain and generate differentiated progeny; the decision to do one or the other is called a cell fate decision. How the microenvironment that surrounds stem cells influences cell fate decisions is poorly understood. This is because the compositions of microenvironments are complex and stem cells are extremely scarce. Dissecting a human stem cell niche, understandably, is even more difficult because of the inability to do experiments in vivo. To begin to overcome these obstacles, we utilized a new technology that enables highly parallel functional analysis of combinatorial microenvironments, image analysis of 3D organotypic cultures and micropatterned culture substrata. Here we have identified combinations of components in the human mammary microenvironment that impose distinct cell fate decisions and could putatively direct mammary progenitor cell functions in vivo.
Results
Characterization of bipotent human mammary progenitor cells
Human mammary stem or progenitor cells that express keratin (K)14, K19, K15, SSEA4,8 and have high levels of ALDH activity11 give rise to the basic functional units of the mammary gland, termed terminal ductal lobular units (TDLU).8,12,13 TDLUs are bilayered, branching, and secretory structures composed of an inner layer of K8- and K19-expressing luminal epithelial cells (LEPs; K14−/K19+/K8+) and an outer layer of K14-expressing myoepithelial cells (MEPs; K14+/K19−/K8−). In order to study many unique microenvironment–cell interactions using the same human stem/progenitor cells and circumvent problems of access to primary tissue and genetic variability as well as survival, we utilized the human mammary progenitor-derived cell line, D920. Importantly, however, our principal findings were confirmed also with primary mammary progenitor cells (discussed below). Briefly, D920s were derived when the ESA+/Muc1− subpopulation was isolated from a normal reduction mammoplasty specimen and immortalized with HPV E6 and E7 proteins. The cells which comprise the D920 cultures are non-malignant, and contain bipotent human mammary progenitor cells that are capable of forming TDLUs in vivo in cleared fat pads of NOD/SCID mice and in 3D Matrigel cultures.14 Here we show that the CD29hi subpopulation of D920 (Fig. 1A(a)) was enriched for K14+/K19+ cells that stochastically gave rise to differentiated LEP and MEP cells when cultured on collagen1-coated plastic (Fig. 1A(b)). In three dimensional (3D) Matrigel cultures, CD29hi cells were enriched also for TDLU-forming activity (Fig. 1A(c and d); Fig. S1†) and self-renewal (Fig. S1†). Hereafter they will be referred to as D920 progenitors.
Fig. 1.
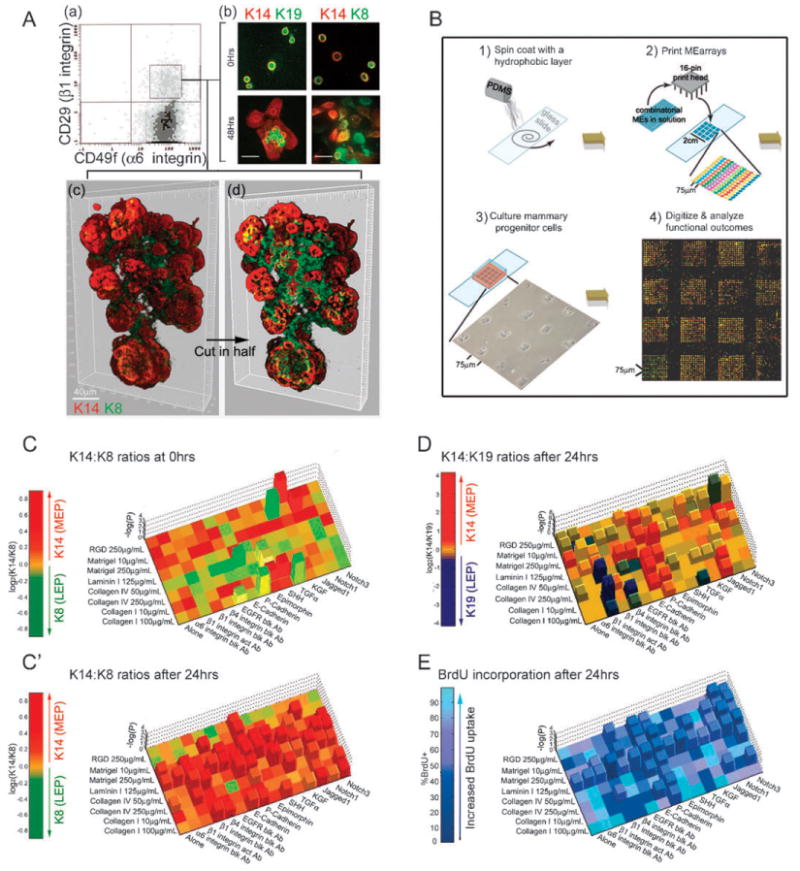
Mammary progenitor cells make distinctive cell fate decisions in response to different combinatorial microenvironments. (A) CD49f and CD29 expression in D920 parental cells cultured on collagen-coated plates was determined by FACS: (a) A distinctive CD29hi subpopulation (box, 1–3% of total) can be identified, although most of the cells in the population express at least some CD29. The CD29hi population (designated D920 progenitors) was FACS-sorted and stained to determine (b) expression of keratin (K) 8, 14, and 19 as a function of time in culture, immediately after isolation (0 h) and 48 h later on 2D cultures. (c) A deconvoluted 3D reconstruction is shown of a TDLU grown from a single CD29hi cell that was embedded in 3D Matrigel for 20 days. Staining for markers of myoepithelial cells (K14, red) and of luminal epithelial cells (K8, green) is shown in an opaque representation of the TDLU). (d) A digital slice through (c) to enable visualization of the inner K8+ cells. Bars represent 40 μm. (B) Microenvironment microarrays (MEArrays) were fabricated as described in material and methods, steps 1 and 2. D920 progenitor cells were isolated as described in Fig. 1A and cultured on MEArrays, step 3. The arrays were immuno-stained for K8/K14, K19/K14 or BrdU incorporation after 24 h, and digitized with a microarray scanner, step 4. (C–D) Results are expressed as log2 of the K14 : K8 or K14 : K19 ratio of mean intensity (e.g. log2(1/1) = 0), where increases in K14 (myoepithelial-like, red) or in K8 or K19 (luminal-like, green or blue, respectively) expression on any given microenvironment are compared to the keratin ratios in cells grown on collagen I microenvironments, which serves as a control baseline. Two-color heat maps used to represent ratiometric data were modified by also including a z-axis (–log10(p)), only for those microenvironments that induced a significant (at least p < 0.05 by Dunnette's test) change in the mean keratin ratios. (E) Progenitors were treated with BrdU for the final 4 h of a 24 h incubation to determine the microenvironmental effects on cell proliferation. The percent BrdU pixels (stained with anti-BrdU) of total DNA pixels (stained with Topro3) for all of the pair-wise combinations.
Combinatorial microenvironments impose distinct cell fate decisions on bipotent progenitors
We know little of what the signals originating from combinations of the molecules comprising the breast microenvironment are, or of the discrete cell fate decisions they are translated into. To shed light on this question, we fabricated what we have coined microenvironment microarrays (MEArrays™) (Fig. 1B) by robotically printing 192 unique combinatorial microenvironments with 12 replicates each (2304 total features) composed of many of the ECM and signaling molecules that are expressed in mammary gland, but which hitherto were not associated necessarily with stem cell function. Following culture of D920 progenitors on MEArrays, the ratios of expressed LEP (K8 or K19) versus MEP (K14) protein markers were analyzed using immunofluorescence and image analysis software to determine their lineage. K19 acts as a switch keratin and is expressed in both luminal and progenitor cells,15 whereas K8 is restricted to luminal cells. Thus juxtaposition of the patterns of K14 : K8 and K14 : K19 ratios allowed us to distinguish between conditions that induced K14+/K19+ (the progenitors), K14+/K8−/K19− (MEP), or K14−/K8+/K19+ (LEP) cell fates. Each unique pair-wise combinatorial microenvironment contained a constituent that was shared in common with at least four other microenvironments. In this way, dominant components that induced a phenotype in progenitors, irrespective of other constituents, were identified based on differentiation trends that crossed multiple unique-but related-substrata. D920 progenitors which normally account for 0.5–4% of the total cells based on colony-forming, by 24 h on collagen1-alone stochastically give rise to LEP (25–57%) and MEP (40–74%). Therefore the keratin ratios in progenitors on each unique combinatorial microenvironment were compared to ratios in D920 progenitors on collagen1-alone microenvironments.
There were no significant trends in binding to the substrata among K14 : K8 subtypes 30 min after cell adhesion (n = 3) (Fig. 1C), indicating that for the most part progenitors were binding evenly across the different substrata and that particular constituents of a given microenvironment were not “panning” for a subpopulation among the progenitor-enriched cells. After 24 h, however, patterns of statistically significant shifts in K14 : K8 (n = 4) (Fig. 1C′) and K14 : K19 (n = 3) (Fig. 1D) phenotypes emerged, indicating that some microenvironments induced differentiation towards a particular lineage. Patterns of changes in cell proliferation, as measured by BrdU incorporation, often correlated with the cell fate trends either positively or negatively (n = 3) (Fig. 1E). In collagen1-alone features located randomly within the arrays, phenotypes were similar to each other, suggesting that paracrine signals were not strong cell fate effectors within this time frame. However, the activities of three individual microenvironmental components generated notable and significant trends in keratin and proliferation phenotypes: 1-laminin1, a basement membrane (BM) protein which is a crucial signaling ligand for functional differentiation in the mammary gland;16,17 2-Jagged1, a Notch pathway ligand expressed essentially in MEP cells (by in situ hybridization),18 and which we now show to be also present in the stem cells in vivo (Fig. 2B); and 3-P-cadherin, a MEP adherens junction protein.19 That each of these proteins is expressed in the ducts proximal to putative mammary stem cells was verified in tissue sections from normal reduction mammoplasty specimens (Fig. 2).
Fig. 2.
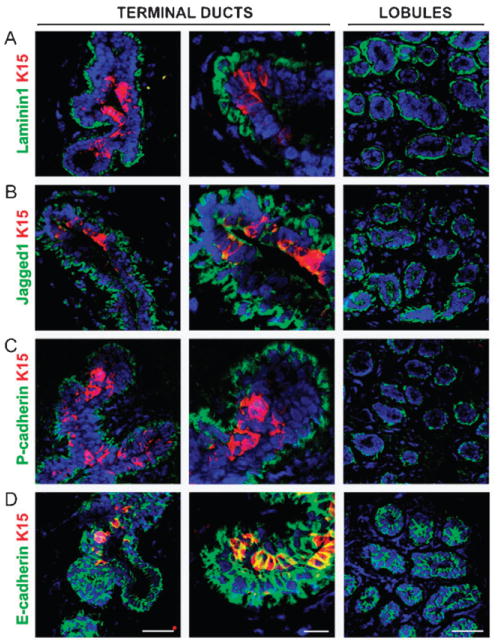
The MEArray-identified proteins are expressed proximal to the putative stem cell niche in vivo. Normal reduction mammoplasty specimens were sectioned and evaluated by immunofluorescence to determine where the MEArray-identified proteins are expressed relative to cells expressing the putative mammary stem cell marker K15 (red, in all images) in the terminal ducts. For all antibody combinations, images are shown of terminal ducts, which contain K15+ cells, and of lobules, which do not contain K15+ cells. (A) Laminin1 (laminin-111) (green) surrounds the glands as part of the basement membrane, K15+ cells can be seen forming occasional protrusions that contact the laminin1. (B) Jagged1 (green) is expressed in the myoepithelial cells, and based on the yellow-appearing overlap, also in some K15+ ductal cells. (C) P-cadherin (green) is expressed on the basal and lateral edges of myoepithelial cells. (D) E-cadherin (green) is strongly expressed in the luminal layer including K15+ cells.
Laminin1 maintains progenitors in a quiescent state
Laminin1-containing gels are known to inhibit the growth of mammary epithelial cells20 and to prevent them from apoptosing,21 thus maintaining tissue homeostasis. On laminin1-containing features, BrdU incorporation was decreased in all cases (40–70% less than collagen1) (Fig. 1E and S2A′†), irrespective of which other molecule was paired with laminin1 and which keratin phenotype was induced (Fig. 1C′ and 1D, and S2A and A′†). Thus, the major function of laminin1 with respect to progenitor cell functions is to maintain a quiescent or growth-suppressed state (Table 1).
Table 1.
Summary of conditions that have functional consequences in mammary progenitors
| Signaling proteins that also exist in vivo | Cell fate | ECM | BrdU incorporation | Antagonists | Proposed function for progenitors |
|---|---|---|---|---|---|
| Laminin1 | Dependent on partner | Decrease | None identified | Maintains quiescence suppresses growth | |
| Jagged1 | aK14+/K19+ progenitor | Collagen4-rich Laminin1-rich (e.g. Matrigel) | Dependent on partner | GSI, AG, LY anti-β1, α6 integrins anti-EGFR | Maintains the progenitor cell pool |
| P-Cadherin | K14+/K19−/K8− MEP | All | Decrease | GSI, LY, anti-α6,-β4,-β1 integrins | Guides differentiation into MEP |
| E-Cadherin (or a cell–cell contact) |
K14−/K19+/K8+ GATA3+ LEP |
Collagen1 | ND |
bEGTA bCa++-free media |
Facilitates differentiation into LEP |
MEP = myoepithelial, LEP = luminal epithelial, GSI = γ-secretase inhibitor, AG = AG1478, LY = LY294002, ND = no data.
Paired with collagen1, Jagged1 induced an K14+/K19−/K8− MEP- or basal-like phenotype that was not consistently growth suppressed.
5 mM EGTA for 24 h or Ca ++ -free media prevented generation of the K14−/K8 +. LEP cells under conditions where cell–cell contact was permitted (data not shown).
The role of the Notch pathway is context-dependent in mammary progenitors
Dontu et al. showed previously that stimulation of the Notch pathway by a synthetic “DSL-peptide” (Delta-Serrate-LAG2 motif-containing peptide) in putative human mammary progenitors, cultured as ‘mammospheres’, would lead to self-renewal.9 This would suggest that a Notch receptor ligand in the mammary gland could function in a similar role in vivo. In the same report it was noted that DSL-peptide stimulation of mammospheres followed by culture on collagen1-plates lead to differentiation into MEP cells.9 In another report, Notch activity was shown to be required for maintenance of LEP cells.22 Our parallel combinatorial approach demonstrates that different cell fate decisions can be induced in a context-dependent manner by the Notch ligand, Jagged1. In combination with collagen4-rich or laminin1-rich microenvironments—including Matrigel—Jagged1 maintained a K14+/K19+/K8− progenitor-like phenotype with few exceptions (Fig. 1C′ and 1D, and S2B and S2D†); however the BrdU profiles were not different from controls suggesting that there were no additional proliferation-suppressive roles for Jagged1 under those conditions (Fig. 1E and S2E†). The Jagged1-induced keratin phenotype was dose-dependent (Fig. S3†) and was blocked by γ-secretase inhibitors (GSI) (Fig. S2B†), which prevents intracellular cleavage of activated Notch receptors.23 When combined with collagen1, Jagged1 induced a K14+/K19−/K8− MEP-like phenotype (Fig. 1C′ and 1D, and S2B and S2D†). In vivo Jagged1 was expressed primarily by the MEPs, but some cells within the nests of stem and progenitors in the terminal ducts also expressed the protein (Fig. 2B). Taken together, the functional role of Jagged1 is context-dependent (Table 1).
P-Cadherin imposes differentiation into the myoepithelial phenotype
One way in which cell experience their neighbors is through adherins junction proteins. P-Cadherin is thought to negatively regulate cell growth in the mammary gland;24 however, its role with respect to stem and progenitor cells is unknown. Progenitors cultured on P-cadherin-containing substrata incorporated 50–80% less BrdU than cells on collagen1 (Fig. 1E and S2E†), consistent with P-cadherin's proposed role as a negative growth regulator. In addition to growth control, P-cadherin also directed differentiation towards a MEP-like cell fate when combined with any of the ECM molecules tested: it induced a dose dependent K14+/K19−/K8− phenotype (Fig. 1C′ and 1D, Fig. S2C and S2D, and Fig. S3†). This was blocked by GSI which prevents the intracellular cleavage of cadherin receptors23 (Fig. S3C†). That the P-cadherin-induced phenotype in D920 progenitors was antagonized by inclusion of integrin-blocking antibodies (Fig. S2C, D, and E†) suggested that the ECM molecules play an active signaling role in the differentiation process25 (Table 1). It was previously shown that cell–ECM contact increases P-cadherin expression.26 The combined data suggest that we have uncovered a self-reinforcing loop that maintains the MEP cell fate, in which P-cadherin engagement imposes the MEP phenotype, and cell–ECM engagement maintains P-cadherin expression.
Cell–cell contact facilitates differentiation into the luminal phenotype
Surprisingly, the predominant phenotypes induced on the MEArrays under all the conditions tested so far were MEP- or progenitor-phenotypes with almost no increases in the LEP-phenotype. We reasoned that every MEArray feature we utilized so far incorporated some form of ECM molecule to mediate cell attachment, whereas in vivo, LEPs reside essentially in a cell–cell context surrounded by MEPs and other LEPs. To determine the effect of cell–cell plus cell–ECM contacts versus only cell–ECM contacts, cell fate was measured as a function of time in progenitors that were cultured on micropatterned arrays of 1600 μm2 square-shaped features of collagen1 at 0 (Fig. 3A) and 24 h (Fig. 3B–H) time points. Cell fate specification was determined using immunofluorescence and image analysis, to measure expression of K14, K8, K19 and GATA3, a transcription factor known to be required for LEP induction in rodents,27,28 in single cells. Micropatterns allowed cell–ECM interaction while controlling cell–cell contact. Immediately following progenitor enrichment and adhesion the progenitors shared a similar keratin phenotype (Fig. 3A). After 24 h, Single progenitors that were alone on a feature differentiated into K14+/K8− MEP cells (Fig. 3B and E) and did not express GATA3 (Fig. 3G). Excitingly, however, when multiple cells were touching, the GATA3+ K14−/K8+ LEP phenotype emerged (Fig. 3C and E), suggesting that cell–cell contact facilitated LEP differentiation. K19 and K14 expression were also evaluated: single cells were either K14+/K19− MEP or K14+/K19+ progenitors, but K14−/K19+ LEP were observed only when cell–cell contact was permitted (Fig. S4). To determine whether E-cadherin, an important adherens junction protein, participated in mediating the LEP phenotype, we subjected single cells with E-cadherin-conjugated beads and showed that this treatment also induced the GATA3+ K14−/K8+ LEP phenotype (Fig. 3D, F and H). Therefore, E-cadherin junctions between two epithelial cells could themselves be the signal that induces GATA3, allowing the LEP-phenotype to emerge (Table 1). Because we observed that E-cadherin is expressed not only by LEP, but also by the mammary stem cells in vivo (Fig. S2†), we suggest that homotypic E-cadherin interactions must be counter balanced by inhibitors within the niche to prevent premature specification of the LEP cell fate.
Fig. 3.
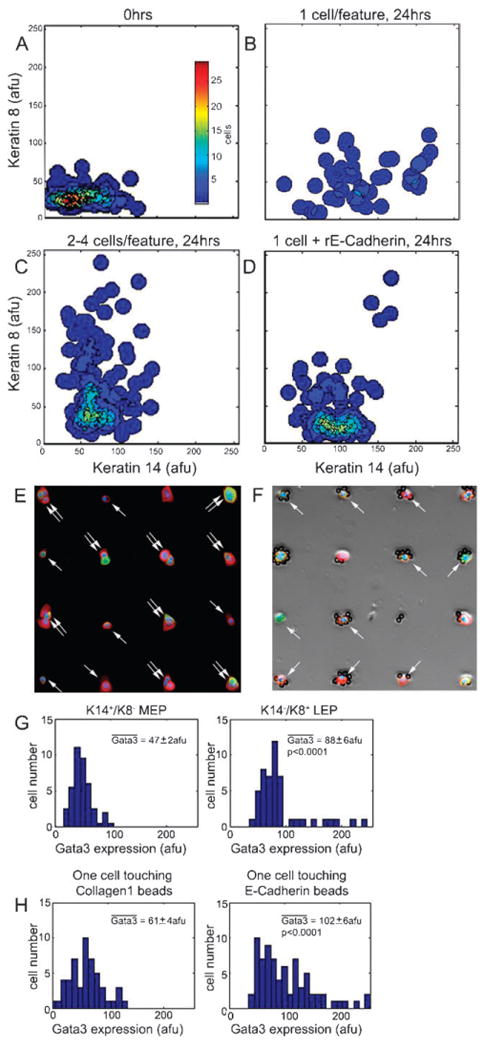
Cell–cell contact is necessary for induction of the luminal-like phenotype. To control cell–cell contact, D920 progenitor cells were cultured on collagen I that was micropatterned into arrays of 1600 μm2 square-shaped features for 24 h. (A–D) Contour plots are shown of the absolute intensities of K8 and K14 expressed in individual cells. These range from 0 to 255 arbitrary fluorescence units (afu) (150 cells/plot/experiment unless otherwise indicated, n = 3). (A) K8/K14 profiles at 0 h immediately after adhesion, (B) after 24 h where only one cell was bound per feature (73 cells), (C) after 24 h where 2–4 cells were bound to each feature enabling cell–cell contact, and (D) after 24 h with one cell per feature plus the addition of recombinant E-cadherin-conjugated silicon beads to mimic cell-cell contact. Representative immunoflourescence images of DAPI stain (blue), and K8 (green) and K14 (red) expression in D920 progenitors (E) after 24 h of culture on collagen patterns, and (F) after 24 h on patterns with E-cadherin beads, fluorescence image is merged with a phase image to aid in visualization of the beads. (E) Single arrows identify features with only one cell, two arrows identify features with >2 cells. (F) Arrows identify single cells touching beads. (G–H) Histograms represent the absolute fluorescence intensity of Gata3 expression per cell, which ranges in value from 0 to 255 afu (75 single cells/histogram/experiment, n = 2). (G) Gata3 expression in D920 progenitor cells grown on collagen patterns that were permitted cell-cell contact. (H) Gata3 expression in cells 24 h after single D920 progenitors were grown together with either collagen I-coated or E-cadherin-coated beads. p values were determined with Mann–Whitney tests.
MEArray-based predictions were validated in a 3D organotypic culture model
To elucidate whether the MEArray profiles were indicative of the activities of Jagged1 and P-cadherin in producing TDLUs, D920 progenitors were cultured in 3D Matrigel, but in the presence of the recombinant proteins. Cells with K14 and K19 phenotypes were then determined both qualitatively in 10 day cultures, and quantitatively in 24 h cultures using confocal microscopy. After 10 days progenitors in control cultures began to generate bilayered branching structures (Fig. 4A, left), Jagged1 protein maintained a K14+/K19+ progenitor phenotype (Fig. 4A, middle), and P-cadherin induced K14+/K19− MEP phenotype (Fig. 4A, right). Using image analysis software, we quantified changes in K14 : K19 ratios after only 24 h, a time-point chosen to approximate the MEArray experiments. In addition, the progenitors were mainly still single cells at this time, and thus were more amenable to image quantification. The image analysis tools were required to detect changes in keratin expression that were not obvious to the eye. Cells in control cultures exhibited a wide distribution of K14 : K19 phenotypes suggesting they stochastically differentiated (Fig. 4B, left). When treated with Jagged1, the distribution shifted towards a K14+/K19+ progenitor phenotype (Fig. 4B, middle), and P-cadherin induced a K14+/K19− MEP-like phenotype (Fig. 4B, right). Additionally, the number of progenitors that expressed the putative mammary stem cell marker, SSEA4, increased after Jagged1 treatment, and the number of cells that expressed the myoepithelial marker, α-smooth muscle actin, increased after P-cadherin treatment (Fig. S5†). The cell fates measured after 24 h were predictive of differentiation trajectories that would be sustained for as long as 10 days in the presence of inducing molecules. Importantly, the basic trends observed on the MEArrays were reproduced also in 3D cultures.
Fig. 4.
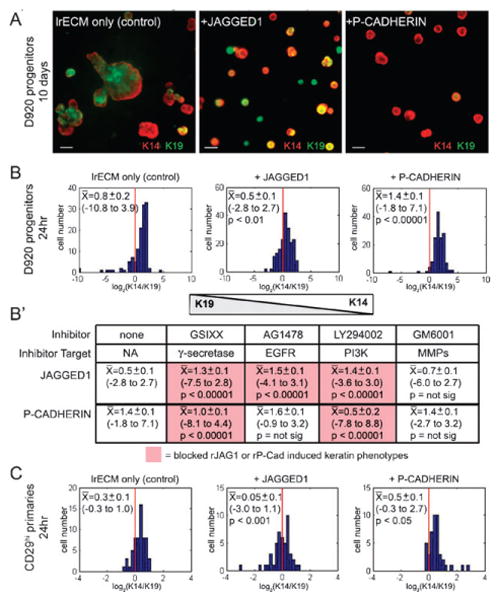
Jagged1- and P-cadherin-induced keratin phenotypes in D920 and in primary human breast progenitor cells results from the integration of multiple pathways. To determine the fate decision outcomes after induction of Notch and P-cadherin pathways in physiological culture conditions, D920 progenitor cells were cultured in 3D Matrigel. (A) K14 (red)/K19 (green) expression in D920 progenitor cells cultured for 10 days with or without 50 μg mL−1 of the recombinant Jagged1 or P-cadherin proteins. Bars represent 40 μm. (B–B′) D920 progenitors were cultured in 3D Matrigel for 24 h in the presence or absence of 10 μg mL−1 Jagged1 or P-cadherin. Fluorescence intensity values for K14 and K19 expression were determined for each cell (150 cells per condition). (B) Histograms of log2(K14/K19) expression displays the distribution of keratin phenotypes that are achieved by D920 progenitors in control, Jagged1- or P-cadherin-containing cultures. (n = 3). (B′) Small molecule inhibitors were added to the 3D cultures to identify those which blocked the Jagged1- and P-cadherin-directed cell-fate decisions (n = 2). (C) The CD29hi subpopulation of primary human mammary epithelial cells from a reduction mammoplasty specimen were cultured in 3D Matrigel for 24 h (n = 2). Histograms of log2(K14/K19) expression displays the distribution of keratin phenotypes that are achieved by primary progenitors in control, Jagged1- or P-cadherin-containing cultures. Mean log2(K14/K19) values (X̄); the absolute range of values are in parenthesis. p values were determined with Mann–Whitney tests.
Microenvironment-directed cell fate in finite-lifespan mammary progenitors
To ascertain that D920 progenitors can indeed recapitulate what happens in primary cultures, we isolated cells from normal reduction mammoplasty that could be and cultured them in a newly designed medium (included in Materials and methods) with little or no senescence for up to eight passages without the use of immortalizing agents. The CD29hi subpopulation was 4–5 fold enriched for TDLU-forming activity in 3D Matrigel relative to non-CD29 enriched cultures (data not shown). From two successive passages (6 and 7) we then determined the K14 : K19 phenotypes when cultured in 3D Matrigel in the presence of recombinant ligands. Relative to the distribution of K14 : K19 phenotypes of the untreated controls (Fig. 4C, left), addition of Jagged1 induced a K14+/K19+ phenotype (Fig. 4C, middle), and P-cadherin induced a K14+/K19− phenotype (Fig. 4C, right). That both finite life-span and these E6/E7 immortalized human mammary progenitors responded similarly to the microenvironmental variations we exposed them to confirms our previous assertion that breast progenitor cells immortalized with E6–E714 remain similar to primary non-immortalized cultures for up to 30 generations and are still subject to similar controls by their microenvironment.
Multiple pathways are required for each microenvironment-imposed cell fate decision
Having determined key regulatory molecules that mediate cell fate decisions, we then concentrated on the pathways involved. To do so a number of small molecule inhibitors were added to the 3D cultures. AG1478 (EGFR inhibitor) did not antagonize the P-cadherin-induced MEP-phenotype suggesting that active EGFR signaling was not required; indeed, it has been reported for a mouse cell line that EGFR activity may impair differentiation into the MEP-phenotype.26 PI3K acts downstream from both EGFR and integrins, and the PI3K inhibitor, LY294002, blocked the MEP-phenotype (Fig. 4B′), underscoring the probable importance of integrin signaling in establishing the MEP cell fate. The Jagged1-induced phenotype was attenuated by addition of GSI, AG1478, and LY294002. Thus integrins likely play also an important role in context-dependent Notch signaling. These data also suggest that Notch pathway signaling is linked to EGFR signaling, a relationship that has been described in lower organisms during development,29 but is not well-studied in humans. Addition of the broad spectrum matrix metalloproteinase (MMP) inhibitor, GM6001, even at 40 μm did not block the effects of either of the inducing molecules (Fig. 4B′), suggesting that MMP or sheddase activities were not required for differentiation at the single cell stage. These data demonstrate that the mammary stem cell niche must simultaneously regulate at least both E- and P-cadherins, Notch, EGFR, and integrin pathways to solicit even relatively simple decisions such as cell fate even before cells became part of tissues or organs.
Discussion
An evolving view of tissue-specific stem cell regulation suggests that stem cells do not follow pre-determined programs that direct their cell fate decisions, but that microenvironments impose specific behavior upon them.4–6 Here, we have demonstrated that human mammary progenitors indeed display considerable flexibility in cell fate decisions when they are exposed in parallel to hundreds of unique microenvironments. The results have allowed us to ascribe putative functional roles, with respect to cell fate determination and lineage maintenance, to extracellular proteins in the human mammary gland. We demonstrate that each discrete cell fate decision requires the integration of multiple pathways. These data may provide additional clues as to how the developmental hierarchy in mammary gland is governed in vivo.
The self-organizing model of tissue-specific stem cell regulation posits that stem cells reside inside a specialized microenvironment, a niche, where they give rise to daughter cells (i.e. progenitor cells), which differ primarily by their ability to leave the niche and subsequently differentiate and function according to the needs of their new microenvironment.3,6 For the model to be valid, progenitor cells should exhibit flexibility in their response to the requirements of the tissue, and their more mature progeny with distinctly different functional roles should reside within microenvironments that are different from the niche. Our functional findings with culture appear to be supported by localization of the relevant signaling molecules in vivo. In our functional assays, P-cadherin imposed the K14+/K19−/K8− MEP phenotype (Fig. 1 and Fig. 4), and is expressed in the MEP layer in vivo, typically at least one cell-diameter away from the nests of K15+ stem/progenitor cells (Fig. 2). Laminin1, which maintained the cells in a quiescent state (Fig. 1), is made by MEP cells16 and in vivo it is about one cell-diameter from K15+-containing nests (Fig. 2). The Notch ligand, Jagged1, in our assays imposed both the MEP phenotype as well as the maintenance of the K14+/K19+ progenitor phenotype, and did so in a context-dependent manner (Fig. 1 and Fig. 4). This was also true for Jagged1, which is expressed both by the MEP as well as inside the nests of putative progenitor cells in vivo (Fig. 2). E-Cadherin-conjugated beads facilitated differentiation of progenitors into the K8+/K14− LEP phenotype; however, the E-cadherin-beads were qualitatively less efficient at LEP-induction than actual cell-cell contact (Fig. 3). Because E-cadherin was expressed also by the putative stem/progenitor cells as well by the LEP cells in vivo (Fig. 2), we hypothesize that there are yet-to-be described constituents in the LEP cells or as the result of cell–E-cadherin contact that would tip the balance in favor of LEP differentiation. Thus, the microenvironment constituents that functionally demonstrated the greatest ability to impose quiescence, MEP or LEP phenotypes in our culture assays, were expressed in proximity of the nests of putative stem cells in vivo. Assuming that the functional phenotypes of the cells and molecules that we measured in our assays were approximations of their abilities in vivo, these experiments suggest that the self-organizing model could be applied to explain how stem and progenitor cells are directed to maintain homeostasis in human mammary gland.
Approaches such as ours that combine bioengineered substrata and organotypic culture models will be crucial for dissecting components of human stem cell regulation for the reasons that, relative to mammalian model systems, humans are difficult to study and impossible to manipulate, and that mouse models will not always faithfully recapitulate human biology.30–33 Array-type approaches were used to identify molecules that differentiated human embryonic stem cells and neural progenitor cells,34,35 and bioengineered substrata were used to understand how physical properties of the microenvironment can affect tissue-specific stem cell differentiation.36 Due to the complexity of microenvironments in vivo, these types of bioengineered approaches allow for combinatorial analysis of the components of the microenvironment and are useful for deconstructing basic mechanisms and functional motifs, especially for human tissues. MEArrays and similar technologies will not only be useful for identifying microenvironment components that direct stem cell differentiation, but can also identify components that are required for maintaining cell fates. Should therapeutic implications for stem cells come to fruition in cancer and regenerative medicine, comprehending the nuanced control mechanisms of human stem cells will be critical. For as we have demonstrated here, even the activities of seemingly dominant factors within a given microenvironment are ultimately the results of signals initiated from multiple points of origin.
Experimental
Cell culture
D920 cells at passage 63 were cultured in H14 medium37 on Vitrogen-coated (Cohesion) flasks for no more than 8 additional passages before starting from a fresh vial. CD29hi D920 cells were either enriched by FACS (EPICS Elite ESP, Beckman-Coulter) or with magnetic columns (Miltenyi Biotech). Both methods yielded comparable results in the TDLU-forming assay. Due to speed and improved recovery and survival we used magnetic enrichment after the initial comparison of the two methods. D920 cells were grown to ∼90% confluency prior to beginning either of the sorting protocols. For FACS, D920 cultures were incubated at 107 cells mL−1 with anti-CD49f-PE (1 : 100, Becton Dickinson) and anti-CD29-APC (1 : 1000, Becton Dickinson) for 45 min in ice. For magnetic enrichment, D920 cultures were resuspended at 107 cells mL−1 in separation buffer (DMEM–2%FBS–1 mM EDTA) and incubated with anti-CD29-APC (1 : 20, Becton Dickinson) for 30 min in ice. The cells were then incubated with MACS beads conjugated to anti-APC (1 : 20, Miltenyi Biotech) for 30 min on ice, and passed over two magnetic columns to enrich for CD29-expressing cells.
Finite life span mammary epithelial cells were isolated from reduction mammoplasty tissue from specimen 184, a 21 year old woman. The cells were cultured in M87A medium (1 : 1 mix of MEBM with 5.0 μg mL−1 insulin, 70.0 μg mL−1 bovine pituitary extract, 0.5 μg mL−1 Hydrocortisone, 5.0 ng mL−1 EGF, 5.0 μg mL−1 transferrin, 10 nM isoproterenol, 2.0 mM glutamine, and DMEM–F12 with 10 μg mL−1 insulin, 10 nM tri-iodothyronine, 1.0 nM 17β-estradiol, 0.1 μg mL−1 hydrocortisone, 5 ng mL−1 EGF, 2 mM glutamine, 0.5% fetal calf serum, 0.1% AlbuMAX) supplemented with cholera toxin (1 ng mL−1) and oxytocin (0.1 nM).38 The CD29hi subpopulation was enriched as above.
3D Matrigel assays were performed for as long as 20 days as previously described.14 Briefly, 8-well chamber slides were coated with 50 μL of pure Matrigel (Becton Dickinson) and allowed to gel at 37 °C. 5000 cells were added in a small volume of H14 medium (or M87+ medium for the primary cells) and allowed to adhere to the coating for 2–5 min, then H14/20% Matrigel, with the appropriate amount of recombinant proteins (10 μg mL−1 for 24 h assays, and 50 μg mL−1 for 10 day assays) or pathway inhibitors, was added by dripping on top. Inhibitors used were: AG1478 80 nM (Sigma), peptidomimetic GSI 100 μM (Sigma) or GSIxx 100 nM (EMD Biosciences) (both yielded similar results), LY294002 10 nM (Calbiochem), and GM6001 40 μM (a kind gift from Dr R. Galardy, Glycomed, Alameda, CA). Based on previous studies the 40μm concentration of GM6001 was sufficient to inhibit all MMPs in addition to sheddases.
Immunofluorescence of cells inside Matrigel
Matrigel supernatants were aspirated, then fixed with methanol : acetone (1:1) for 30 min at −20 °C. Cells were blocked for 3–4 h in PBS–10%NGS–0.1% Triton-X-100 with gentle rocking. Primary antibodies were added and incubated overnight at 4 °C with gentle rocking, followed by 2 h of washing with many buffer changes of PBS–0.1% Triton-X-100. Secondary antibodies were then added overnight at 4 °C with gentle rocking, followed by at least 2 h of washing with PBS–0.1% Triton-X-100, and one PBS wash at 4 °C overnight. Cells were then imaged with a spinning disc confocal microscope (Carl Zeiss) with samples under PBS.
Immunofluorescence of reduction mammoplasty specimens
Cryostat sections from biopsies were fixed in methanol or formaldehyde as described previously.8 Primary antibodies used were keratin 15 diluted 1 : 25 (LHK15, Neomarkers), E-cadherin 1 : 25 (HECD-1) and P-cadherin 1 : 5 (NCC-CAD299, kindly provided by A. Ochiai, National Cancer Center Research Institute, Tokyo, Japan), laminin α1 1 : 2 (161 EB7,39 kindly provided by I. Virtanen, University of Helsinki, Helsinki, Finland), Jagged1 1 : 25 (Abcam). Isotype-specific goat anti-mouse or goat anti-rabbit antibodies (AlexaFluor 488 and –568, Molecular Probes) were used as secondary antibodies. Nuclei were counterstained with TO-PRO-3 iodide (Molecular Probes).
Expression analysis in 3D
Images from three random fields of mammary progenitor cells after 24 h of culture were captured with a spinning disc confocal microscope (Carl Zeiss). RGB images of K14 (Covance) and K19 (DSHB) expression were then analyzed with Image J software (NIH); using the elliptical selection tool and the RGB measure function, the fluorescence intensity corresponding to each protein was determined. Log2(K14/K19) was then determined for each cell and the output was graphically represented as a histogram with MatLab7 software (The Mathworks inc) to show the distribution of possible phenotypes.
MEArray™ fabrication and use in culture
To facilitate adsorption of proteins onto a microscope slide surface, microscope slides are spin coated with polydimethyl-siloxane (PDMS, Sylgard 184) (Ellsworth Adhesives) prepared at a base : cure ratio of 10 : 1 for 1 min at 6000 rpm, then cured for at least 1 h in a 60 °C oven. Combinatorial microenvironments were prepared in 384-well plates in solutions of printing buffer (100 mM acetate, 20% glycerol, 0.05% Triton-X-100, pH5.2). The ECM molecules used were: collagen I (Cohesion), collagen IV (Sigma), laminin I (Sigma), RGD (Sigma), and Matrigel (Becton Dickinson). Recombinant purified proteins used were: rhNotch3, rhNotch1, rrJagged1, rhSHH, rhP-cadherin, and rhE-cadherin (R&D systems); KGF and TGFα (Sigma); rEpimorphin (courtesy of Dr Hidetoshi Mori). Antibodies included were: β1 integrin activating MAb (MAB1951Z, Chemicon), β1 integrin blocking MAb (MAB1965, Chemicon), α6 integrin blocking MAb (MAB1378, Chemicon), β4 integrin blocking MAb (MAB2058, Chemicon), and EGFR blocking MAb (MAB88910, Chemicon). Arrays were printed on a linear servo motor-powered microarrayer (UCSF Center for Advanced Technology) with a 16-pin silicon tip (Parallel Synthesis) printing arrays. Features were 2304 in number and spaced 250 μm apart. After printing, a plastic culture chamber was attached with silicone aquarium sealant (DAP) to encircle the arrayed portion of the slide and reduce the volume required for culture. To prevent spurious cell adhesion in the regions between printed features, the slides were blocked with Pluronics F108 (gift from BASF) for 60 min and then thrice rinsed in PBS. 1 mL of H14 media was added to the chamber, then cells were added such that the final concentration was 104–105 cells mL−1. Although unincorporated PDMS monomers can affect cell physiology,40 due to the washing and high volume we do not anticipate significant impact here. To account for differences in rates of binding to the different substrata, we allowed the enough time for cell binding that they reached a steady state, as determined by phase microscopy when >95% of features were covered with cells (10–15 per feature). Unbound cells were then washed away and the remaining adherent cells were incubated for 24 h at 37 °C, 5% CO2. When necessary, 100 μM peptidomimetic γ-secretase inhibitor (Sigma) was added at time 0, or 2 μM BrdU was added 4 h prior to the experimental endpoint. Cells were fixed with methanol : actetone (1 : 1) at −20 °C for 15 min, blocked with PBS–10%NGS–0.1% Triton-X-100, then stained for expression of K14 (Covance), K8 (Troma I, DSHB), or K19 (Troma III, DSHB), and visualized with secondaries conjugated to Alexa-568 or Alexa-633 (Molecular Probes). Slides were scanned with an Axon 4200A microarray scanner (Axon Instruments) and the images were analyzed with Genepix Pro software (Axon Instruments).
MEArray™ analysis
For MEArrays, fluorescence information for each feature was extracted by use of the irregular feature definition tool in Genepix; fluorescence was normalized to collagen I alone features. To assess the effect of microenvironments on differentiation, log2 of the ratio of mean (LRM) fluorescence intensities of K8 (633 nm) and K14 (568 nm), or of K19 (633 nm) and K14 (568 nm) was determined. Cells on features with LRM values greater than 0 indicated more K14 expression and could be considered more MEP-like than for cells cultured on collagen I alone. Cells on features with LRM values less than 0 indicated more K8 or K19 expression, and were considered to be more LEP-like than for cells cultured on collagen I alone. To assess the effect of microenvironments on DNA synthesis, total BrdU (568 nm) pixels per feature were divided by total DNA (633 nm, Topro3, Molecular Probes) pixels and the mean BrdU incorporation was determined for each microenvironment combination. Graphic output as 3D color maps was generated with MatLab 7 software (The Mathworks Inc).
Statistical methods
Each MEArray incorporates control microenvironments, which can be defined as a microenvironment that is either known to induce a certain phenotype or that is known not to induce any particular phenotype. Each unique microenvironment is represented in a MEArray by at least 12 replicates. Microenvironments that impose phenotypes statistically different from the control features were detected by associating a p value to the control paired with each unique microenvironment by Dunnette's T-test. Variance of the means is confirmed by ANOVA. p < 0.05 is considered significantly different.
Statistically significant differences between distributions of lineage marker ratios or GATA3 expression in the single cell image analyses were detected by associating a p value by comparison of the distributions using Mann–Whitney tests, where significance is p < 0.05.
Micropatterning experiments
Micropatterned substrata were made according to Tan et al.41 Briefly, PDMS stamps were formed by curing prepolymer with base : cure ratio of 10 : 1 against a prepatterned master. The stamps were peeled away and coated with 100 μg mL−1 collagen I (Koken), then washed in water, and blown dry with compressed nitrogen. Culture chambers were attached to PDMS-coated microscope slides, which were then UV oxidized for 7 min (UVO-Cleaner 42, Jelight Co.), stamped with collagen I, blocked with Pluronic F108 for 1 h, and rinsed with H14 medium. Cells were allowed to bind for 15–60 min, depending on desired outcome, before washing away the unattached cells. BSA 250 μg mL−1 (Sigma), Collagen1 250 μg mL−1 (Koken), or rhE-cadherin 250 μg mL−1 (R&D Systems) was conjugated to >1.0 mm silicone polybeads (Polysciences), conjugated beads were added to the 400 μm2 patterns immediately after cell adhesion. For analysis, cells were fixed with methanol : acetone (1 : 1) at −20 °C for 15 min, blocked with PBS–10%NGS–0.1% Triton-X-100–0.5% λ-carrageenan for 2 h, then in a buffer composed of PBS–10%NGS–0.1% Triton-X-100 cells were stained for expression of K14 (Covance), K8 (DSHB), K19 (DSHB) and GATA3 (BD); visualized with secondaries conjugated to Alexa-633, Alexa-568 or Alexa-488 (Molecular Probes). Following image acquisition with a spinning disc confocal microscope (Carl Zeiss), Image J software (NIH) was used to outline single cells, classified as either cells alone on a feature or as single cells juxtaposed to other cells, and then measure the fluorescence intensity from all channels with the RGB measure tool. The output was graphed as FACS-like plots or as histograms using MatLab 7 software (The Mathworks inc).
Conclusion
In principle, one stem cell can give rise to an entire organ through a hierarchical series of differentiation. Tissue-specific stem cells are thought self-renew and also to give rise to the differentiated cell types that comprise a given tissue. However, relatively little is known about how such multipotent cells acquire specific cell fates. One explanation is that the stem and progenitor cells intrinsically “know” which cell fate to acquire based on a stochastic mechanism. A second explanation is that stem and progenitor cell behavior is instructed by their microenvironments. That stem cells and all of their progeny share identical genomes, and that stem cells reside inside of microenvironments that are completely unique as compared to those of the surrounding tissue, suggests that microenvironments exert a tremendous influence over stem cell behavior. Stem cells must be able to respond to a wide variety of physiological needs in order to maintain tissue homeostasis throughout an organism's lifetime, and on the basis of mathematical modeling it was proposed that stem cells are instructed to respond by cell-extrinsic cues.6 We show here that cell fate decisions by human mammary progenitor cells are quite plastic when viewed in the context of hundreds of unique microenvironments in parallel. At a minimum, these data suggest that progenitor cell fate decisions are dictated by cell-extrinsic cues, or more likely, that a dynamic and reciprocal relationship exists between stem cell genomes and their microenvironments, whereby microenvironmental constituents elicit particular stem cell functions that are partly explained by the fidelity of the genome.1,5
Supplementary Material
Acknowledgments
We thank Drs Charles C. Kim and Adam Carroll from the University of California, San Francisco for lending their expertise in robot operation and microarray analysis. This work was supported by the grants from OBER Office of Biological and Environmental Research of the U.S. Department of Energy (SC-BSO and a Distinguished Fellow Award to MJB), by National Institute of Health (R01CA064786 to MJB and OWP, and the U.S. Department of Defense (DAMD170210438, an Innovator Award to MJB, and W81XWH-04-1-0582 to CMN). OWP, RV, and AF are supported by The European Commission Research Directorates (contract HPRN-CT-2002-00246), The Danish Research Agency (contract 2107-05-0006), and the Danish Cancer Society (DP0763). CMN holds a Career Award at the Scientific Interface from the Burroughs Wellcome Fund. MAL is supported by a postdoctoral fellowship from the American Cancer Society.
List of abbreviations
- LEP
Luminal epithelial cell
- MEP
Myoepithelial cell
- GSI
γ-secretase inhibitor
- K
Keratin
- EGFR
Epidermal growth factor receptor
- TDLU
Terminal ductal lobular unit
- ALDH
Aldehyde dehydrogenase
- SSEA4
Stage specific embryonic antigen 4
Footnotes
Electronic supplementary information (ESI) available: Supplementary figures. See DOI: 10.1039/b816427j
Contributor Information
Mark A. LaBarge, Email: MALabarge@lbl.gov.
Mina J. Bissell, Email: MJBissell@lbl.gov.
References
- 1.Bissell MJ. Int Rev Cytol. 1981;70:27–100. doi: 10.1016/s0074-7696(08)61130-4. [DOI] [PubMed] [Google Scholar]
- 2.Scadden DT. Nature. 2006;441:1075–1079. doi: 10.1038/nature04957. [DOI] [PubMed] [Google Scholar]
- 3.Potten CS, Loeffler M. Development. 1990;110:1001–1020. doi: 10.1242/dev.110.4.1001. [DOI] [PubMed] [Google Scholar]
- 4.Bissell MJ, Labarge MA. Cancer Cell. 2005;7:17–23. doi: 10.1016/j.ccr.2004.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.LaBarge MA, Petersen OW, Bissell MJ. Stem Cell Rev. 2007;3:137–146. doi: 10.1007/s12015-007-0024-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loeffler M, Roeder I. Cells Tissues Organs. 2002;171:8–26. doi: 10.1159/000057688. [DOI] [PubMed] [Google Scholar]
- 7.Nelson CM, Bissell MJ. Annu Rev Cell Dev Biol. 2006;22:287–309. doi: 10.1146/annurev.cellbio.22.010305.104315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villadsen R, Fridriksdottir AJ, Ronnov-Jessen L, Gudjonsson T, Rank F, Labarge MA, Bissell MJ, Petersen OW. J Cell Biol. 2007;177:87–101. doi: 10.1083/jcb.200611114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dontu G, Jackson KW, McNicholas E, Kawamura MJ, Abdallah WM, Wicha MS. Breast Cancer Res. 2004;6:R605–R615. doi: 10.1186/bcr920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ginestier C, Wicha MS. Breast Cancer Res. 2007;9:109. doi: 10.1186/bcr1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deome KB, Faulkin LJ, Jr, Bern HA, Blair PB. Cancer Res. 1959;19:515–520. [PubMed] [Google Scholar]
- 13.Kordon EC, Smith GH. Development. 1998;125:1921–1930. doi: 10.1242/dev.125.10.1921. [DOI] [PubMed] [Google Scholar]
- 14.Gudjonsson T, Villadsen R, Nielsen HL, Ronnov-Jessen L, Bissell MJ, Petersen OW. Genes Dev. 2002;16:693–706. doi: 10.1101/gad.952602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stasiak PC, Purkis PE, Leigh IM, Lane EB. J Invest Dermatol. 1989;92:707–716. doi: 10.1111/1523-1747.ep12721500. [DOI] [PubMed] [Google Scholar]
- 16.Gudjonsson T, Ronnov-Jessen L, Villadsen R, Rank F, Bissell MJ, Petersen OW. J Cell Sci. 2002;115:39–50. doi: 10.1242/jcs.115.1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Streuli CH, Bailey N, Bissell MJ. J Cell Biol. 1991;115:1383–1395. doi: 10.1083/jcb.115.5.1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reedijk M, Odorcic S, Chang L, Zhang H, Miller N, McCready DR, Lockwood G, Egan SE. Cancer Res. 2005;65:8530–8537. doi: 10.1158/0008-5472.CAN-05-1069. [DOI] [PubMed] [Google Scholar]
- 19.Shimoyama Y, Hirohashi S, Hirano S, Noguchi M, Shimosato Y, Takeichi M, Abe O. Cancer Res. 1989;49:2128–2133. [PubMed] [Google Scholar]
- 20.Petersen OW, Ronnov-Jessen L, Howlett AR, Bissell MJ. Proc Natl Acad Sci U S A. 1992;89:9064–9068. doi: 10.1073/pnas.89.19.9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boudreau N, Sympson CJ, Werb Z, Bissell MJ. Science. 1995;267:891–893. doi: 10.1126/science.7531366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buono KD, Robinson GW, Martin C, Shi S, Stanley P, Tanigaki K, Honjo T, Hennighausen L. Dev Biol. 2006;293:565–580. doi: 10.1016/j.ydbio.2006.02.043. [DOI] [PubMed] [Google Scholar]
- 23.Selkoe DJ. Proc Natl Acad Sci U S A. 2001;98:11039–11041. doi: 10.1073/pnas.211352598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Radice GL, Ferreira-Cornwell MC, Robinson SD, Rayburn H, Chodosh LA, Takeichi M, Hynes RO. J Cell Biol. 1997;139:1025–1032. doi: 10.1083/jcb.139.4.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Gumbiner BM. Curr Opin Cell Biol. 2006;18:572–578. doi: 10.1016/j.ceb.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 26.Deugnier MA, Faraldo MM, Rousselle P, Thiery JP, Glukhova MA. J Cell Sci. 1999;112(Pt 7):1035–1044. doi: 10.1242/jcs.112.7.1035. [DOI] [PubMed] [Google Scholar]
- 27.Asselin-Labat ML, Sutherland KD, Barker H, Thomas R, Shackleton M, Forrest NC, Hartley L, Robb L, Grosveld FG, van der Wees J, Lindeman GJ, Visvader JE. Nat Cell Biol. 2007;9:201–209. doi: 10.1038/ncb1530. [DOI] [PubMed] [Google Scholar]
- 28.Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. Cell. 2006;127:1041–1055. doi: 10.1016/j.cell.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yoo AS, Bais C, Greenwald I. Science. 2004;303:663–666. doi: 10.1126/science.1091639. [DOI] [PubMed] [Google Scholar]
- 30.Anisimov VN, Ukraintseva SV, Yashin AI. Nat Rev Cancer. 2005;5:807–819. doi: 10.1038/nrc1715. [DOI] [PubMed] [Google Scholar]
- 31.Ginis I, Luo Y, Miura T, Thies S, Brandenberger R, Gerecht-Nir S, Amit M, Hoke A, Carpenter MK, Itskovitz-Eldor J, Rao MS. Dev Biol. 2004;269:360–380. doi: 10.1016/j.ydbio.2003.12.034. [DOI] [PubMed] [Google Scholar]
- 32.Rangarajan A, Weinberg RA. Nat Rev Cancer. 2003;3:952–959. doi: 10.1038/nrc1235. [DOI] [PubMed] [Google Scholar]
- 33.Vargo-Gogola T, Rosen JM. Nat Rev Cancer. 2007;7:659–672. doi: 10.1038/nrc2193. [DOI] [PubMed] [Google Scholar]
- 34.Flaim CJ, Chien S, Bhatia SN. Nat Methods. 2005;2:119–125. doi: 10.1038/nmeth736. [DOI] [PubMed] [Google Scholar]
- 35.Soen Y, Mori A, Palmer TD, Brown PO. Mol Syst Biol. 2006;2:37. doi: 10.1038/msb4100076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 37.Briand P, Petersen OW, Van Deurs B. In Vitro Cell Dev Biol. 1987;23:181–188. doi: 10.1007/BF02623578. [DOI] [PubMed] [Google Scholar]
- 38.Garbe JC, Bhattacharya S, Merchant B, Bassett E, Swisshelm K, Feiler H, Wyrobek AJ, Stampfer MR. The molecular distinctions between the stasis and the telomere attrition senescence barriers demonstrated by long-term cultures of normal pre-stasis human mammary epithelial calls. doi: 10.1158/0008-5472.CAN-09-0270. in prep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maatta M, Virtanen I, Burgeson R, Autio-Harmainen H. J Histochem Cytochem. 2001;49:711–726. doi: 10.1177/002215540104900605. [DOI] [PubMed] [Google Scholar]
- 40.Paguirigan AL, Beebe DJ. Bioessays. 2008;30:811–821. doi: 10.1002/bies.20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tan JL, Liu W, Nelson CM, Raghavan S, Chen CS. Tissue Eng. 2004;10:865–872. doi: 10.1089/1076327041348365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.