

Abstract
Oxidative stress, resulting from mitochondrial dysfunction, excitotoxicity, or neuroinflammation, is implicated in numerous neurodegenerative conditions. Damage due to superoxide, hydroxyl radical, and peroxynitrite has been observed in diseases such as Alzheimer's disease, Parkinson's disease, and amyotrophic lateral sclerosis, as well as in acute conditions that lead to neuronal death, such as stroke and epilepsy. Antioxidant therapies to remove these toxic compounds have been of great interest in treating these disorders. Catalytic antioxidants mimic the activities of superoxide dismutase or catalase or both, detoxifying superoxide and hydrogen peroxide, and in some cases, peroxynitrite and other toxic species as well. Several compounds have demonstrated efficacy in in vitro and in animal models of neurodegeneration, leading to optimism that catalytic antioxidants may prove to be useful therapies in human disease. Antioxid. Redox Signal. 11, 555–569.
Oxidative damage is implicated in the etiology of numerous neurodegenerative disorders, including Alzheimer's disease (AD), Parkinson's disease (PD), amyotrophic lateral sclerosis (ALS), Huntington's disease, and ataxia telangiectasia, as well as acute disorders resulting in neuron loss, such as stroke and epilepsy. Antioxidant compounds have therefore been of great interest as potential therapies for these disorders. Catalytic antioxidants, which are small, molecular mimics of superoxide dismutase or catalase or both, and some of which are also potent detoxifiers of lipid peroxides and peroxynitrite (reviewed in 34), hold particular promise. Because they are catalytic and not merely free radical scavengers, these compounds are much more potent antioxidants than dietary additives such as vitamin E that act stoichiometrically. Additionally, these synthetic compounds can be chemically modified to increase their ability to cross the blood–brain barrier, as well as their availability to various subcellular compartments. This review discusses reactive oxygen species (ROS) and oxidative stress, evidence for the role of oxidative stress in neurodegenerative disease, the classes of catalytic antioxidants, and their application to neurodegenerative disease.
Reactive Oxygen Species, Reactive Nitrogen Species, and Oxidative Stress
Reactive oxygen species (ROS) are generated in various cellular compartments as a consequence of normal metabolism, and under homeostatic conditions are kept in check by endogenous antioxidant defenses that include detoxifying enzymes such as superoxide dismutase (SOD), catalase, and glutathione peroxidase. Oxidative stress describes the pathologic condition in which the balance of oxidant generation and detoxification is tipped toward a prooxidant state, antioxidant defenses are overwhelmed, reactive species accumulate, and damage to nucleic acids, proteins, and membrane lipids ensues. Three phenomena leading to oxidative stress in the brain, and implicated in neurodegenerative disease, include the inhibition of mitochondrial metabolism (Fig. 1), neuronal excitotoxicity (Fig. 2), and neuroinflammation (Fig. 3).
FIG. 1.
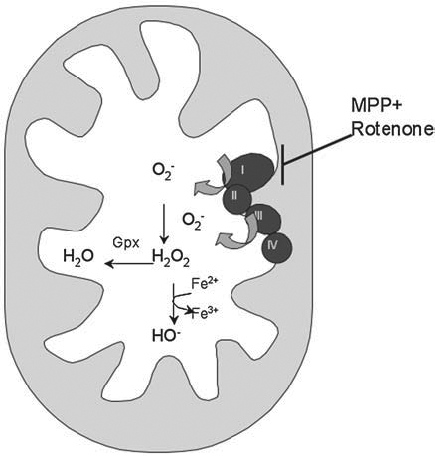
Mitochondrial oxidative stress in neurodegeneration. Inhibition of mitochondrial complexes, especially complex I, by compounds such as MPP+ and rotenone, can cause an increase in superoxide production by the mitochondria. Superoxide is converted to hydrogen peroxide by SOD. The resulting hydrogen peroxide, if not detoxified by glutathione peroxidase (Gpx) in the mitochondria, or catalase in the cytosol, and if free iron is present, can be converted into the highly reactive hydroxyl radical via the Fenton reaction.
FIG. 2.
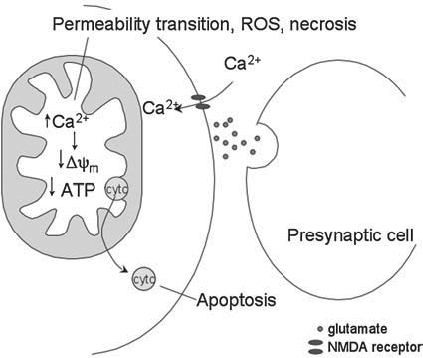
Excitotoxicity and neurodegeneration. Excitoxicity results from stimulation of NMDA receptors. Calcium can flow into the cell, where it is sequestered in the mitochondria. The mitochondria's ability to buffer calcium is overwhelmed, and a decrease in membrane potential and reduction in ATP production can result, causing cytochrome c (CytC) to be released, and leading to apoptosis. Alternatively, the mitochondria may undergo the permeability transition, producing ROS, and leading to cell death by necrosis.
FIG. 3.
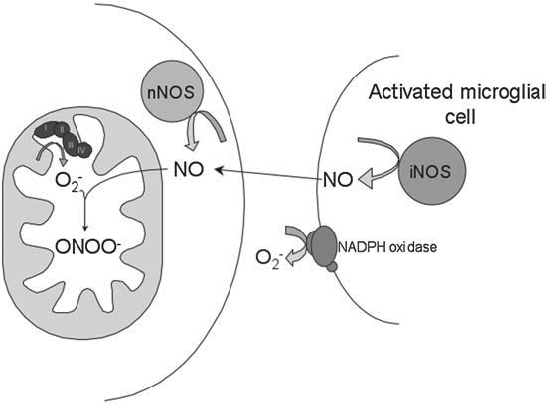
Neuroinflammation and neurodegeneration. During neuroinflammation, activated microglia release NO from iNOS and superoxide from NADPH oxidase. NO can diffuse to neighboring neurons, where it can react in a diffusion-limited reaction with superoxide, forming peroxynitrite.
In the mitochondrion, superoxide is formed as a result of electron “leak,” which allows the single-electron reduction of molecular oxygen. The level of superoxide formed under physiologic conditions, when mitochondria are respiring primarily on NAD-linked substrates, has been estimated to account for 0.1% to 0.4% of electron flow (23, 65). This superoxide is normally removed by the mitochondrial superoxide dismutase, MnSOD, which dismutates superoxide to hydrogen peroxide and oxygen. The significance of detoxifying endogenous mitochondrial superoxide is made clear by the severe pathologies present in the MnSOD-null mouse (86). Lacking MnSOD is either embryonic or neonatal lethal, depending on the genetic background (69). On the CD1 background, mutants survive ~8 days, and develop fatty liver, metabolic acidosis, and cardiomyopathy (44, 86, 99). If treated with the catalytic antioxidant MnTBAP, the life span of the mice can be extended, and the liver and heart pathology can be prevented, but a neurodegenerative phenotype is uncovered, indicating the importance of detoxifying endogenous mitochondrial superoxide in the brain (57, 98). Other catalytic oxidants that are better able to cross the blood–brain barrier are able to rescue the spongiform encephalopathy (57, 100).
It is possible to increase the rate of mitochondrial superoxide production by increasing the oxidation state of the electron-transport chain (ETC), either by severely inhibiting ETC complexes or by otherwise increasing the mitochondrial membrane potential. Superoxide production increases dramatically when electron flow is inhibited at either complex I or complex III. This is of particular interest with regard to Parkinson disease (PD), as reduced activity of complex I has been observed in patients with PD (130, 131).
In addition to being a by-product of mitochondrial metabolism, superoxide is produced at the plasma membrane by NADPH oxidase, an enzyme found in many cell types and especially important to the function of immune cells. NADPH oxidase assembly is induced by exposure to growth factors and cytokines, and the resulting superoxide is dismutated to hydrogen peroxide by cytosolic or extracellular SOD or both. Mice null for the cytosolic and extracellular forms of SOD do not display the severe phenotypes observed in MnSOD-null mice, but do demonstrate reduced life span, increased oxidative damage to DNA, increased cancer rates, and increased sensitivity to some stresses (18, 19, 42, 69, 120).
Superoxide readily inactivates aconitase, releasing iron from the enzyme's labile Fe-S cluster (54, 85). However, superoxide has a short half-life and does not readily react with DNA or polyunsaturated lipids (51). Despite this, MnSOD-null mice have a severe neonatal lethal phenotype, which indicates that mitochondrially generated superoxide is highly toxic. The conversion of mitochondrially generated superoxide into more-reactive species, such as hydroxyl radical or peroxynitrite, probably accounts for much of its toxicity.
Superoxide spontaneously dismutes to hydrogen peroxide, a reaction catalyzed by SOD. Hydrogen peroxide is more stable than superoxide, with a longer half-life and the ability to diffuse through membranes, allowing it to function as a signaling molecule (for review, see 149). In the presence of free iron, hydrogen peroxide can react via the Fenton reaction to form hydroxyl radical, a very potent oxidant. Hydroxyl radical readily reacts with DNA, membrane lipids, and protein.
Reactive nitrogen species (RNS) also play a role in oxidative stress. Nitric oxide (NO) acts as a signal-transduction molecule in vasodilation (71, 107) and neuronal signaling (8), and nitrosylation is proposed to be a redox-sensitive protein modification involved in signal transduction (reviewed in 119, 141). NO also reacts with superoxide in a reaction that is diffusion limited, forming peroxynitrite (ONOO—) (Fig. 4a), a powerful oxidant. This reaction can serve as a mechanism to control levels of superoxide or NO or both, affecting cell signaling by NO. Additionally, peroxynitrite and its breakdown products can react with proteins, resulting in nitration of tyrosine residues (Fig. 4b) (73). Both superoxide and nitric oxide are produced in the course of the inflammatory response, leading to the formation of nitrotyrosine, which can be used as a molecular footprint of nitrosative stress in inflammation (80). Nitrotyrosine has been considered a hallmark of oxidative stress and is increased in many disease states, including Parkinson's disease (59) and Alzheimer's disease (67).
FIG. 4.

Formation of peroxynitrite and nitrative damage. (a) Peroxynitrite is formed by the diffusion-limited reaction of nitric oxide, generated by nitric oxide synthase, with superoxide. (b) Peroxynitrite can react with protein, resulting in the nitration of tyrosine residues. This modification can interfere with phosphorylation of tyrosine.
Catalytic Antioxidants
Currently, three major classes of metal-containing catalytic antioxidants are known: metalloporphyrins, macrocyclics, and salen compounds (Fig. 5). Each of these contains a manganese moiety that catalyzes the dismutation of superoxide in a mechanism analogous to that found in native SOD.
FIG. 5.

Three classes of metal-containing catalytic antioxidants. (a) Metalloporphyrin AEOL 10150. (b) Salen mimetic EUK-134. (c) Macrocyclic mimetic M40403.
Metalloporphyrins [AEOL series, developed by Aeolus Pharmaceuticals, Inc. (Laguna Niguel, CA) a subsidiary of Incuara Pharmaceuticals] contain manganese coordinated by four axial nitrogen ligands. These compounds have been demonstrated to scavenge superoxide, hydrogen peroxide, lipid peroxyl radicals, and peroxynitrite (reviewed in 112). In the dismutation of superoxide, the manganese changes its valence state between Mn(III) and Mn(II) in a series of alternate reduction and oxidation steps, similar to the alternating reduction and oxidation that occurs in native SODs. In the dismutation of hydrogen peroxide, the conjugated ring system may undergo reversible one-electron oxidation, analogous to the heme prosthetic groups of catalases (38). In general, SOD and catalase activities have been correlated in the metalloporphyrins, but recently developed members of the family have relatively high catalase and lipid peroxidation inhibitory activity and low SOD activity (146), allowing investigation of new hypotheses regarding in vivo activity. The older generation of metalloporphyrins (i.e., pyridine and carboxylic acid derivatives) have only <1% of the catalase activity of native catalases, yet they are able to protect against hydrogen peroxide (33). The scavenging of lipid peroxyl radicals and of peroxynitrite may involve formation of an oxo-Mn(IV) complex, which is reduced by endogenous antioxidants such as ascorbate and glutathione (83).
Salen compounds (EUK series, developed by Eukarion, Inc., Bedford, MA, acquired by Tyrian Diagnostics, North Ryde, Sydney, Australia, in 2005) are similar to the metalloporphyrins in that they contain a manganese coordinated by four axial ligands, but the four axial ligands of salen compounds consist of two oxygen and two nitrogen atoms rather than four nitrogen atoms. EUK compounds have been demonstrated to scavenge superoxide and hydrogen peroxide, to react with ONOO− (133), and may react with lipid peroxides as well. Their mechanisms of action are thought to be similar to those of the metalloporphyrins.
Macrocyclics (M series developed by MetaPhore Pharmaceuticals, Inc., Fort Lee, NJ and St. Louis, MO) contain a manganese atom coordinated to five nitrogen ligands. The pentavalent coordination allows the manganese to participate only in single-electron transfers, and so makes these compounds specific for the scavenging of superoxide, relative to hydrogen peroxide or peroxynitrite, which require two-electron transfers. Other compounds, such as flavins and ubiquinones, can participate in one-electron transfers, so the macrocyclics may react with compounds besides superoxide in vivo.
The carboxyfullerenes are an additional class of compounds that can function as radical traps, but one member of which (C3; Fig. 6) has been described as having SOD activity with a rate-constant ~100-fold slower than endogenous SOD (5). These compounds do not contain metal centers, but are thought to attract a superoxide molecule to an electron-deficient area of the C3 molecule, holding the superoxide there until a second superoxide molecule approaches and the dismutation reaction can occur with help from carboxyl groups on C3 or from water molecules (5). The compounds can also scavenge hydroxyl radical and other free radicals, probably because of their highly conjugated double-bond systems. C3 is concentrated in the mitochondria.
FIG. 6.

The structure of the carboxyfullerene derivative C3.
The antioxidant properties of each of these families of compounds have been studied in vitro, and, as is discussed later, several compounds have been demonstrated to have biologic activity in vivo. A question remains, however, regarding which antioxidant activity is most important to protection in a given system. Compound activities in vivo do not always correlate with the activities measured in vitro, making it difficult to predict activity in a disease model. A compound may participate in additional reactions or demonstrate novel activities in a biologic context. In vivo experiments comparing different classes of compounds in the same model have not been done, but would be informative in determining mechanisms.
Additional modification of compound activities is achieved by localization, both within the cell and within the organism. Compounds are being engineered to improve their ability to cross the blood–brain barrier, to increase their concentration in hydrophobic membranes, or to target the mitochondria. Regardless of its activities as measured in vitro, the concentration of a compound in the correct compartment or tissue may dramatically affect its ability to demonstrate efficacy in a disease model.
Catalytic Antioxidants and Neurodegeneration
Parkinson's disease
Parkinson's disease (PD) is a progressive disorder characterized by the selective death of dopaminergic neurons in the substantia nigra. Approximately 15% of cases are familial, with five causal genes identified to date (reviewed in 108). The functions of the proteins encoded by these genes implicate protein degradation, mitochondrial function, and other processes in the disease. How these inherited forms of the disease relate to sporadic PD is unclear. PD patients have reduced complex I activity in the substantia nigra relative to age-matched controls (75, 130). Focus on complex I is further supported by the discovery that MPTP, a compound that induces a Parkinson-like syndrome in humans and other organisms, including mice, is an inhibitor of complex I (27, 68, 91, 138).
Inhibition of complex I could result in reduced ATP production, leading to an energy-starved state that would interfere with the maintenance of membrane ion gradients and neuronal function. Additionally, inhibition of complex I might lead to an increase in ROS, which could damage the dopaminergic neurons and lead to cell death. Evidence of oxidative stress is seen in PD brain, including increased oxidative modification of DNA (4), proteins (3) and membrane lipids (35, 36). The hypothesis that inhibition of complex I may contribute to the development of PD has led to a search for environmental toxins that possess this function. Epidemiologic studies implicate exposure to paraquat, an herbicide, as a risk factor in acquiring PD (68, 91. For review, see 16).
Another hallmark of PD is increased inflammation. Microglia, the resident immune cells of the brain, are activated and proliferate in the substantia nigra in PD. The cells are likely responding to the death of dopamine neurons, and their proliferation and activation contribute to additional death, essentially resulting in an amplification of the pathology. Antiinflammatory drugs have been found to be protective in an MPTP mouse model of PD (39, 156). Additionally, population studies suggested that nonsteroidal antiinflammatory drug use may reduce the chance of developing PD (24, 151), although other studies found no protective effect (13, 46).
Catalytic antioxidants have been found to be neuroprotective in several animal models of PD. Two EUK compounds, EUK-134 and EUK-189, were demonstrated to protect against apoptotic cell death in dopaminergic cells treated with paraquat and to protect against neuronal death in primary neuronal cultures (113). In the same study, pretreatment with EUK-189 prevented the loss of nigral dopaminergic neurons in mice exposed to paraquat (113), and it was proposed that the EUK compounds exerted their effect by inhibiting the activation of JNK-mediated apoptosis. Pretreatment of primary dopaminergic cultures with EUK-134 prevented the accumulation of nitrotyrosine in tyrosine hydroxylase in response to either MPP+ or 6-hydroxydopamine (117). Carboxyfullerene (94) and MnTBAP (a metalloporphyrin) (26) were able to protect against 6-hydroxydopamine, but not against MPP + toxicity.
A major advancement in the field of catalytic antioxidants was the demonstration that AEOL 11207, a lipophilic metalloporphyrin, protected against MPTP toxicity in vivo when administered orally (89). This compound is a member of a new series of metalloporphyrins, the AEOL 112 series of glyoxylate metalloporphyrins (146), which were designed to have greater lipid solubility, oral bioavailability, and ability to cross the blood–brain barrier. In vitro assays were used to rank compounds from the AEOL 112 series with respect to ability to scavenge hydrogen peroxide generated by paraquat-treated mitochondria (21). The IC50 measured for AEOL 11207 in the in vitro assay, 104 nM, agreed well with the brain concentration reached in vivo (∼200 nM) at which antioxidant effects were observed (89). Treatment of MPTP-intoxicated mice with the compound AEOL 11207 reduced the death of dopaminergic neurons in the substantia nigra, the decrease in dopamine levels in the striatum, and markers of oxidative damage including 3-nitrotyrosine levels, 4-hydroxy-2-nonenol levels, and a decrease in the ratio of oxidized to reduced glutathione (89). The oral efficacy of AEOL 11207 in a well-established model of PD suggests the potential clinical utility of this class of compounds.
The future applicability of catalytic antioxidants to PD is promising, but substantial issues remain. First, in animal and cell-culture models, catalytic antioxidants have been administered before or concurrently with exposure to the toxin or both, which cannot mimic the situation in human beings. Individuals do not demonstrate parkinsonian symptoms until they have lost a large percentage of their dopaminergic neurons, at which time interventions to prevent cell death may be too late. Additionally, the precise identity of the species against which the compounds are acting is unknown, because the compounds have demonstrated activity against superoxide, peroxide, and peroxynitrite. The compounds may be blocking ROS resulting from an initial inflammatory response, in addition to removing ROS resulting from mitochondrial dysfunction. In the case of AEOL 11207, it likely primarily targets membrane peroxides because the compound is lipophilic and has high relative catalase activity and weak SOD activity (21).
Alzheimer's disease
Alzheimer's disease (AD) is a progressive dementing disease involving loss of cortical and hippocampal neurons and characterized by the accumulation of extracellular amyloid plaques and intracellular neurofibrillary tangles. The majority of cases are sporadic, but early-onset familial AD cases have led to the identification of three genes encoding amyloid precursor protein (APP) and presenilins 1 and 2. The amyloid plaques observed in AD brain are composed of a 4-kDa peptide, β-amyloid (Aβ), which is a cleavage product of APP. The presenilins are involved in APP processing, and the convergence of the three known genetic determinants of AD on this one peptide has led to the amyloid hypothesis of AD, which proposes that the accumulation of Aβ is responsible for AD (56; for review, see 145). The only genetic risk factor yet confirmed for late-onset or sporadic AD is the ApoE4 allele of apolipoprotein E, which may indicate a role for cholesterol metabolism or vascular function in the disease.
A great deal of evidence has been accumulated for oxidative stress in AD, with increased damage detected to lipids (129), protein (139), nuclear DNA (53), mitochondrial DNA (97), and RNA (104) in AD relative to those in age-matched controls. Whether oxidative damage is responsible for the disease or merely associated with it has been difficult to determine. Increased oxidative damage was found to precede pathology in one study (105), but the lesions associated with AD can also be a source of ROS (see later). In some studies, diets rich in antioxidants have been associated with a decreased AD risk, but in others, no effect has been seen (reviewed in 140). Treatment with dietary antioxidants has not been demonstrated to slow disease progression (55, 127), although it has been hypothesized that the intervention may have been introduced too late into disease onset (reviewed in 140).
Table 1.
Summary of In Vivo and In Vitro Trials of Catalytic Antioxidants in Neurodegenerative Disease
| Disease | Compound | Treatment protocol | Results |
|---|---|---|---|
| Stroke/ischemia | AEOL 10150 | 60-min middle cerebral artery occlusion (MCAO) in mouse. Compound given intravenously (IV) 5 min after initiation of reperfusion | Microarray used to examine gene-expression changes; increased expression of inflammatory genes reduced by treatment (14) |
| AEOL 10150 AEOL 10113 |
90-min MCAO in mouse. Compounds administered intracerebroventricularly (ICV) 90 min or 6 h after initiation of reperfusion | Both compounds reduced infarct size at 90 min. AEOL 10150 reduced infarct size when 2 administered 6 h after reperfusion (134) | |
| EUK-189 EUK-207 |
2-h Oxygen-glucose deprivation (OGD) and 2.5-h reoxygenation in hippocampal slices from 2-mo or p10 rats. Compound applied during OGD and reoxygenation | Compounds increased cell viability as assayed by LDH release and PI uptake. Compounds also reduced ATP depletion, blocked ROS production, and reduced ERK1/2 dephosphorylation. Increased cell survival related to ATP and ROS levels, not MEK/ERK or p38 pathways, or calpain activation (159) | |
| EUK-134 EUK-8 |
MCAO in rat. Compounds administered 3 h after focal ischemia | Reduced infarct size in dose-dependent manner. EUK-134 more effective, suggesting H2O2 important to injury (10) | |
| M40401 | 90-min MCAO in rat. Compound administered by injection 30 min before, 60 min into, or 90 min after initiation of MCAO | Infarct volume reduced only when compound administered before MCAO (136) | |
| M40401 M40403 |
Carotid ligation and 3-h 8% hypoxia in immature rats. Compounds administered 2 h before hypoxia/ischemia | M40403 and M40401 reduced infarct size (135) | |
| Carboxyfullerene | 60-min MCAO in rat. Compound administered via IV or ICV 30 min before ischemia/reperfusion | ICV infusion reduced infarct size and prevented glutathione depletion and lipid peroxidation (90) | |
| Alzheimer's disease | EUK-8 EUK-134 EUK-189 |
Primary rat cortical neurons, peroxynitrite generated by SIN-1 | Compounds protect primary cortical neurons against cell death due to SIN-1 or H2O2 (126) |
| EUK-189 | Tg2567 (APP) mice. Compound administered via IP 3 times weekly from 90 to 300–400 days of age | Cataract severity reduced by treatment with EUK-189 (101) | |
| EUK-134 | Primary rat microglial cells activated with LPS and INF-γ | EUK-134 reduced PGE2 production by activated microglia (6) | |
| EUK-8 | Organotypic rat hippocampal slice cultures treated with Aβ (1-40 or 1-42). Compound added simultaneously | EUK-8 protected against Aβ-induced cell death and also blocked free radical accumulation and lipid peroxidation (17) | |
| EUK-8 EUK-134 |
Microglia purified from primary mixed glial cultures from 7-d rats. Activated with fibrillar Aβ(1–40) | Compounds blocked proliferation, and blocked the increase in TNF-α production by Aβ-activated microglia (76) | |
| Parkinson's disease | EUK-134 EUK-189 |
Adult animals, N27 cell line, and primary mesencephalic cultures treated with paraquat. Compounds administered before paraquat exposure | Both compounds reduce cell death in paraquat-treated N27 cells and primary mesencephalic cultures. EUK-189 decreased SNpc dopaminergic cell death in response to paraquat in vivo (113) |
| EUK-134 | Primary dopaminergic neuronal cultures, exposed to MPP+ or 6-OHDA. Pretreated with EUK-134 | Pretreatment with EUK-134 reduced neurotoxicity, and prevented nitration of tyrosine residues in tyrosine hydroxylase caused by MPP+ or 6-OHDA treatment (117) | |
| C3 | Primary mesencephalic cultures treated with MPP+ or 6-OHDA | C3 rescued dopminergic neurons from 6-OHDA–meditated, and partially protected against MPP+–mediated cell death (94) | |
| MnTBAP | Murine dopaminergic cell line, treated with MPP+ or 6-OHDA | 6-OHDA caused ROS-mediated JNK activation, which was attenuated by MnTBAP. MnTBAP reduced cell death induced by 6-OHDA, but not by MPP+ (26) | |
| AEOL 11207 | MPTP mouse model of PD. Compound administered subcutaneously and orally | Compound protected against MPTP induced cell death, dopamine depletion, glutathione depletion, and 3-NT formation when administered SC or orally (89) | |
| Epilepsy/excitotoxicity | EUK-134 | Kainic acid (KA)-treated rats. Compound administered before KA treatment | Pretreatment with EUK-134 reduced protein nitration, AP-1 and NF-ϰB binding activity, spectrin proteolysis, and neuronal damage, but did not reduce latency or duration of seizures (125) |
| MnTBAP | KA-treated rats. Compound administered via osmotic minipump starting 48 h before KA treatment | Pretreatment with MnTBAP improved neuronal survival, maintained aconitase activity, and reduced oxidative DNA damage, but did not affect behavioral seizures (87) | |
| ALS | AEOL 10150 | G93A mouse model of ALS, compound administered at symptom onset | Compound extended survival from onset up to threefold, increased motor neuron survival, and reduced astrogliosis and markers of oxidative stress (31) |
| AEOL 10150 + PBA | G93A mouse model, of ALS, compounds administered alone or together at time of disease onset | AEOL 10150 increased survival by 11%, PBA by 13%, and combined treatment by 19%. Treatment reduced markers of oxidative stress, and increased expression of protective genes (115) | |
| EUK-8 EUK-134 |
G93A mouse model of ALS. Compound administered via IP injection starting at 60 days of age | Treatment reduced markers of oxidative stress and increased survival, but did not affect disease onset (78) | |
| C3 | G93A mouse model of ALS. Compound continuously infused via osmotic minipump starting at 73 days of age | C3 treatment resulted in a 10-day delay in symptom onset, and 8-day improvement in survival (41) |
Aβ may directly contribute to the oxidative stress observed in AD. The peptide accumulates in mitochondria and can inhibit the electron-transport chain, particularly complex IV, cytochrome oxidase (COX) (30). However, decreased COX activity reduced plaque load and ROS in another study (52), challenging the proposition that COX inhibition may be a source of ROS in AD. Aβ interacts with ABAD, resulting in ROS production and apoptosis (95). Additionally, Aβ itself may enzymatically generate hydrogen peroxide and hydroxyl radical in the presence of copper (9, 30, 70, 106). Alternatively, it has been hypothesized that APP or Aβ or both could function as antioxidants under nonpathologic conditions (7).
Substantial evidence exists for neuroinflammation in AD (reviewed in 11, 124). Microglia are found within and around amyloid plaques and can be activated in vitro by Aβ peptide (76). Epidemiology studies indicate that NSAID therapy reduces the risk of AD (15, 142; reviewed in 143, 150). However, this protection may be significant only for those individuals with at least one ApoE4 allele (144). Except for one early positive result (123), clinical trials of NSAIDs have not demonstrated protection against disease progression (1, 2, 121, 132), or against disease incidence (62).
Catalytic antioxidants have demonstrated some efficacy against Aβ-induced toxicity in vitro. EUK-8 inhibited cell death in response to Aβ in organotypic hippocampal cultures (17). Microglial proliferation induced by fibrillar Aβ was blocked by catalase, EUK-8, and EUK-134 (76), leading to the conclusion that hydrogen peroxide resulting from activity of the NADPH oxidase mediates microglial activation by Aβ. Additionally, ROS from the NADPH oxidase have been implicated in mediating the effects of Aβ on vascular dysfunction (110). The Aβ peptide was found to accumulate as supranuclear cataracts in AD (58) and a mouse model of AD (101). Cataract development in the mouse model was reduced by treatment with EUK-189 (101), which provides the first example of a catalytic antioxidant protecting against an Aβ-related pathology in vivo.
Stroke
In ischemic stroke, blood flow is interrupted to a portion of the brain, usually by a clot blocking an artery. The subsequent reperfusion of the affected brain area when blood flow is resumed is accompanied by a burst of oxidative damage. A region of acute cell death results, with a surrounding penumbra of damaged tissue that degenerates more slowly. Rescuing the cells in the penumbra from death is the goal of most stroke-treatment models.
Reactive oxygen and nitrogen species have been implicated in the death of cells in the penumbra. A decrease in ascorbate levels, indicative of increased free radical damage, was detected after cerebral ischemia (50), and glutathione levels are decreased during reperfusion after ischemia (103, 109). Overexpression of Cu/Zn SOD protects against focal ischemic injury in adult mice (81), transient focal ischemia in mice (158), global ischemia and reperfusion injury in rats (22), and transient focal ischemia in rats (32), implying that superoxide is a toxic species in stroke. However, protection provided by SOD1 overexpression was dependant on the presence of glutathione peroxidase in a mouse model of focal ischemia/reperfusion (29), meaning that it is essential to detoxify the resulting hydrogen peroxide as well. Nitric oxide levels also are increased in ischemia (155). These levels, when generated by nNOS, are protective (43). However, a further increase in NO levels produced by iNOS can be damaging if NOS is converted to peroxynitrite in the presence of superoxide (43).
More than 100 clinical trials have been carried out with compounds proposed to work as neuroprotectants in stroke (61). In 1999, a series of guidelines was published for preclinical and clinical testing of drugs for acute ischemic stroke (AIS) by the Stroke Therapy Academic Industry Roundtable (STAIR). The guidelines for preclinical testing include the demonstration of efficacy in permanent and transient ischemia, in rodent and primate models, from multiple laboratories and with long-term follow-up, before clinical trials are warranted. A spin-trap agent, NXY-059, was believed to be a promising neuroprotective agent after animal studies and generated excitement after a positive effect in initial clinical trials [SAINT I, (84)]. However, a second, larger trial failed to demonstrate any protection by NXY-059 in acute ischemic stroke (137), demonstrating the difficulty in extending animal research to human trials, and leading some to question the feasibility of neuroprotection as an approach in treating stroke (152). Some authors, however, have used this as an example of the costs of not adhering closely enough to the 1999 STAIR criteria (128), after a reexamination of the initial animal studies.
Several catalytic antioxidants have proven effective in stroke models. The macrocyclics M40403 and M40401 decreased infarct volume when administered 2 h before hypoxia/ischemia injury (135). Especially promising with regard to treatment of human disease are models in which catalytic antioxidants protect against damage when administered several hours after the stroke event, mirroring the situation in which the patients cannot receive treatment until they reach the hospital and are diagnosed as having had a stroke. The macrocyclics were not protective when administered after transient focal ischemia (136), which may indicate that detoxifying superoxide is not sufficient to provide protection in the absence of added hydrogen peroxide detoxification. A single injection of EUK-134 3 h after middle cerebral artery occlusion (permanent regional cerebral ischemia) prevented further increase in infarct volume (10). In this study, EUK-134 was found to be more effective than EUK-8, a related compound with lesser catalase activity, consistent with the genetic study indicating that hydrogen peroxide is especially important to ischemia-related injury (29). A panel of related EUK compounds was tested in the same model, as well as a cell-culture model (37). The ability to protect in the culture model was related to the catalase activity of the compound, but interestingly, the activities of the compounds in the two assays were not always correlated, pointing out the difficulty in predicting in vivo activities (37). AEOL 10113 reduced infarct size when administered up to 6 h after focal ischemia (96). AEOL 10150 also reduced infarct size in a transient ischemia model (134) and attenuated the increase in inflammatory gene expression in response to transient middle cerebral artery occlusion (14). These results have led to optimism that catalytic antioxidants might have therapeutic value in stroke.
Epilepsy
Epilepsy is not classified as a neurodegenerative disease per se, but seizure activity leads to cell loss and neurodegeneration, which contribute to disease progression. Epilepsy can be age-related, developing in response to age-related diseases such as stroke. Evidence exists of oxidative stress accumulating in epilepsy. Status epilepticus causes a depletion of glutathione (88). Prolonged seizure activity results in excitotoxicity, which leads to an influx of extracellular calcium, a dysregulation of intracellular calcium, and mitochondrial ROS production (25, 40, 111, 122). The cell death that results is dependant on superoxide production (111), and that superoxide is produced by the mitochondria (85). The cell death may activate an inflammatory response, resulting in more oxidative damage (125).
Catalytic antioxidants have reduced oxidative damage in epileptic animals, although they have not been demonstrated to reduce seizure duration or latency. Pretreatment with EUK-134 prevented neuronal damage and decreased markers of oxidative damage, including protein nitration, resulting from kainite-induced seizures, although the compound did not affect the latency or duration of seizures (125). The catalytic antioxidant MnTBAP also inhibited kainite-induced oxidative DNA damage, aconitase inactivation, and neuronal death (87). Whether these compounds would reduce disease severity or progression remains to be tested.
Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS), or Lou Gehrig disease, involves the progressive loss of motor neurons, resulting in muscle atrophy. The majority of cases are sporadic, but 15–20% of familial cases are associated with mutations in SOD1. This led to the hypothesis that ALS was caused by an increase in superoxide; however, many of the mutations associated with the disease do not change the activity of SOD1, and sod-1–null mice do not develop ALS. Therefore, a gain of toxic function is proposed for the SOD1 mutations, although that function has not yet been identified. Hypotheses include a copper-dependent ROS-generating function of mutant SOD1 (45, 63), a propensity of mutant SOD1 to aggregate (74, 77), increased apoptosis due to Bcl-2 sequestration by aggregated SOD1 (118), and an ability of mutant SOD1 to bind to rac, activating the NADPH oxidase on microglial cells and enhancing superoxide production (66).
The mutation acts non–cell-autonomously, in that astrocytes carrying the mutation can induce apoptosis in nonmutant neurons (102, 148), and nonmutant astrocytes can extend the survival of mutant neurons (28), although with high enough expression levels, expression of mutant SOD1 only in neurons is enough to cause motor neuron disease (74). Interestingly, overexpression of wild-type SOD1 results in the formation of aggregates in astrocytes (74), and oxidation of wild-type SOD1 can cause it to misfold and gain some of the toxic properties of mutant SOD1 (47), leading to the hypothesis that oxidative stress and oxidative damage of wild-type SOD1 could be behind sporadic ALS (79).
Despite the lack of support for the hypothesis that SOD1 mutations lead to an increase in superoxide because of a decrease in SOD activity, substantial evidence exists for increased oxidative stress in ALS. Mice carrying a mutant human form of SOD1 accumulate damaged DNA, protein, and lipid (64, 92, 154), and neural tissue from ALS patients also demonstrates oxidative damage (48, 49, 72). In a cell-culture model, expression of several mutant forms of SOD1 was found to cause an increase in mitochondrial superoxide and neuronal cell death, and cell survival was increased by a concomitant overexpression of SOD2 (160). Expression of mutant SOD1 resulted in mitochondrial dysfunction and increased oxidative and nitrative damage in astrocytes, which could be rescued by mitochondrially targeted antioxidants (20). Overexpression of SOD2 and phospholipid glutathione peroxidase (93) and treatment with a mitochondrially targeted antioxidant peptide (114) extended the life span in a mouse model of ALS. Copper chelators can also extend survival, consistent with a metal-catalyzed oxidative-stress component of ALS (116).
Deleting the gene for gp91, the catalytic component of the microglial NADPH oxidase, blocked neuronal cell death in a mouse ALS model (157), supporting a role for neuroinflammation. However, a clinical trial of minocycline, an antiinflammatory drug, did not slow disease progression (60), although it was protective in an animal model of the disease (82, 147). This points out a long-running issue of lack of correlation between in vitro and animal and human studies in ALS (reviewed in 12), analogous to that seen for neuroprotective drugs in stroke.
Catalytic antioxidants have shown promise in animal models of ALS. AEOL 10150 extended survival from symptom onset up to threefold, when the drug was administered to each mouse on first day of muscle weakness in the mouse G93A model of ALS (31). In this same study, AEOL 10150 was combined with dietary creatine and rofecoxib, with no additional increase in survival. In another study, AEOL 10150 was administered alone or in combination with the histone deacetylase inhibitor phenylbutyrate (PBA) at symptom onset in the G93A mouse model (115). All three paradigms were found to extend survival, but the combination therapy was the most effective, extending survival by 19%. EUK-8 and EUK-134 also extended survival when administered before disease onset (78), and C3 similarly delayed symptom onset and death (41). Human trials with a reactive oxygen and reactive nitrogen species scavenger R+ pramipexole demonstrate the drug is well tolerated and hint at positive effect on disease progression (153), providing hope that antioxidant strategies may translate into treating human disease.
Conclusions
Oxidative stress is a component of and plays a complex role in many neurodegenerative diseases. In some cases, oxidative stress is secondary to other pathology (e.g., oxidative damage occurs as the result of an immune response initiated by cell death). In other cases, oxidative damage due to mitochondrial dysfunction is a primary cause of cell death and neuronal loss. Catalytic antioxidants can intervene at each of these steps, preventing cell death in response to mitochondrial dysfunction or excitotoxicity and preventing the immune-response–driven vicious cycle of increased oxidative and nitrative damage, which can contribute to subsequent cell death and disease progression. Therefore, multiple ways exist in which catalytic antioxidants can exert the protective effects observed in the in vitro and in vivo models of neurodegeneration, and much promise is seen that these compounds may present useful therapies in human disease.
Abbreviations
Aβ, amyloid beta; AD, Alzheimer's disease; AIS, acute ischemic stroke; ALS, amyotrophic lateral sclerosis; ApoE, apolipoprotein E; APP, amyloid precursor protein; DNA, deoxyribonucleic acid; ETC, electron-transport chain; iNOS, inducible nitric oxide synthase; MnSOD, manganese superoxide dismutase; MnTBAP, manganese (III) tetrakis (4-benzoic acid)porphyrin; MPP+, 1-methyl-4-phenylpyridinium; MPTP, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; NAD, nicotineamide adenine dinucleotide; NADPH, nicotineamide adenine dinucleotide phosphate; nNOS, neuronal nitric oxide synthase; NO, nitric oxide; NSAID, nonsteroidal antiinflammatory drug; ONOO−, peroxynitrite; PD, Parkinson's disease; RNA, ribonucleic acid; RNS, reactive nitrogen species; ROS, reactive oxygen species; SOD, superoxide dismutase; SOD1, intracellular Cu/Zn superoxide dismutase; SOD2, mitochondrial manganese superoxide dismutase; STAIR, Stroke Therapy Academic Industry Roundtable.
Acknowledgments
Dr. Patel has received grant funding from the National Institutes of Health (RONS45748, R01NS39587, R21NS53548, and Aeolus Pharmaceuticals, a company that develops metallopophyrin antioxidants for human disease.
References
- 1.Aisen PS. Schmeidler J. Pasinetti GM. Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology. 2002;58:1050–1054. doi: 10.1212/wnl.58.7.1050. [DOI] [PubMed] [Google Scholar]
- 2.Aisen PS. Schafer KA. Grundman M. Pfeiffer E. Sano M. Davis KL. Farlow MR. Jin S. Thomas RG. Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;28921:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- 3.Alam ZI. Daniel SE. Lees AJ. Marsden DC. Jenner P. Halliwell B. A generalised increase in protein carbonyls in the brain in Parkinson's but not incidental Lewy body disease. J Neurochem. 1997;69:1326–1329. doi: 10.1046/j.1471-4159.1997.69031326.x. [DOI] [PubMed] [Google Scholar]
- 4.Alam ZI. Jenner A. Daniel SE. Lees AJ. Cairns N. Marsden CD. Jenner P. Halliwell B. Oxidative DNA damage in the parkinsonian brain: an apparent selective increase in 8-hydroxyguanine levels in substantia nigra. J Neurochem. 1997;69:1196–1203. doi: 10.1046/j.1471-4159.1997.69031196.x. [DOI] [PubMed] [Google Scholar]
- 5.Ali SS. Hardt JI. Quick KL. Kim-Han JS. Erlanger BF. Huang TT. Epstein CJ. Dugan LL. A biologically effective fullerene (C60) derivative with superoxide dismutase mimetic properties. Free Radic Biol Med. 2004;37:1191–1202. doi: 10.1016/j.freeradbiomed.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 6.Anderson I. Adinolfi C. Doctrow S. Huffman K. Joy KA. Malfroy B. Soden P. Rupniak HT. Barnes JC. Oxidative signalling and inflammatory pathways in Alzheimer's disease. Biochem Soc Symp. 2001;67:141–149. doi: 10.1042/bss0670141. [DOI] [PubMed] [Google Scholar]
- 7.Andorn AC. Kalaria RN. Factors affecting pro- and antioxidant properties of fragments of the b-protein precursor (bPP): implication for Alzheimer's disease. J Alzheimers Dis. 2000;2:69–78. doi: 10.3233/jad-2000-2201. [DOI] [PubMed] [Google Scholar]
- 8.Arancio O. Kiebler M. Lee CJ. Lev-Ram V. Tsien RY. Kandel ER. Hawkins RD. Nitric oxide acts directly in the presynaptic neuron to produce long-term potentiation in cultured hippocampal neurons. Cell. 1996;87:1025–1035. doi: 10.1016/s0092-8674(00)81797-3. [DOI] [PubMed] [Google Scholar]
- 9.Atamna H. Heme binding to amyloid-beta peptide: mechanistic role in Alzheimer's disease. J Alzheimers Dis. 2006;10:255–266. doi: 10.3233/jad-2006-102-310. [DOI] [PubMed] [Google Scholar]
- 10.Baker K. Marcus CB. Huffman K. Kruk H. Malfroy B. Doctrow SR. Synthetic combined superoxide dismutase/catalase mimetics are protective as a delayed treatment in a rat stroke model: a key role for reactive oxygen species in ischemic brain injury. J Pharmacol Exp Ther. 1998;284:215–221. [PubMed] [Google Scholar]
- 11.Bamberger ME. Landreth GE. Microglial interaction with beta-amyloid: implications for the pathogenesis of Alzheimer's disease. Microsc Res Tech. 2001;54:59–70. doi: 10.1002/jemt.1121. [DOI] [PubMed] [Google Scholar]
- 12.Benatar M. Lost in translation: treatment trials in the SOD1 mouse and in human ALS. Neurobiol Dis. 2007;26:1–13. doi: 10.1016/j.nbd.2006.12.015. [DOI] [PubMed] [Google Scholar]
- 13.Bornebroek M. de Lau LM. Haag MD. Koudstaal PJ. Hofman A. Stricker BH. Breteler MM. Nonsteroidal antiinflammatory drugs and the risk of Parkinson disease. Neuroepidemiology. 2007;28:193–196. doi: 10.1159/000108110. [DOI] [PubMed] [Google Scholar]
- 14.Bowler RP. Sheng H. Enghild JJ. Pearlstein RD. Warner DS. Crapo JD. A catalytic antioxidant (AEOL 10150) attenuates expression of inflammatory genes in stroke. Free Radic Biol Med. 2002;33:1141–1152. doi: 10.1016/s0891-5849(02)01008-0. [DOI] [PubMed] [Google Scholar]
- 15.Breitner JC. Gau BA. Welsh KA. Plassman BL. McDonald WM. Helms MJ. Anthony JC. Inverse association of anti-inflammatory treatments and Alzheimer's disease: initial results of a co-twin control study. Neurology. 1994;44:227–232. doi: 10.1212/wnl.44.2.227. [DOI] [PubMed] [Google Scholar]
- 16.Brown TP. Rumsby PC. Capleton AC. Rushton L. Levy LS. Pesticides and Parkinson's disease: is there a link? Environ Health Perspect. 2006;114:156–164. doi: 10.1289/ehp.8095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bruce AJ. Malfroy B. Baudry M. beta-Amyloid toxicity in organotypic hippocampal cultures: protection by EUK-8, a synthetic catalytic free radical scavenger. Proc Natl Acad Sci U S A. 1996;93:23120–23126. doi: 10.1073/pnas.93.6.2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Busuttil RA. Garcia AM. Cabrera C. Rodriguez A. Suh Y. Kim WH. Huang TT. Vijg J. Organ-specific increase in mutation accumulation and apoptosis rate in CuZn-superoxide dismutase-deficient mice. Cancer Res. 2005;65:11271–11275. doi: 10.1158/0008-5472.CAN-05-2980. [DOI] [PubMed] [Google Scholar]
- 19.Carlsson LM. Jonsson J. Edlund T. Marklund SL. Mice lacking extracellular superoxide dismutase are more sensitive to hyperoxia. Proc Natl Acad Sci U S A. 1995;92:6264–6268. doi: 10.1073/pnas.92.14.6264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cassina P. Cassina A. Pehar M. Castellanos R. Gandelman M. de Leon A. Robinson KM. Mason RP. Beckman JS. Barbeito L. Radi R. Mitochondrial dysfunction in SOD1G93A-bearing astrocytes promotes motor neuron degeneration: prevention by mitochondrial-targeted antioxidants. J Neurosci. 2008;28:4115–4122. doi: 10.1523/JNEUROSCI.5308-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castello PR. Drechsel DA. Day BJ. Patel M. Inhibition of mitochondrial hydrogen peroxide production by lipophilic metalloporphyrins. J Pharmacol Exp Ther. 2008;324:970–976. doi: 10.1124/jpet.107.132134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chan PH. Kawase M. Murakami K. Chen SF. Li Y. Calagui B. Reola L. Carlson E. Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chance B. Sies H. Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 24.Chen H. Jacobs E. Schwarzschild MA. McCullough ML. Calle EE. Thun MJ. Ascherio A. Nonsteroidal antiinflammatory drug use and the risk for Parkinson's disease. Ann Neurol. 2005;58:963–967. doi: 10.1002/ana.20682. [DOI] [PubMed] [Google Scholar]
- 25.Choi DW. Koh JY. Peters S. Pharmacology of glutamate neurotoxicity in cortical cell culture: attenuation by NMDA antagonists. J Neurosci. 1988;1:185–196. doi: 10.1523/JNEUROSCI.08-01-00185.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi WS. Yoon SY. Oh TH. Choi EJ. O'Malley KL. Oh YJ. Two distinct mechanisms are involved in 6-hydroxydopamine- and MPP+-induced dopaminergic neuronal cell death: role of caspases, ROS, and JNK. J Neurosci Res. 1999;57:86–94. doi: 10.1002/(SICI)1097-4547(19990701)57:1<86::AID-JNR9>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 27.Cleeter MW. Cooper JM. Schapira AH. Irreversible inhibition of mitochondrial complex I by 1-methyl-4-phenylpyridinium: evidence for free radical involvement. J Neurochem. 1992;58:786–789. doi: 10.1111/j.1471-4159.1992.tb09789.x. [DOI] [PubMed] [Google Scholar]
- 28.Clement AM. Nguyen MD. Roberts EA. Garcia ML. Boillee S. Rule M. McMahon AP. Doucette W. Siwek D. Ferrante RJ. Brown RH., Jr Julien JP. Goldstein LS. Cleveland DW. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science. 2003;302:113–117. doi: 10.1126/science.1086071. [DOI] [PubMed] [Google Scholar]
- 29.Crack PJ. Taylor JM. de Haan JB. Kola I. Hertzog P. Iannello RC. Glutathione peroxidase-1 contributes to the neuroprotection seen in the superoxide dismutase-1 transgenic mouse in response to ischemia/reperfusion injury. J Cereb Blood Flow Metab. 2003;23:19–22. doi: 10.1097/01.WCB.0000035181.38851.71. [DOI] [PubMed] [Google Scholar]
- 30.Crouch PJ. Harding SM. White AR. Camakaris J. Bush AI. Masters CL. Mechanisms of A beta mediated neurodegeneration in Alzheimer's disease. Int J Biochem Cell Biol. 2008;40:181–198. doi: 10.1016/j.biocel.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 31.Crow JP. Calingasan NY. Chen J. Hill JL. Beal MF. Manganese porphyrin given at symptom onset markedly extends survival of ALS mice. Ann Neurol. 2005;58:258–265. doi: 10.1002/ana.20552. [DOI] [PubMed] [Google Scholar]
- 32.Davis AS. Zhao H. Sun GH. Sapolsky RM. Steinberg GK. Gene therapy using SOD1 protects striatal neurons from experimental stroke. Neurosci Lett. 2007;411:32–36. doi: 10.1016/j.neulet.2006.08.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Day BJ. Fridovich I. Crapo JD. Manganic porphyrins possess catalase activity and protect endothelial cells against hydrogen peroxide-mediated injury. Arch Biochem Biophys. 1977;347(2):256–262. doi: 10.1006/abbi.1997.0341. [DOI] [PubMed] [Google Scholar]
- 34.Day BJ. Catalytic antioxidants: a radical approach to new therapeutics. Drug Discov Today. 2004;9:557–566. doi: 10.1016/S1359-6446(04)03139-3. [DOI] [PubMed] [Google Scholar]
- 35.Dexter DT. Carter CJ. Wells FR. Javoy-Agid F. Agid Y. Lees A. Jenner P. Marsden CD. Basal lipid peroxidation in substantia nigra is increased in Parkinson's disease. J Neurochem. 1998;52:381–389. doi: 10.1111/j.1471-4159.1989.tb09133.x. [DOI] [PubMed] [Google Scholar]
- 36.Dexter DT. Holley AE. Flitter WD. Slater TF. Wells FR. Daniel SE. Lees AJ. Jenner P. Marsden CD. Increased levels of lipid hydroperoxides in the parkinsonian substantia nigra: an HPLC and ESR study. Mov Disord. 1994;9:92–97. doi: 10.1002/mds.870090115. [DOI] [PubMed] [Google Scholar]
- 37.Doctrow SR. Huffman K. Marcus CB. Tocco G. Malfroy E. Adinolfi CA. Kruk H. Baker K. Lazarowych N. Mascarenhas J. Malfroy B. Salen-manganese complexes as catalytic scavengers of hydrogen peroxide and cytoprotective agents: structure-activity relationship studies. J Med Chem. 2002;45:4549–4558. doi: 10.1021/jm020207y. [DOI] [PubMed] [Google Scholar]
- 38.Dolphin D. Forman A. Borg DC. Fajer J. Felton RH. Compounds I of catalase and horseradish peroxidase: pication radicals. Proc Natl Acad Sci U S A. 1971;68:614–618. doi: 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Du Y. Ma Z. Lin S. Dodel RC. Gao F. Bales KR. Triarhou LC. Chernet E. Perry KW. Nelson DL. Luecke S. Phebus LA. Bymaster FP. Paul SM. Minocycline prevents nigrostriatal dopaminergic neurodegeneration in the MPTP model of Parkinson's disease. Proc Natl Acad Sci U S A. 2001;98:14669–14674. doi: 10.1073/pnas.251341998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dugan LL. Gabrielsen JK. Yu SP. Lin TS. Choi DW. Buckminsterfullerenol free radical scavengers reduce excitotoxic and apoptotic death of cultured cortical neurons. Neurobiol Dis. 1996;3:129–135. doi: 10.1006/nbdi.1996.0013. [DOI] [PubMed] [Google Scholar]
- 41.Dugan LL. Turetsky DM. Du C. Lobner D. Wheeler M. Almli CR. Shen CK. Luh TY. Choi DW. Lin TS. Carboxyfullerenes as neuroprotective agents. Proc Natl Acad Sci U S A. 1997;94:9434–9439. doi: 10.1073/pnas.94.17.9434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Elchuri S. Oberley TD. Qi W. Eisenstein RS. Jackson Roberts L. Van Remmen H. Epstein CJ. Huang TT. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 43.Eliasson MJ. Huang Z. Ferrante RJ. Sasamata M. Molliver ME. Snyder SH. Moskowitz MA. Neuronal nitric oxide synthase activation and peroxynitrite formation in ischemic stroke linked to neural damage. J Neurosci. 1999;19:5910–5918. doi: 10.1523/JNEUROSCI.19-14-05910.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Esposito LA. Melov S. Panov A. Cottrell BA. Wallace DC. Mitochondrial disease in mouse results in increased oxidative stress. Proc Natl Acad Sci U S A. 1999;96:4820–4825. doi: 10.1073/pnas.96.9.4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Estevez AG. Crow JP. Sampson JB. Reiter C. Zhuang Y. Richardson GJ. Tarpey MM. Barbeito L. Beckman JS. Induction of nitric oxide-dependent apoptosis in motor neurons by zinc-deficient superoxide dismutase. Science. 1999;286:2498–2500. doi: 10.1126/science.286.5449.2498. [DOI] [PubMed] [Google Scholar]
- 46.Etminan M. Carleton BC. Samii A. Non-steroidal antiinflammatory drug use and the risk of Parkinson disease: a retrospective cohort study. J Clin Neurosci. 2008;15:576–577. doi: 10.1016/j.jocn.2007.02.095. [DOI] [PubMed] [Google Scholar]
- 47.Ezzi SA. Urushitani M. Julien JP. Wild-type superoxide dismutase acquires binding and toxic properties of ALS-linked mutant forms through oxidation. J Neurochem. 2007;102:170–178. doi: 10.1111/j.1471-4159.2007.04531.x. [DOI] [PubMed] [Google Scholar]
- 48.Ferrante RJ. Browne SE. Shinobu LA. Bowling AC. Baik MJ. MacGarvey U. Kowall NW. Brown RH., Jr Beal MF. Evidence of increased oxidative damage in both sporadic and familial amyotrophic lateral sclerosis. J Neurochem. 1997;69:2064–2074. doi: 10.1046/j.1471-4159.1997.69052064.x. [DOI] [PubMed] [Google Scholar]
- 49.Ferrante RJ. Shinobu LA. Schulz JB. Matthews RT. Thomas CE. Kowall NW. Gurney ME. Beal MF. Increased 3-nitrotyrosine and oxidative damage in mice with a human copper/zinc superoxide dismutase mutation. Ann Neurol. 1997;42:326–334. doi: 10.1002/ana.410420309. [DOI] [PubMed] [Google Scholar]
- 50.Flamm ES. Demopoulos HB. Seligman ML. Poser RG. Ransohoff J. Free radicals in cerebral ischemia. Stroke. 1978;9:445–447. doi: 10.1161/01.str.9.5.445. [DOI] [PubMed] [Google Scholar]
- 51.Fridovich I. Superoxide anion radical (O2−), superoxide dismutases, and related matters. J Biol Chem. 1997;272:18515–18517. doi: 10.1074/jbc.272.30.18515. [DOI] [PubMed] [Google Scholar]
- 52.Fukui H. Diaz F. Garcia S. Moraes CT. Cytochrome coxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2007;104:14163–14168. doi: 10.1073/pnas.0705738104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gabbita SP. Lovell MA. Markesbery WR. Increased nuclear DNA oxidation in the brain in Alzheimer's disease. J Neurochem. 1998;71:2034–2040. doi: 10.1046/j.1471-4159.1998.71052034.x. [DOI] [PubMed] [Google Scholar]
- 54.Gardner PR. Raineri I. Epstein LB. White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995;270:13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 55.Gilgun-Sherki Y. Melamed E. Offen D. Antioxidant treatment in Alzheimer's disease: current state. J Mol Neurosci. 2003;21:1–11. doi: 10.1385/JMN:21:1:1. [DOI] [PubMed] [Google Scholar]
- 56.Glenner GG. Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 57.Golden TR. Hubbard A. Morten KJ. Hinerfeld D. Melov S. Pharmacogenomic profiling of an oxidative stress-mediated spongiform encephalopathy. Free Radic Biol Med. 2005;39:152–163. doi: 10.1016/j.freeradbiomed.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 58.Goldstein LE. Muffat JA. Cherny RA. Moir RD. Ericsson MH. Huang X. Mavros C. Coccia JA. Faget KY. Fitch KA. Masters CL. Tanzi RE. Chylack LT., Jr Bush AI. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer's disease. Lancet. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- 59.Good PF. Hsu A. Werner P. Perl DP. Olanow CW. Protein nitration in Parkinson's disease. J Neuropathol Exp Neurol. 1998;57:338–342. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 60.Gordon PH. Moore DH. Miller RG. Florence JM. Verheijde JL. Doorish C. Hilton JF. Spitalny GM. MacArthur RB. Mitsumoto H. Neville HE. Boylan K. Mozaffar T. Belsh JM. Ravits J. Bedlack RS. Graves MC. McCluskey LF. Barohn RJ. Tandan R. Efficacy of minocycline in patients with amyotrophic lateral sclerosis: a phase III randomised trial. Lancet Neurol. 2007;6:1045–1053. doi: 10.1016/S1474-4422(07)70270-3. [DOI] [PubMed] [Google Scholar]
- 61.Green AR. Pharmacological approaches to acute ischaemic stroke: reperfusion certainly, neuroprotection possibly. Br J Pharmacol. 2008;153(suppl 1):S325–S338. doi: 10.1038/sj.bjp.0707594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Group AR. Lyketsos CG. Breitner JC. Green RC. Martin BK. Meinert C. Piantadosi S. Sabbagh M. Naproxen and celecoxib do not prevent AD in early results from a randomized controlled trial. Neurology. 2007;68:1800–1808. doi: 10.1212/01.wnl.0000260269.93245.d2. [DOI] [PubMed] [Google Scholar]
- 63.Gunther MR. Donahue JA. Bicarbonate and active site zinc modulate the self-peroxidation of bovine copper-zinc superoxide dismutase. Free Radic Res. 2007;41:1005–1016. doi: 10.1080/10715760701516308. [DOI] [PubMed] [Google Scholar]
- 64.Hall ED. Andrus PK. Oostveen JA. Fleck TJ. Gurney ME. Relationship of oxygen radical-induced lipid peroxidative damage to disease onset and progression in a transgenic model of familial ALS. J Neurosci Res. 1998;53:66–77. doi: 10.1002/(SICI)1097-4547(19980701)53:1<66::AID-JNR7>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 65.Hansford RG. Hogue BA. Mildaziene V. Dependence of H2O2 formation by rat heart mitochondria on substrate availability and donor age. J Bioenerg Biomembr. 1997;29:89–95. doi: 10.1023/a:1022420007908. [DOI] [PubMed] [Google Scholar]
- 66.Harraz MM. Marden JJ. Zhou W. Zhang Y. Williams A. Sharov VS. Nelson K. Luo M. Paulson H. Schoneich C. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest. 2008;118:659–670. doi: 10.1172/JCI34060. others. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hensley K. Maidt ML. Yu Z. Sang H. Markesbery WR. Floyd RA. Electrochemical analysis of protein nitrotyrosine and dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci. 1998;18:8126–8132. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hertzman C. Wiens M. Bowering D. Snow B. Calne D. Parkinson's disease: a case-control study of occupational and environmental risk factors. Am J Indian Med. 1990;17:349–355. doi: 10.1002/ajim.4700170307. [DOI] [PubMed] [Google Scholar]
- 69.Huang TT. Carlson EJ. Kozy HM. Mantha S. Goodman SI. Ursell PC. Epstein CJ. Genetic modification of prenatal lethality and dilated cardiomyopathy in Mn superoxide dismutase mutant mice. Free Radic Biol Med. 2001;31:1101–1110. doi: 10.1016/s0891-5849(01)00694-3. [DOI] [PubMed] [Google Scholar]
- 70.Huang X. Atwood CS. Hartshorn MA. Multhaup G. Goldstein LE. Scarpa RC. Cuajungco MP. Gray DN. Lim J. Moir RD. The A beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38:7609–7616. doi: 10.1021/bi990438f. others. [DOI] [PubMed] [Google Scholar]
- 71.Ignarro LJ. Buga GM. Wood KS. Byrns RE. Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ilieva EV. Ayala V. Jove M. Dalfo E. Cacabelos D. Povedano M. Bellmunt MJ. Ferrer I. Pamplona R. Portero-Otin M. Oxidative and endoplasmic reticulum stress interplay in sporadic amyotrophic lateral sclerosis. Brain. 2007;130:3111–3123. doi: 10.1093/brain/awm190. [DOI] [PubMed] [Google Scholar]
- 73.Ischiropoulos H. Zhu L. Chen J. Tsai M. Martin JC. Smith CD. Beckman JS. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 74.Jaarsma D. Teuling E. Haasdijk ED. De Zeeuw CI. Hoogenraad CC. Neuron-specific expression of mutant superoxide dismutase is sufficient to induce amyotrophic lateral sclerosis in transgenic mice. J Neurosci. 2008;28:2075–2088. doi: 10.1523/JNEUROSCI.5258-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Janetzky B. Hauck S. Youdim MB. Riederer P. Jellinger K. Pantucek F. Zochling R. Boissl KW. Reichmann H. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson's disease. Neurosci Lett. 1994;169:126–128. doi: 10.1016/0304-3940(94)90372-7. [DOI] [PubMed] [Google Scholar]
- 76.Jekabsone A. Mander PK. Tickler A. Sharpe M. Brown GC. Fibrillar beta-amyloid peptide Abeta1–40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J Neuroinflamm. 2006;3:24. doi: 10.1186/1742-2094-3-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Johnston JA. Dalton MJ. Gurney ME. Kopito RR. Formation of high molecular weight complexes of mutant Cu, Zn-superoxide dismutase in a mouse model for familial amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A. 2000;97:12571–12576. doi: 10.1073/pnas.220417997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jung C. Rong Y. Doctrow S. Baudry M. Malfroy B. Xu Z. Synthetic superoxide dismutase/catalase mimetics reduce oxidative stress and prolong survival in a mouse amyotrophic lateral sclerosis model. Neurosci Lett. 2001;304:157–160. doi: 10.1016/s0304-3940(01)01784-0. [DOI] [PubMed] [Google Scholar]
- 79.Kabashi E. Valdmanis PN. Dion P. Rouleau GA. Oxidized/misfolded superoxide dismutase-1: the cause of all amyotrophic lateral sclerosis? Ann Neurol. 2007;62:5533–5539. doi: 10.1002/ana.21319. [DOI] [PubMed] [Google Scholar]
- 80.Kaur H. Halliwell B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation: nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994;350:9–12. doi: 10.1016/0014-5793(94)00722-5. [DOI] [PubMed] [Google Scholar]
- 81.Kinouchi H. Epstein CJ. Mizui T. Carlson E. Chen SF. Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci U S A. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kriz J. Nguyen MD. Julien JP. Minocycline slows disease progression in a mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2002;10:268–278. doi: 10.1006/nbdi.2002.0487. [DOI] [PubMed] [Google Scholar]
- 83.Lee J. Hunt JA. Groves JT. Manganese porphyrins as redox-coupled peroxynitrite reductases. J Am Chem Soc. 1998;120:6053–6061. [Google Scholar]
- 84.Lees KR. Zivin JA. Ashwood T. Davalos A. Davis SM. Diener HC. Grotta J. Lyden P. Shuaib A. Hardemark HG. Wasiewski WW. NXY-059 for acute ischemic stroke. N Engl J Med. 2006;354:588–600. doi: 10.1056/NEJMoa052980. [DOI] [PubMed] [Google Scholar]
- 85.Li QY. Pedersen C. Day BJ. Patel M. Dependence of excitotoxic neurodegeneration on mitochondrial aconitase inactivation. J Neurochem. 2001;78:746–755. doi: 10.1046/j.1471-4159.2001.00457.x. [DOI] [PubMed] [Google Scholar]
- 86.Li Y. Huang TT. Carlson EJ. Melov S. Ursell PC. Olson JL. Noble LJ. Yoshimura MP. Berger C. Chan PH. Wallace DC. Epstein CJ. Dilated cardiomyopathy and neonatal lethality in mutant mice lacking manganese superoxide dismutase. Nat Genet. 1995;11:376–381. doi: 10.1038/ng1295-376. [DOI] [PubMed] [Google Scholar]
- 87.Liang LP. Ho YS. Patel M. Mitochondrial superoxide production in kainate-induced hippocampal damage. Neuroscience. 2000;101:563–570. doi: 10.1016/s0306-4522(00)00397-3. [DOI] [PubMed] [Google Scholar]
- 88.Liang LP. Patel M. Seizure-induced changes in mitochondrial redox status. Free Radic Biol Med. 2006;40:316–322. doi: 10.1016/j.freeradbiomed.2005.08.026. [DOI] [PubMed] [Google Scholar]
- 89.Liang LP. Huang J. Fulton R. Day BJ. Patel M. An orally active catalytic metalloporphyrin protects against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in vivo. J Neurosci. 2007;27:4326–4333. doi: 10.1523/JNEUROSCI.0019-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lin AM. Fang SF. Lin SZ. Chou CK. Luh TY. Ho LT. Local carboxyfullerene protects cortical infarction in rat brain. Neurosci Res. 2002;43:317–321. doi: 10.1016/s0168-0102(02)00056-1. [DOI] [PubMed] [Google Scholar]
- 91.Liou HH. Tsai MC. Chen CJ. Jeng JS. Chang YC. Chen SY. Chen RC. Environmental risk factors and Parkinson's disease: a case-control study in Taiwan. Neurology. 1997;48:1583–1588. doi: 10.1212/wnl.48.6.1583. [DOI] [PubMed] [Google Scholar]
- 92.Liu D. Wen J. Liu J. Li L. The roles of free radicals in amyotrophic lateral sclerosis: reactive oxygen species and elevated oxidation of protein, DNA, and membrane phospholipids. FASEB J. 1999;13:2318–2328. doi: 10.1096/fasebj.13.15.2318. [DOI] [PubMed] [Google Scholar]
- 93.Liu R. Li B. Flanagan SW. Oberley LW. Gozal D. Qiu M. Increased mitochondrial antioxidative activity or decreased oxygen free radical propagation prevent mutant SOD1-mediated motor neuron cell death and increase amyotrophic lateral sclerosis-like transgenic mouse survival. J Neurochem. 2002;80:488–500. doi: 10.1046/j.0022-3042.2001.00720.x. [DOI] [PubMed] [Google Scholar]
- 94.Lotharius J. Dugan LL. O'Malley KL. Distinct mechanisms underlie neurotoxin-mediated cell death in cultured dopaminergic neurons. J Neurosci. 1999;19:1284–1293. doi: 10.1523/JNEUROSCI.19-04-01284.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lustbader JW. Cirilli M. Lin C. Xu HW. Takuma K. Wang N. Caspersen C. Chen X. Pollak S. Chaney M. ABAD directly links Abeta to mitochondrial toxicity in Alzheimer's disease. Science. 2004;304:448–452. doi: 10.1126/science.1091230. others. [DOI] [PubMed] [Google Scholar]
- 96.Mackensen GB. Patel M. Sheng H. Calvi CL. Batinic-Haberle I. Day BJ. Liang LP. Fridovich I. Crapo JD. Pearlstein RD. Warner DS. Neuroprotection from delayed postischemic administration of a metalloporphyrin catalytic antioxidant. J Neurosci. 2001;21:4582–4592. doi: 10.1523/JNEUROSCI.21-13-04582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mecocci P. MacGarvey U. Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer's disease. Ann Neurol. 1994;36:747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 98.Melov S. Schneider JA. Day BJ. Hinerfeld D. Coskun P. Mirra SS. Crapo JD. Wallace DC. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genet. 1998;18:159–163. doi: 10.1038/ng0298-159. [DOI] [PubMed] [Google Scholar]
- 99.Melov S. Coskun P. Patel M. Tuinstra R. Cottrell B. Jun AS. Zastawny TH. Dizdaroglu M. Goodman SI. Huang TT. Miziorko H. Epstein CJ. Wallace DC. Mitochondrial disease in superoxide dismutase 2 mutant mice. Proc Natl Acad Sci U S A. 1999;96:846–851. doi: 10.1073/pnas.96.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Melov S. Doctrow SR. Schneider JA. Haberson J. Patel M. Coskun PE. Huffman K. Wallace DC. Malfroy B. Lifespan extension and rescue of spongiform encephalopathy in superoxide dismutase 2 nullizygous mice treated with superoxide dismutase-catalase mimetics. J Neurosci. 2001;21:8348–8353. doi: 10.1523/JNEUROSCI.21-21-08348.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Melov S. Wolf N. Strozyk D. Doctrow SR. Bush AI. Mice transgenic for Alzheimer disease beta-amyloid develop lens cataracts that are rescued by antioxidant treatment. Free Radic Biol Med. 2005;38:258–261. doi: 10.1016/j.freeradbiomed.2004.10.023. [DOI] [PubMed] [Google Scholar]
- 102.Nagai M. Re DB. Nagata T. Chalazonitis A. Jessell TM. Wichterle H. Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Namba K. Takeda Y. Sunami K. Hirakawa M. Temporal profiles of the levels of endogenous antioxidants after four-vessel occlusion in rats. J Neurosurg Anesthesiol. 2001;13:131–137. doi: 10.1097/00008506-200104000-00010. [DOI] [PubMed] [Google Scholar]
- 104.Nunomura A. Perry G. Pappolla MA. Wade R. Hirai K. Chiba S. Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer's disease. J Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nunomura A. Perry G. Aliev G. Hirai K. Takeda A. Balraj EK. Jones PK. Ghanbari H. Wataya T. Shimohama S. Chiba S. Atwood CS. Petersen RB. Smith MA. Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:759–767. doi: 10.1093/jnen/60.8.759. [DOI] [PubMed] [Google Scholar]
- 106.Opazo C. Huang X. Cherny RA. Moir RD. Roher AE. White AR. Cappai R. Masters CL. Tanzi RE. Inestrosa NC. Bush AI. Metalloenzyme-like activity of Alzheimer's disease beta-amyloid: Cu-dependent catalytic conversion of dopamine, cholesterol, and biological reducing agents to neurotoxic H(2)O(2) J Biol Chem. 2002;277:40302–40308. doi: 10.1074/jbc.M206428200. [DOI] [PubMed] [Google Scholar]
- 107.Palmer RM. Ferrige AG. Moncada S. Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature. 1987;327:524–526. doi: 10.1038/327524a0. [DOI] [PubMed] [Google Scholar]
- 108.Pankratz N. Foroud T. Genetics of Parkinson disease. Genet Med. 2007;9:801–811. doi: 10.1097/gim.0b013e31815bf97c. [DOI] [PubMed] [Google Scholar]
- 109.Park EM. Choi JH. Park JS. Han MY. Park YM. Measurement of glutathione oxidation and 8-hydroxy-2′-deoxyguanosine accumulation in the gerbil hippocampus following global ischemia. Brain Res Brain Res Protoc. 2000;6:25–32. doi: 10.1016/s1385-299x(00)00033-7. [DOI] [PubMed] [Google Scholar]
- 110.Park L. Anrather J. Zhou P. Frys K. Pitstick R. Younkin S. Carlson GA. Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci. 2005;25:1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Patel M. Day BJ. Crapo JD. Fridovich I. McNamara JO. Requirement for superoxide in excitotoxic cell death. Neuron. 1996;16:345–355. doi: 10.1016/s0896-6273(00)80052-5. [DOI] [PubMed] [Google Scholar]
- 112.Patel M. Day BJ. Metalloporphyrin class of therapeutic catalytic antioxidants. Trends Pharmacol Sci. 1999;20:359–364. doi: 10.1016/s0165-6147(99)01336-x. [DOI] [PubMed] [Google Scholar]
- 113.Peng J. Stevenson FF. Doctrow SR. Andersen JK. Superoxide dismutase/catalase mimetics are neuroprotective against selective paraquat-mediated dopaminergic neuron death in the substantial nigra: implications for Parkinson disease. J Biol Chem. 2005;280:29194–29198. doi: 10.1074/jbc.M500984200. [DOI] [PubMed] [Google Scholar]
- 114.Petri S. Kiaei M. Damiano M. Hiller A. Wille E. Manfredi G. Calingasan NY. Szeto HH. Beal MF. Cell-permeable peptide antioxidants as a novel therapeutic approach in a mouse model of amyotrophic lateral sclerosis. J Neurochem. 2006;98:1141–1148. doi: 10.1111/j.1471-4159.2006.04018.x. [DOI] [PubMed] [Google Scholar]
- 115.Petri S. Kiaei M. Kipiani K. Chen J. Calingasan NY. Crow JP. Beal MF. Additive neuroprotective effects of a histone deacetylase inhibitor and a catalytic antioxidant in a transgenic mouse model of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;22:40–49. doi: 10.1016/j.nbd.2005.09.013. [DOI] [PubMed] [Google Scholar]
- 116.Petri S. Calingasan NY. Alsaied OA. Wille E. Kiaei M. Friedman JE. Baranova O. Chavez JC. Beal MF. The lipophilic metal chelators DP-109 and DP-460 are neuroprotective in a transgenic mouse model of amyotrophic lateral sclerosis. J Neurochem. 2007;102:991–1000. doi: 10.1111/j.1471-4159.2007.04604.x. [DOI] [PubMed] [Google Scholar]
- 117.Pong K. Doctrow SR. Baudry M. Prevention of 1-methyl-4-phenylpyridinium- and 6-hydroxydopamine-induced nitration of tyrosine hydroxylase and neurotoxicity by EUK-134, a superoxide dismutase and catalase mimetic, in cultured dopaminergic neurons. Brain Res. 2000;881:182–189. doi: 10.1016/s0006-8993(00)02841-9. [DOI] [PubMed] [Google Scholar]
- 118.Puglielli L. Friedlich AL. Setchell KD. Nagano S. Opazo C. Cherny RA. Barnham KJ. Wade JD. Melov S. Kovacs DM. Bush AI. Alzheimer disease beta-amyloid activity mimics cholesterol oxidase. J Clin Invest. 2005;115:2556–2563. doi: 10.1172/JCI23610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Raines KW. Bonini MG. Campbell SL. Nitric oxide cell signaling: S-nitrosation of Ras superfamily GTPases. Cardiovasc Res. 2007;75:229–239. doi: 10.1016/j.cardiores.2007.04.013. [DOI] [PubMed] [Google Scholar]
- 120.Reaume AG. Elliott JL. Hoffman EK. Kowall NW. Ferrante RJ. Siwek DF. Wilcox HM. Flood DG. Beal MF. Brown RH., Jr Scott RW. Snider WD. Motor neurons in Cu/Zn superoxide dismutase-deficient mice develop normally but exhibit enhanced cell death after axonal injury. Nat Genet. 1996;13:43–47. doi: 10.1038/ng0596-43. [DOI] [PubMed] [Google Scholar]
- 121.Reines SA. Block GA. Morris JC. Liu G. Nessly ML. Lines CR. Norman BA. Baranak CC. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- 122.Reynolds IJ. Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rogers J. Kirby LC. Hempelman SR. McGeer PL. Kaszniak AW. Zalinski J. Cofield M. Mansukhani L. Willson P. Kogan F. Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993;43:1609–1611. doi: 10.1212/wnl.43.8.1609. [DOI] [PubMed] [Google Scholar]
- 124.Rogers J. Webster S. Lue LF. Brachova L. Civin WH. Emmerling M. Shivers B. Walker D. McGeer P. Inflammation and Alzheimer's disease pathogenesis. Neurobiol Aging. 1996;17:681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- 125.Rong Y. Doctrow SR. Tocco G. Baudry M. EUK-134, a synthetic superoxide dismutase and catalase mimetic, prevents oxidative stress and attenuates kainate-induced neuropathology. Proc Natl Acad Sci U S A. 1999;96:9897–9902. doi: 10.1073/pnas.96.17.9897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rupniak HT. Joy KA. Atkin C. Brown G. Barnes JC. Doctrow SR. Malfroy B. Wong T. Anderson IK. Molloy CR. Mills GI. Soden P. Oxidative neuropathology and putative chemical entities for Alzheimer's disease: neuroprotective effects of salen-manganese catalytic anti-oxidants. Neurotox Res. 2000;2:167–178. doi: 10.1007/BF03033792. [DOI] [PubMed] [Google Scholar]
- 127.Rutten BP. Steinbusch HW. Korr H. Schmitz C. Antioxidants and Alzheimer's disease: from bench to bedside (and back again) Curr Opin Clin Nutr Metab Care. 2002;5:645–651. doi: 10.1097/00075197-200211000-00006. [DOI] [PubMed] [Google Scholar]
- 128.Savitz SI. A critical appraisal of the NXY-059 neuroprotection studies for acute stroke: a need for more rigorous testing of neuroprotective agents in animal models of stroke. Exp Neurol. 2007;205:20–25. doi: 10.1016/j.expneurol.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 129.Sayre LM. Zelasko DA. Harris PL. Perry G. Salomon RG. Smith MA. 4-Hydroxynonenal-derived advanced lipid peroxidation end products are increased in Alzheimer's disease. J Neurochem. 1997;68:2092–2097. doi: 10.1046/j.1471-4159.1997.68052092.x. [DOI] [PubMed] [Google Scholar]
- 130.Schapira AH. Cooper JM. Dexter D. Clark JB. Jenner P. Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem. 1990;54:823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 131.Schapira AH. Mann VM. Cooper JM. Dexter D. Daniel SE. Jenner P. Clark JB. Marsden CD. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson's disease. J Neurochem. 1990;55:2142–2145. doi: 10.1111/j.1471-4159.1990.tb05809.x. [DOI] [PubMed] [Google Scholar]
- 132.Scharf S. Mander A. Ugoni A. Vajda F. Christophidis N. A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999;53:197–201. doi: 10.1212/wnl.53.1.197. [DOI] [PubMed] [Google Scholar]
- 133.Sharpe MA. Ollosson R. Stewart VC. Clark JB. Oxidation of nitric oxide by oxomanganese-salen complexes: a new mechanism for cellular protection by superoxide dismutase/catalase mimetics. Biochem J. 2002;366:97–107. doi: 10.1042/BJ20020154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sheng H. Enghild JJ. Bowler R. Patel M. Batinic-Haberle I. Calvi CL. Day BJ. Pearlstein RD. Crapo JD. Warner DS. Effects of metalloporphyrin catalytic antioxidants in experimental brain ischemia. Free Radic Biol Med. 2002;33:947–961. doi: 10.1016/s0891-5849(02)00979-6. [DOI] [PubMed] [Google Scholar]
- 135.Shimizu K. Rajapakse N. Horiguchi T. Payne RM. Busija DW. Neuroprotection against hypoxia-ischemia in neonatal rat brain by novel superoxide dismutase mimetics. Neurosci Lett. 2003;346:41–44. doi: 10.1016/s0304-3940(03)00558-5. [DOI] [PubMed] [Google Scholar]
- 136.Shimizu K. Rajapakse N. Horiguchi T. Payne RM. Busija DW. Protective effect of a new nonpeptidyl mimetic of SOD, M40401, against focal cerebral ischemia in the rat. Brain Res. 2003;963:8–14. doi: 10.1016/s0006-8993(02)03796-4. [DOI] [PubMed] [Google Scholar]
- 137.Shuaib A. Lees KR. Lyden P. Grotta J. Davalos A. Davis SM. Diener HC. Ashwood T. Wasiewski WW. Emeribe U. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357:562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 138.Singer TP. Castagnoli N., Jr Ramsay RR. Trevor AJ. Biochemical events in the development of parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem. 1987;49:1–8. doi: 10.1111/j.1471-4159.1987.tb03384.x. [DOI] [PubMed] [Google Scholar]
- 139.Smith MA. Sayre LM. Monnier VM. Perry G. Oxidative posttranslational modifications in Alzheimer disease: a possible pathogenic role in the formation of senile plaques and neurofibrillary tangles. Mol Chem Neuropathol. 1996;28:41–48. doi: 10.1007/BF02815203. [DOI] [PubMed] [Google Scholar]
- 140.Staehelin HB. Micronutrients and Alzheimer's disease. Proc Nutr Soc. 2005;64:565–570. doi: 10.1079/pns2005459. [DOI] [PubMed] [Google Scholar]
- 141.Stamler JS. Lamas S. Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- 142.Stewart WF. Kawas C. Corrada M. Metter EJ. Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997;48:626–632. doi: 10.1212/wnl.48.3.626. [DOI] [PubMed] [Google Scholar]
- 143.Szekely CA. Thorne JE. Zandi PP. Ek M. Messias E. Breitner JC. Goodman SN. Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004;23:159–169. doi: 10.1159/000078501. [DOI] [PubMed] [Google Scholar]
- 144.Szekely CA. Breitner JC. Fitzpatrick AL. Rea TD. Psaty BM. Kuller LH. Zandi PP. NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008;70:17–24. doi: 10.1212/01.wnl.0000284596.95156.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tanzi RE. Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 146.Trova MP. Gauuan PJ. Pechulis AD. Bubb SM. Bocckino SB. Crapo JD. Day BJ. Superoxide dismutase mimetics, Part 2: synthesis and structure-activity relationship of glyoxylate- and glyoxamide-derived metalloporphyrins. Bioorg Med Chem. 2003;11:2695–2707. doi: 10.1016/s0968-0896(03)00272-4. [DOI] [PubMed] [Google Scholar]
- 147.Van Den Bosch L. Tilkin P. Lemmens G. Robberecht W. Minocycline delays disease onset and mortality in a transgenic model of ALS. Neuroreport. 2002;13:1067–1070. doi: 10.1097/00001756-200206120-00018. [DOI] [PubMed] [Google Scholar]
- 148.Vargas MR. Pehar M. Cassina P. Beckman JS. Barbeito L. Increased glutathione biosynthesis by Nrf2 activation in astrocytes prevents p75NTR-dependent motor neuron apoptosis. J Neurochem. 2006;97:687–696. doi: 10.1111/j.1471-4159.2006.03742.x. [DOI] [PubMed] [Google Scholar]
- 149.Veal EA. Day AM. Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 150.Vlad SC. Miller DR. Kowall NW. Felson DT. Protective effects of NSAIDs on the development of Alzheimer disease. Neurology. 2008;70:1672–1677. doi: 10.1212/01.wnl.0000311269.57716.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wahner AD. Bronstein JM. Bordelon YM. Ritz B. Nonsteroidal anti-inflammatory drugs may protect against Parkinson disease. Neurology. 2007;69:1836–1842. doi: 10.1212/01.wnl.0000279519.99344.ad. [DOI] [PubMed] [Google Scholar]
- 152.Wang CX. Shuaib A. Neuroprotective effects of free radical scavengers in stroke. Drugs Aging. 2007;24:537–546. doi: 10.2165/00002512-200724070-00002. [DOI] [PubMed] [Google Scholar]
- 153.Wang H. Larriviere KS. Keller KE. Ware KA. Burns TM. Conaway MA. Lacomis D. Pattee GL. Phillips LH., 2nd Solenski NJ. Zivkovic SA. Bennett JP., Jr R+ pramipexole as a mitochondrially focused neuroprotectant: initial early phase studies in ALS. Amyotroph Lateral Scler. 2008;9:50–58. doi: 10.1080/17482960701791234. [DOI] [PubMed] [Google Scholar]
- 154.Warita H. Hayashi T. Murakami T. Manabe Y. Abe K. Oxidative damage to mitochondrial DNA in spinal motoneurons of transgenic ALS mice. Brain Res Mol Brain Res. 2001;89:147–152. doi: 10.1016/s0169-328x(01)00029-8. [DOI] [PubMed] [Google Scholar]
- 155.Wei G. Dawson VL. Zweier JL. Role of neuronal and endothelial nitric oxide synthase in nitric oxide generation in the brain following cerebral ischemia. Biochim Biophys Acta. 1999;1455:23–34. doi: 10.1016/s0925-4439(99)00051-4. [DOI] [PubMed] [Google Scholar]
- 156.Wu DC. Teismann P. Tieu K. Vila M. Jackson-Lewis V. Ischiropoulos H. Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A. 2003;100:6145–6150. doi: 10.1073/pnas.0937239100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wu DC. Re DB. Nagai M. Ischiropoulos H. Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A. 2006;103:12132–12137. doi: 10.1073/pnas.0603670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Yang G. Chan PH. Chen J. Carlson E. Chen SF. Weinstein P. Epstein CJ. Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:1655–1670. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 159.Zhou M. Dominguez R. Baudry M. Superoxide dismutase/catalase mimetics but not MAP kinase inhibitors are neuroprotective against oxygen/glucose deprivation-induced neuronal death in hippocampus. J Neurochem. 2007;103:2212–2223. doi: 10.1111/j.1471-4159.2007.04906.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zimmerman MC. Oberley LW. Flanagan SW. Mutant SOD1-induced neuronal toxicity is mediated by increased mitochondrial superoxide levels. J Neurochem. 2007;102:609–618. doi: 10.1111/j.1471-4159.2007.04502.x. [DOI] [PubMed] [Google Scholar]