

Abstract
Purpose
To examine the biochemical characteristics of rod and cone arrestin with respect to their ability to quench the activity of light-activated rhodopsin in transgenic mice.
Methods
The mouse rod opsin promoter was used to drive expression of mouse cone arrestin in rod photoreceptor cells of rod arrestin knockout (arr1−/−) mice. Suction electrode recordings from single rods were performed to investigate cone arrestin’s ability to quench the catalytic activity of light-activated rhodopsin. In addition, the ability of cone arrestin to prevent light-induced retinal damage caused by prolonged activation of the phototransduction cascade was assessed.
Results
Two independent lines of transgenic mice were obtained that expressed cone arrestin in rod photoreceptors, and each was bred into the arr1−/− background. Flash responses measured by suction electrode recordings showed that cone arrestin reduced signaling from photolyzed rhodopsin but was unable to quench its activity completely. Consistent with this observation, expression of mouse cone arrestin conferred dose-dependent protection against photoreceptor cell death caused by low light exposure to arr1−/− retinas, but did not appear to be as effective as rod arrestin.
Conclusions
Cone arrestin can partially substitute for rod arrestin in arr1−/− rods, offering a degree of protection from light-induced damage and increasing the extent of rhodopsin deactivation in response to flashes of light. Although earlier work has shown that rod arrestin can bind and deactivate cone pigments efficiently, the results suggest that cone arrestin binds light-activated, phosphorylated rhodopsin less efficiently than does rod arrestin in vivo. These results suggest that the structural requirements for high-affinity binding are fundamentally distinct for rod and cone arrestins.
Rod photoreceptor cells operate in dim light and can signal the absorption of single photons. In contrast, cone photoreceptor cells function in bright light. In rods and cones, the ability to detect light is conferred by rhodopsin and cone opsins, respectively. Rhodopsin and cone opsins are visual pigment molecules that, on photon absorption, become catalytically active (metarhodopsin II, or R*) and initiate signaling cascades that ultimately lead to changes in membrane current.1–3 Compared with rods, cones are less sensitive, and their light responses are faster.4 Although much is known about the molecular mechanisms that shape rod responses, little is known about the mechanisms that determine the time course of cone responses. Because rods and cones express a set of related visual transduction proteins, it is thought that their signaling cascades are qualitatively similar and that the differences in their light responses arise primarily from differences in the amount or activity of the transduction proteins.
One such difference between rods and cones is the expression of unique forms of arrestin, which differ significantly in the amino acid sequence of key functional domains.5–7 In rods, rod arrestin (ARR1 for the protein and arr1 for the gene) binds to light-activated, phosphorylated rhodopsin (R*-P) and is essential to deactivate R* completely.8–10 In rods of arr1−/− mice, the recovery of the light response is greatly slowed and biphasic.11 The initial phase of recovery, which occurs over hundreds of milliseconds, is probably due to phosphorylation of R*, in as much as phosphorylation is known to reduce, but not completely quench, the ability of R* to activate the G protein transducin.8,12,13 The second phase of recovery is much more gradual, occurring over a period of tens of seconds and is most likely due to the thermal decay of R*-P to opsin and all-trans retinal.11 Therefore, the lack of ARR1 results in prolonged signaling in rod cells after photon absorption and is thought to be responsible for photoreceptor cell death observed in arr1−/− retinas under low light exposure.14
Like ARR1, cone arrestin binds light-activated and phosphorylated cone opsins in vitro,7,15 but the role of cone arrestin in deactivating visual pigments has not been demonstrated in intact photoreceptors. This is largely because cone cells represent only 3% of the photoreceptors in the murine retina,16 which makes biochemical studies or suction electrode recordings from mouse cones challenging. In this study, we circumvent these challenges by expressing mouse cone arrestin (mCAR) in rods of mice lacking ARR1.11 This has allowed us to compare ARR1 and cone arrestin in their ability to quench light-activated, phosphorylated rhodopsin, R*-P, in intact rods.
Materials and Methods
The study was conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research as well as University of Southern California (USC) and University of California (UC) Davis Institutional Animal Care and Use Committee (IACUC) Guidelines.
Expression of Cone Arrestin in Rod Photoreceptor Cells of Transgenic Mice
The full-length mouse cone arrestin cDNA was obtained by RT-PCR with total RNA isolated from mouse retinas. The cDNA fragment was cloned and its nucleotide sequence was verified. The 4.4-kb 5′ region of the mouse rhodopsin promoter was used to direct its expression in rod photoreceptors,17 and the 0.6-kb mouse protamine 1 sequence was placed downstream to provide the polyadenylation signal as well as a splice site. The expression vector was excised from the plasmid backbone, purified, and injected into single-cell embryos. Founder mice were analyzed for transgene integration by PCR and Southern blot analysis. PCR was performed to genotype mice by using genomic DNA isolated from tail biopsy specimens. PCR-positive founder mice were mated with arr1−/− mice11 to express mouse cone arrestin (mCAR) in the absence of endogenous ARR1. To estimate the level of mCAR expression, mouse retinas from transgene-positive and transgene-negative littermate controls were homogenized in 100 μL of 50 mM Tris (pH 7.2) with protease inhibitor cocktail (one tablet per 10 mL of buffer; Complete Mini Protease Inhibitor Cocktail; Roche Molecular Biochemicals, Mannheim, Germany). Serial dilutions of retinal homogenates were applied and electrophoretically separated by 12% SDS-PAGE. Proteins were transferred to Western polyvinylidene difluoride (PVDF) membranes (Hybond-ECL; GE Healthcare, Buckinghamshire, UK). Blots were incubated with cone arrestin-specific primary antibody (LUMI-J, 1:5000 dilution18) followed by incubation with anti-rabbit IgG coupled to horseradish peroxidase (Vector Laboratories, Burlingame, CA) and visualized by chemiluminescence (Enhanced Chemiluminescence [ECL] kit; GE Healthcare). Signals from mCAR were quantified through analyzing the optical densities of scanned bands on computer (Quantity One software; Bio-Rad). Two lines, mCAR-L and mCAR-H that expressed mouse cone arrestin at low and high levels, respectively, were used in the study.
Estimation of Rod and Cone Arrestin Levels in Wild-Type Mice and in Transgenic Mice
To estimate how the content of cone arrestin in retinas of transgenic mice compares with the level of endogenous ARR1 in wild-type retinas, retinas from wild-type, arr1−/−, or mCAR-Harr1−/− mice were homogenized in either homogenization buffer (80 mM Tris [pH 8], 10 mM EDTA, 4 mM MgCl2, and protease inhibitors cocktail), or in 5 M urea/homogenization buffer. Samples were normalized per total protein content, as determined by Bradford assay (Bio-Rad Laboratories, Hercules, CA). Total retinal homogenates (20 μg total protein) or supernatant fractions from urea-treated samples (40 μg total protein) were resolved in a Tris/glycine 12% SDS-PAGE. (6.2 × 7′ gel, run for 14 hours at 12 mA constant current). Proteins were visualized by Coomassie staining (Coomassie G-250 stain; Bio-Rad Laboratories).
Estimation of rod and cone arrestin levels was also performed by using recombinantly expressed proteins as standards in Western blot analysis. Mouse rod (NM_009118) and cone (AF156979) arrestin cDNAs were subcloned in frame between the NcoI and HindIII or EcoRI sites, respectively, of the pTrcHisB vector (Invitrogen), eliminating the His tag in the original vector. Arrestins were expressed in Escherichia coli strain BL21 codon plus (Invitrogen) and purified by sequential heparin-Sepharose, Q-Sepharose, and SP-Sepharose chromatography, essentially as described.7,19 Retinal homogenate and serial dilutions of recombinant proteins were separated on SDS-PAGE, transferred onto nitrocellulose and incubated with the respective primary antibodies and secondary antibody conjugated to an infrared dye terminator mix (IRDye 800; LI-COR Biosciences, Lincoln, NE). The protein bands were visualized and quantified (Odyssey Infrared Imaging System; LI-COR Biociences).
Indirect Immunofluorescence Microscopy
Wild-type and mCAR-Harr1−/− mice were dark adapted overnight. The next day, mice were either killed and processed in complete darkness (dark) or exposed to 2000-lux white fluorescent light for 20 minutes with their pupils dilated and then processed in light. Eyes were enucleated, and the cornea and lens were removed. Eye cups were fixed in 4% paraformaldehyde (Polysciences, Inc., Warrington, PA) and 0.5% glutaraldehyde (Ted Pella, Inc., Redding, CA) in 0.1 M cacodylate buffer (Electron Microscopy Sciences, Hatfield, PA) for 1.5 hours at room temperature. The eye cups were rinsed, embedded in OCT (VWR Scientific, West Chester, PA) and 10 μm frozen sections were obtained. Sections were incubated in phosphate-buffered saline (PBS) with 1 mM CaCl2, 1 mM MgCl2, and 1 mg/mL bovine serum albumin (BSA) for 15 minutes at room temperature followed by incubation with anti-cone arrestin LUMI-J antibody diluted with PBS/BSA in a humidified chamber for 1 hour. The sections were then rinsed and incubated with secondary anti-rabbit antibody conjugated to fluorescein (1:100 dilution; Vector Laboratories). Micrographs were obtained by confocal microscope (LSM 510; Carl Zeiss Meditec, Inc., Dublin, CA). All slides were scanned under the same conditions for magnification, laser intensity, brightness, gain, and pinhole size. Images were processed using the microscope software (LSM 510 software ver. 3.2 SP2; Carl Zeiss Meditec, Inc.).
Light Exposure and Morphologic Analysis
Dark-reared 4-week-old C57/BL/6J, arrestin knockout, mCAR-Larr1−/−, and mCAR-Harr1−/− mice were maintained in the dark or exposed to white fluorescent light of ~3000, ~2000, or ~1000 lux for 72 hours. Results for the 2000-lux condition are not shown. Animals were killed immediately after exposure to light, and the superior apex of the eye was marked by cauterization before enucleation. Eye cups were dissected, leaving a flap of cornea marking the superior apex of the eye, and fixed overnight in 2.5% glutaraldehyde, 2% paraformaldehyde, and 0.1 M cacodylate buffer (pH 7.2). Eye cups were rinsed, embedded in epoxy resin, and sectioned at 1 μm thickness along the vertical meridian from the apex of the superior pole through the optic nerve and stained with Richardson’s stain (1% azure blue, 1% methyl blue, and 1% borax).
Outer nuclear layer (ONL) thickness was measured by using a previously described protocol.20 Briefly, the total length of the ONL was first measured from the optic nerve head (designated 0) to the superior pole and the inferior pole (negative for superior and positive for inferior). The length for each region was then divided into 11 segments. Each segment was further divided into three points, spaced an equal distance apart, where ONL thickness was measured and the mean calculated. The same procedure was repeated for each of the 22 segments that span the retina. Four to eight mice were used for each experimental condition, providing mean ± SD data for each of the 22 segments.
Single-Cell Electrophysiology
Suction electrode recordings from rods were performed as previously described.21 Briefly, dark-reared adult arr1−/− and mCAR-Harr1−/− mice were euthanatized and their retinas dissected under infrared conditions and stored on ice. The retinas were cut into small pieces with a razor blade in Leibovitz’s L-15 medium (Gibco-Invitrogen, Grand Island, NY) with 10 mM glucose, 0.1 mg/mL BSA (Sigma-Aldrich, St. Louis, MO), and DNase I (~25 U/mL; GE Healthcare). Tissue was then loaded into a recording chamber that was perfused with bicarbonate buffer (pH 7.4) at 35°C to 37°C. Suction electrodes with tips 1 to 2 μm in diameter containing HEPES-buffered Locke’s solution were used to draw in individual rods gently, with an infrared charge-coupled device camera (Stanford Photonics, Palo Alto, CA). Membrane currents from a single rod outer segment (OS) were measured with a current-to-voltage converter (Axopatch 1B; Axon Instruments-Molecular Devices, Union City, CA) and low-pass filtered (eight-pole Bessel; Frequency Devices, Inc., Haverhill, MA) using 30-Hz corner frequency. Data were digitized at 200 Hz (IgorPro for NIDAQ for Windows; Wavemetrics; National Instruments, Austin, TX). Rod cells were stimulated with 10-ms flashes of 500-nm light. The intensity of light was controlled by using neutral-density filters, calibrated after each experiment by a silicon photodiode (United Detector Technology, Hawthorne, CA).
As mentioned, dim flash responses of arr1−/− rods display two phases of recovery: a fast, initial recovery and a slower recovery. The time constant of the initial recovery phase was determined by fitting a single exponential function from a point just after the peak of the dim flash response to the beginning of the plateau (τrec1). The extent of this initial recovery was variable between cells but consistently decreased as the flash strength increased, with the brightest flashes showing only the slower form of recovery, as previously observed.11 We have developed a metric for measuring the extent of this initial recovery phase in a given cell, called “Flstr50.” To determine this value, the plateau amplitude (the amplitude at which the initial fast phase of recovery is complete and the slow phase of recovery begins) was divided by the peak amplitude for each flash strength. This value was then subtracted from 1 and multiplied by 100 to yield the percentage of initial recovery. The percentage of initial recovery was then plotted versus flash strength and the data were fitted with a single exponential, with the Flstr50 corresponding to the flash strength at which the initial phase of recovery reduced the amplitude to half of its peak value. The exponential nature of the relation most likely arises from exponential response compression.22
The second phase of recovery was difficult to measure because of the prolonged recovery times (consistently greater than 30 seconds), which made them particularly susceptible to baseline drift. In several cells with exceptionally stable recordings, the first 20 to 30 seconds of the final falling phase of the average dim flash response was fitted with a single exponential function, forcing the horizontal asymptote to y = 0 (τrec2). Integration time was determined only for the few cells whose responses fully recovered, and was calculated by dividing the time integral of the average dim flash response by the peak amplitude.23 Because recovery of bright flash responses was very slow (many minutes) and possibly incomplete, the saturating flashes used for measuring the dark current were often delivered at the end of the experiment.
Results
mCAR Expression in Photoreceptors
The mouse rod opsin promoter17 was used to direct expression of mouse cone arrestin in rod photoreceptor cells to compare the functional characteristics of cone and rod arrestin in terminating R*-P signaling and protecting against light-induced retinal degeneration. Two independent lines were obtained and were bred into the rod arrestin knockout (arr1−/−) background to yield mCAR-Harr1−/− and mCAR-Larr1−/− lines. Western blot analysis of serial dilutions of whole retinal homogenates was used to compare the level of mCAR expression in the transgenic lines to that in wild-type mice. The mCAR-Larr1−/− and mCAR-Harr1−/− lines expressed cone arrestin at ~120- and ~1500-fold, respectively, over the endogenous level of cone arrestin present in wild-type retinal homogenates (Fig. 1A). Based on our estimates, these values represent a 1:160 and 1:12.5 molar ratio with rhodopsin, respectively (for comparison, the ratio between ARR1 to rhodopsin was recently estimated to be 1:1.324). Photoreceptor-specific expression of the transgene was confirmed by indirect immunofluorescence microscopy of frozen retinal sections (Fig. 2). In the wild-type retina, expression of cone arrestin appeared to be restricted to cones, whereas in mCAR-Harr1−/− (Fig. 2) and mCAR-Larr1−/− (data not shown) retinas, its expression was observed through-out the photoreceptor layer that includes both rods and cones, but not in other layers of the retina (Fig. 2, bottom left).
Figure 1.
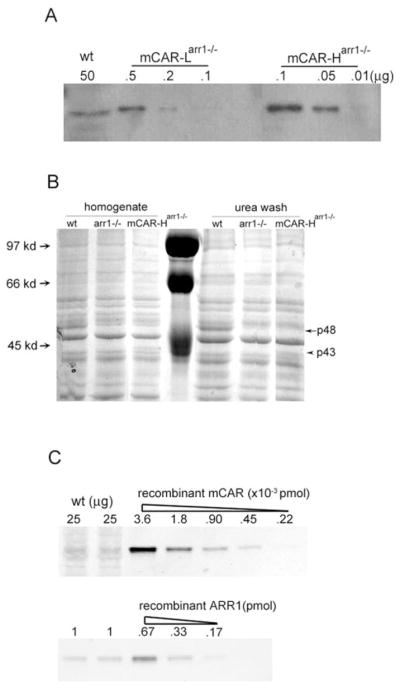
Expression of mCAR in transgenic mouse retinas. (A) Expression level of mCAR was analyzed using Western blot of serial dilution of retinal homogenates from mCAR-Larr1−/− and mCAR-Harr1−/− mice. The amount of protein loaded onto each lane (μg) is indicated. (B) ARR1 (p48, arrow) and mCAR (p43, arrowhead) visualized by Coomassie stain in SDS-PAGE. Twenty micrograms protein from whole retinal homogenate or 40 μg protein from a urea-washed fraction was loaded per lane. Molecular weight markers were shown in the center lane. The gel shown is representative of three independent experiments. (C) Western blot of endogenous cone arrestin (top) and ARR1 (bottom) in whole retinal homogenates from 1-month-old wild-type mice. Amount loaded corresponds to 1/10 (25 μg) or 1/250 (1 μg) of one retina. Serial dilutions of purified recombinant mCAR and ARR1 served as standards. Same results were obtained when the recombinant proteins were mixed with arr1−/− retinal homogenates so that comparable quantity of retinal proteins would be loaded per lane (data not shown).
Figure 2.

Expression pattern and comparison of light-dependent translocation between ARR1 and mCAR in transgenic rods. Top: in the dark, ARR1 was localized primarily in the inner segment and ONL compartments. Light exposure caused ARR1 to move to the outer segment compartment. Middle: the localization of cone arrestin in the dark-adapted retina was more diffuse throughout the cone photoreceptor cells. In response to light, cone arrestin was also observed translocating to the OS compartment. This pattern was closely mirrored by mCAR expressed in rod photoreceptor cells of mCAR-Harr1−/− mice (bottom). Scale bar, 25 μm.
To estimate the relative concentration of mCAR in the transgenic rods compared with rod arrestin in wild-type rods, equal amounts of whole retinal homogenates or the soluble fractions from urea-stripped retinal homogenates of wild-type, arr1−/− and mCAR-Harr1−/− mice were separated on SDS-PAGE and visualized by Coomassie stain (Fig. 1B). ARR1, an abundant visual protein, was evident as a prominent 48-kDa protein band in the wild-type samples (Fig. 1B, arrow). This band was absent in the arr1−/− samples. A 43-kDa protein, corresponding to mCAR, was visible in mCAR-Harr1−/− samples (Fig. 1B, arrowhead), but not in the mCAR-Larr1−/− samples (data not shown). Given that the ratio of rods to cones in the murine retina is 33:1 (97% rods and 3% cones,16 the expression level of cone arrestin in the mCAR-L and mCAR-H lines would represent a ~4-fold (120/33) and ~45-fold (1500/33) overexpression of cone arrestin in rods if rod and cone arrestins are expressed at comparable levels within rod and cone cells. Yet protein analysis using SDS-PAGE shows that mCAR was present at only ~20% of ARR1 in the high-expressing line, mCAR-Harr1−/− (Fig. 1B), suggesting that the amount of cone arrestin in cones is ~225-fold (45/0.2) less than ARR1 in rods. We performed Western blot analyses on whole retinal homogenates from wild-type mice and used recombinant mouse ARR1 and cone arrestin proteins as standards as an independent means of verifying this finding (Fig. 1C). Based on this method, the concentration of mCAR is estimated to be ~6 × 10−3 picomoles per retina, whereas the concentration of ARR1 is estimated to be ~80 picomoles per retina. Because cone arrestin and ARR1 are expressed exclusively in the photoreceptors, we can use these values to estimate the relative levels of cone arrestin in cones to that of ARR1 in rods of wild-type mice. As mentioned, because rods outnumber cones 33:1 in murine retina, the 6 × 10−3 picomoles cone arrestin per retina corresponds to a normalized value (0.006 × 33) of 0.2 picomoles, compared with 80 picomoles of ARR1. This result suggests that in wild-type retina, the level of rod arrestin in rods is ~400-fold higher than is cone arrestin in cones. Thus, both protein and immunoblot analyses indicate a lower expression level of cone arrestin in cones versus rod arrestin in rods.
Light-Dependent Translocation of mCAR
One characteristic of ARR1 is its well documented light-dependent translocation from the ONL and inner segment (proximal) compartments in darkness to the OS (distal) compartment in sustained bright light.25–29 We examined light-dependent translocation of mCAR in rods as another functional test of the heterologously expressed mCAR (Fig. 2). As previously described, in the dark, ARR1 has been localized predominantly to the inner segments, ONL, and the outer plexiform layer of the retina. On exposure to light, ARR1 immunoreactivity dramatically shifted to the OS (Fig. 2, top). The distribution of endogenous cone arrestin was different: It was evenly distributed throughout the entire cone cells in the dark-adapted retina, but like ARR1, mCAR translocated to the OS on light exposure (Fig. 2, middle panels, see also Zhu et al.18). This pattern of cone arrestin distribution in the dark and the light-dependent translocation of mCAR was recapitulated in rod cells of mCAR-Harr1−/− transgenics (Fig. 2, bottom). These results show that ectopically expressed mCAR exhibits functional light-dependent translocation that resembles that of endogenous cone arrestin in native cones.
Flash Responses from mCAR Rods
We recorded from both mCAR-Harr1−/− and arr1−/− rods using suction electrodes to investigate the functional ability of cone arrestin to deactivate rhodopsin. Responses of mCAR-Harr1−/− and arr1−/− rods were qualitatively similar (Figs. 3A, 3B), with dramatically slower recovery phases than responses of wild-type rods (Fig. 3C). Dim flash responses of arr1−/− and mCAR-Harr1−/− rods (Fig. 3D) rose along similar trajectories and reached similar peak amplitudes. The activation kinetics of the dim flash response (time to peak) and the size of the single-photon response (elementary amplitude) did not differ in arr1−/− and mCAR-Harr1−/− rods (Table 1) and were indistinguishable from previously published wild-type values.30 Measures of sensitivity were also unchanged by the expression of mCAR (flash sensitivity and Io, Table 1).
Figure 3.

Flash responses of mCARarr1−/− and arr1−/− rods are similar except that mCAR-Harr1−/− rods show a greater initial recovery. Representative flash response families from arr1−/− (A), mCAR-Harr1−/− (B), and wild-type (C) rods. Flashes were given at t = 0 s. The four traces represent flashes (in photons per square micrometer) of 8, 50, 181, and 607 in the arr1−/− rod, 5, 55, 200, and 668 in the mCAR-Harr1−/− rod and 5, 56, 200, and 671 in the wild-type rod. Responses were normalized by the maximum response amplitude of 12.0 (arr1−/−), 13.6 (mCAR-Harr1−/−) and 16.0 (wild-type) pA. (D) Weighted average of dim flash responses from arr1−/− and mCAR-Harr1−/− rods were normalized by the average flash strength of light used to elicit the responses (9.2 and 10.6 photons · μm−2 for arr1−/− and mCAR-Harr1−/−, respectively). The arr1−/− trace is the average of 458 dim flash responses in 20 cells and has a peak of 0.090 pA/(photons · μm−2). The mCAR-Harr1−/− trace is the average of 733 dim flash responses in 31 cells and has a peak of 0.091 pA/(photons · μm−2). For comparison, the population average dim flash response from 10 wild-type cells of an earlier study (Mendez et al.30) has been scaled so that the peak amplitude matches those of the arr1−/− and mCAR-Harr1−/− responses.
Table 1.
Activation Kinetics of the Dim Flash Response (Time to Peak) and the Size of the Single-Photon Response (Elementary Amplitude)
| Id (pA) | Time to Peak (ms) | Elementary Amplitude (pA) | Flash Sensitivity (pA/photons μm−2) | τrec1 (ms) | τrec2 (s) | Integration Time (s) | Io* (photons μm−2) | Flstr50† (photons μm−2) | |
|---|---|---|---|---|---|---|---|---|---|
| arr1−/− | 10.6 ± 0.6 (19) | 127 ± 21 (21) | 0.41 ± 0.1 (7) | 0.129 ± 0.02 (14) | 243 ± 26 (19) | 29.5 ± 1.9 (9) | 5.3 ± 1.0 (3) | 67.4 ± 4.6 (14) | 187 ± 38 (18) |
| mCAR-Harr1−/− | 12.7 ± 0.5 (29)‡ | 106 ± 7 (34) | 0.40 ± 0.1 (12) | 0.141 ± 0.02 (22) | 273 ± 32 (29) | 23.8 ± 3.7 (8) | 1.5 ± 0.6 (9)‡ | 62.6 ± 8.0 (16) | 884 ± 269 (24)‡ |
All values are mean ± standard error (number of cells).
Flash strength that elicited a half-maximum response.
Flash strength at which the plateau amplitude of the average response after the initial phase of recovery was 50% of the peak response amplitude for that flash strength.
Significant result from t-test versus arr1−/− (P < 0.05).
Like arr1−/− responses, mCAR-Harr1−/− responses also recovered along a biphasic time course. However, the extent of recovery during the initial phase of mCAR-Harr1−/− responses was considerably greater than the initial recovery of arr1−/− responses across all tested flash strengths (Figs. 3A, 3B, 3D). This can be seen in the population mean dim flash response of mCARarr1−/− rods, where the plateau amplitude was roughly two times lower than that of the population mean response from arr1−/− rods (Fig. 3D). Because dim flash responses are approximate linear measures of cascade activity, this two-fold decrease suggests that the presence of mCAR reduces the residual catalytic activity of R*-P by roughly a factor of 2. To quantify the extent of recovery of responses to brighter flashes, we plotted the percent initial recovery (see the Methods section) versus flash strength for arr1−/− and mCAR-Harr1−/− rods (n = 18 and 24, respectively) and fitted these values by single exponential functions. On average, the extent of initial recovery was approximately 20% greater for mCAR-Harr1−/− responses than for arr1−/− responses at all flash strengths (Fig. 4). On average, the flash strength that elicited a response that recovered to 50% of its peak value during the initial phase of recovery was significantly brighter for mCAR-Harr1−/− rods than for arr1−/− rods (884 ± 269 photons/μm2 and 187 ± 38 photons/μm2, respectively; P < 0.05; Table 1).
Figure 4.

mCARarr1−/− rods recover more completely during the initial phase of recovery. The average initial recovery, expressed as a percentage, is plotted versus flash strength from representative arr1−/− and mCAR-Harr1−/− rods. Each point reflects the average of at least five cells, with the error bars representing SEM. The points were fitted with single exponential functions.
In a striking finding, we could detect no significant difference in the time constants of the two phases of recovery (τrec1 and τrec2; Table 1). The first of these, τrec1, was measured by fitting a single exponential to the initial falling phase of the dim flash response, ending where the recovery reaches a plateau, whereas the slower phase of recovery, τrec2, was measured by fitting a single exponential to the wave from this point in the plateau (see Fig. 3D and the Methods section). In several mCAR-Harr1−/− cells (5/34), the initial recovery phase was sufficient to allow for complete recovery of the dim flash response, so τrec2 was not measured in these cells. In contrast, responses of arr1−/− rods always showed a prominent slow component of recovery. Although the average values of these time constants did not differ in mCAR-Harr1−/− and arr1−/− rods, the integration time of the dim flash response, a measure of response duration (see the Methods section), was much longer in arr1−/− rods than in mCAR-Harr1−/− rods (5.3 ± 1.0 and 1.5 ± 0.6 seconds, respectively; see Table 1). This difference can be attributed to the greater extent of initial recovery in mCAR-Harr1−/− rods. Together, these results indicate that mCAR can reduce the catalytic activity of R*-P but is unable to quench R*-P completely, perhaps because it fails to attain high-affinity binding.
On average, mCAR-Harr1−/− rods also showed a small but significant increase in the dark current compared with arr1−/− rods (Id in pA; Table 1). This difference may reflect either the ability of mCAR to quiet partially the spontaneous (thermal) rhodopsin activity in the dark, or it may simply reflect the fact that the bright flashes used to measure Id were usually given only at the end of a recording because of the very long times required for recovery of saturating responses. Imperceptible slow loss of Id may have occurred more commonly during recordings from arr1−/− rods than mCAR-Harr1−/− rods, because recovery is more impaired in the former.
mCAR Protects Arr1−/− Photoreceptors from Light-Induced Damage in a Dose-Dependent Manner
Normally, pigmented mice are highly resistant to light-induced damage, even when they are exposed to constant bright light.31,32 In the absence of ARR1, however, signaling from light-activated rhodopsin is greatly prolonged, rendering the retinas from pigmented arr1−/− mice sensitive to light-induced degeneration, even under low light exposure.14 As another functional test of mCAR, we compared its ability to protect arr1−/− photoreceptors from light-induced damage.
One-month-old dark-reared mice were kept in the dark or exposed to ~3000 lux of white fluorescent light for 72 hours (Fig. 5). Representative photomicrographs of retinal sections taken near the optic nerve are shown in Figure 5A. Retinal morphology of the dark-reared arr1−/−, mCAR-Larr1−/−, and mCAR-Harr1−/− lines were similar to C57/B6 mice. The ONLs of these mice were of similar thickness, and the OS layers showed an organized structure. As expected, exposure to light had no effect on the retinal morphology of wild-type mice, but caused a marked thinning of the ONL and disruption of OS in the arr1−/− retina. Expression of mCAR protected against light-induced damage of arr1−/− retinas in a dose-dependent manner. mCAR-Harr1−/− was protected to a greater degree than mCAR-Larr1−/−, with that line expressing a lower level of the transgene. Morphometric measurements of ONL thickness along the vertical meridian are shown in Figure 5B and confirm the qualitative observations.
Figure 5.
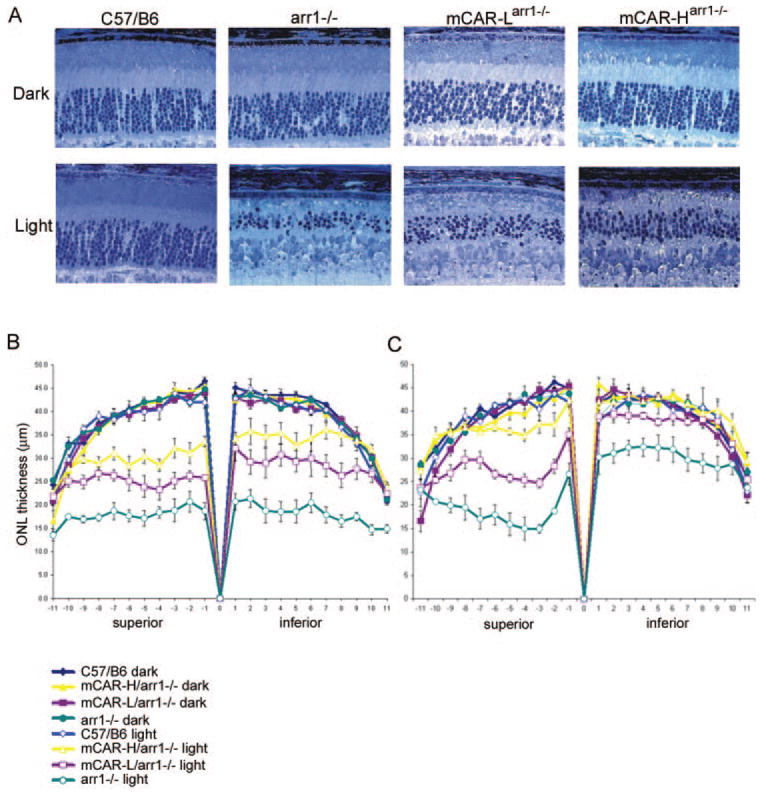
Expression of mCAR partially protects arr1−/− retinas from light-induced damage. (A) Retinal morphology of dark-reared mice and mice exposed to 3000 lux light for 72 hours. Retinal morphology of dark reared arr1−/−, mCAR-Larr1−/− and mCAR-Harr1−/− mice were similar to dark-reared control mice. Light exposure had no discernible effect on C57/B6 mice. In contrast, the ONL, as well as the OS of arr1−/− retina, showed extensive reduction in thickness and length after exposure to light. Expression of mCAR afforded some degree of rescue in the arr1−/− retinas, where more protection was seen in the higher expression line (mCAR-Harr1−/−). (B, C) Morphometric measurements of ONL thickness in dark-reared mice and mice exposed to (B) 3000 lux or (C) 1000 lux light for 72 hours. Scale bar, 25 μm.
Because mCAR expression appeared to protect partially against light-induced damage in a dose-dependent manner at 3000 lux, we sought to determine the light intensity at which mCAR might be fully protective. Morphometric measurements of mice exposed to 1000 lux for 72 hours are shown in Figure 5C. Consistent with our previous report on the effect of acute light exposure on dark-reared arr1−/− mice,33 this light intensity did not cause uniform damage to arr1−/− retinas; rather, the superior region of arr1−/− retinas was more affected. Overall, mCAR was more protective at 1000 lux than at 3000 lux. The ONL thickness of mCAR-Harr1−/− mice was comparable to C57/B6 mice in the inferior region where degeneration in the arr1−/− retina was less severe. The degree of protection at 2000 lux was intermediate between the protection at 3000 and 1000 lux (data not shown). Thus, mCAR demonstrates a dose-dependent ability to protect arr1−/− rods from light-induced photoreceptor cell death. This observation is consistent with the results of suction electrode experiments showing that cone arrestin reduced R*-P signaling but did not fully terminate it.
Discussion
In this study, we expressed cone arrestin in rods of transgenic mice to determine the functional differences between the two visual arrestins. mCAR produced a greater extent of initial recovery of the flash responses from arr1−/− rods and correspondingly offered some degree of protection to arr1−/− retinas from light-induced damage. Strictly speaking, there are two different ways in which the reduced signaling of R*-P in mCARarr1−/− rods could produce the larger initial recovery that we observed in the averaged flash responses. First, mCAR may bind and fully deactivate a subset of the R*-P, with the remaining R*-P molecules undergoing slow thermal decay as in the arr1−/− rods. This effect would be expected to produce, in a given mCARarr1−/− rod, individual dim flash responses that resemble both wild-type and arr1−/− responses, yielding an average response with a greater extent of initial recovery than arr1−/− responses. Alternatively, mCAR may bind to all R*-P with low affinity, partially reducing the ability of each R*-P molecule to activate the phototransduction cascade, which would cause all the dim flash responses of an mCARarr1−/− rod to have a reduced plateau amplitude. In our experiments, we did not observe large trial-to-trial fluctuations in the extent of initial recovery of individual dim flash responses, supporting the idea that mCAR binds to all R*-P with low affinity.
ARR1 exists in a latent inactive conformation that is stabilized by several intramolecular interactions. These include (1) a “polar core,” consisting of five interacting charged residues buried within the molecule and (2) a combination of electrostatic, van der Waals, and hydrogen-bonded interactions provided by its regulatory carboxyl tail.34,35 All these elements are structurally and functionally conserved in cone arrestin.6,7 In its basal state, ARR1 has an “activation-recognition” site and a “phosphorylation-recognition” site that exhibit low-affinity binding to R* and R*-P, respectively.36 When ARR1 encounters R*-P, both of these sites are occupied simultaneously, leading to a disruption of the intramolecular interactions that constrain arrestin in the basal state. The ensuing conformational rearrangement exposes a secondary binding site that permits ARR1 to bind with high affinity to R*-P and thus completely quench its catalytic activity.34 The observation that cone pigments can be deactivated in rods37 suggests that ARR1 is capable of binding cone pigments with high affinity. In contrast, the results of our single-cell recordings suggest that the converse is not true: cone arrestin cannot fully deactivate R*-P in a timely manner, but rather partially reduces its catalytic activity during its slow thermal decay. Previous experiments have shown little equilibrium binding between cone arrestin and R*-P,7 though such in vitro assays reflect an average of all possible interactions between cone arrestin and R*-P, which may include a mixture of high- and low-affinity states. In contrast, analysis of single-photon responses from mCARarr1−/− rods allow measurement of the quantal molecular event that represents the binding of cone arrestin with a single R*-P molecule. Collectively, the results suggest that cone arrestin is not converted into a high-affinity binding conformation by R*-P. A low-affinity interaction with R*-P (rapid binding and dissociation) would be likely to reduce the rate of transducin activation, resulting in a lower plateau in the second phase of recovery when compared with the light responses from arr1−/− rods.
The functional differences between the two visual arrestins in our study are not likely to arise from the relatively lower concentration of mCAR expression in the transgenic rods, because even lower expression (~10% of normal) of a truncated variant of arrestin, p44, restored normal dim flash responses to arr1−/− rods.38 In addition, results from Figure 2 show that the concentration of mCAR in dark-adapted rod OS may be higher than that of ARR1 in rods, due to its partial localization to the OS in darkness. It is also important to note that the functional differences we observed is not due to an indirect compensation effect of gene expression changes between wild-type, arr1−/− and mCAR retinas, because gene chip analysis (GeneChip; Affymetrix, Santa Clara, CA) have shown virtually no difference in retinal transcripts between these retinas other than the absence of arr1 transcripts in the latter two lines of mice (see Ref. 33 and data not shown).
Protein quantification using two independent measurements show a greatly reduced level of cone arrestin in cones when compared with rod arrestin in rods. It is unknown whether such a low level of cone arrestin could support deactivation of phosphorylated cone pigments under high bleach conditions, even though the level of cone pigments is estimated to be 10-fold less than rhodopsin,39 and the volume of the cone OS is ~60% smaller.40 Furthermore, mCAR deactivates S-opsin poorly when both are expressed in transgenic mouse rods, whereas endogenous ARR1 deactivates S-opsin efficiently (Shi G, Yau K-W, Chen J et al., unpublished observations, 2006). Finally, there is evidence that ARR1 is coex-pressed with cone arrestin in murine cones (Zhu X et al. IOVS 2005;46:ARVO E-Abstract 1179),41 and in blue cones of monkey42 and human43 retinas, which may explain why rhodopsin expressed in cones deactivates normally.37 Thus, it seems plausible that ARR1 is potentially essential for the deactivation of cone pigments. However, whereas cone recovery was profoundly slowed in GRK1−/− mice, the lack of ARR1 did not appear to have a discernible phenotype when a double-flash paradigm was used in conjunction with electroretinogram recordings.39 In sum, our results, as well as results of in vitro studies, suggest that rod arrestin and cone arrestin do not play functionally equivalent roles in rods and cones. Future experiments using transgenic approaches in cone photoreceptors and single cell recordings will be useful for determining the mechanisms that regulate deactivation of cone pigments in vivo.
Acknowledgments
The authors thank Bruce Brown for helpful suggestions on the quantification of the visual arrestins and Xumei Zhu for helpful comments on the manuscript.
Supported by the National Eye Institute in the form of a Vision Core Grant to UC Davis (EY12576) and Doheny Eye Institute (EY03040) and individual grants to MEB (EY14047), CMC (EY00395 and EY15851), VVG (EY11500), and JC (EY12155); the Arnold and Mabel Beckman Macular Research Center (JC); and the E. Matilda Ziegler Foundation for the Blind (MEB). CMC is the Mary D. Allen Chair in Vision Research, Doheny Eye Institute.
Footnotes
Disclosure: S. Chan, None; W.W. Rubin, None; A. Mendez, None; X. Liu, None; X. Song, None; S.M. Hanson, None; C.M. Craft, None; V.V. Gurevich, None; M.E. Burns, None; J. Chen, None
References
- 1.Arshavsky VY, Lamb TD, Pugh EN., Jr G proteins and phototransduction. Annu Rev Physiol. 2002;64:153–187. doi: 10.1146/annurev.physiol.64.082701.102229. [DOI] [PubMed] [Google Scholar]
- 2.Burns ME, Baylor DA. Activation, deactivation, and adaptation in vertebrate photoreceptor cells. Annu Rev Neurosci. 2001;24:779–805. doi: 10.1146/annurev.neuro.24.1.779. [DOI] [PubMed] [Google Scholar]
- 3.Lagnado L. Signal amplification: let’s turn down the lights. Curr Biol. 2002;12:R215–R217. doi: 10.1016/s0960-9822(02)00754-6. [DOI] [PubMed] [Google Scholar]
- 4.Korenbrot JI, Rebrik TI. Tuning outer segment Ca2+ homeostasis to phototransduction in rods and cones. Adv Exp Med Biol. 2002;514:179–203. doi: 10.1007/978-1-4615-0121-3_11. [DOI] [PubMed] [Google Scholar]
- 5.Craft CM, Whitmore DH, Wiechmann AF. Cone arrestin identified by targeting expression of a functional family. J Biol Chem. 1994;269:4613–4619. [PubMed] [Google Scholar]
- 6.Smith WC, Gurevich EV, Dugger DR, et al. Cloning and functional characterization of salamander rod and cone arrestins. Invest Ophthalmol Vis Sci. 2000;41:2445–2455. [PubMed] [Google Scholar]
- 7.Sutton RB, Vishnivetskiy SA, Robert J, et al. Crystal structure of cone arrestin at 2.3A: evolution of receptor specificity. J Mol Biol. 2005;354:1069–1080. doi: 10.1016/j.jmb.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Wilden U. Duration and amplitude of the light-induced cGMP hydrolysis in vertebrate photoreceptors are regulated by multiple phosphorylation of rhodopsin and by arrestin binding. Biochemistry. 1995;34:1446–1454. doi: 10.1021/bi00004a040. [DOI] [PubMed] [Google Scholar]
- 9.Bennett N, Sitaramayya A. Inactivation of photoexcited rhodopsin in retinal rods: the roles of rhodopsin kinase and 48-kDa protein (arrestin) Biochemistry. 1988;27:1710–1715. doi: 10.1021/bi00405a049. [DOI] [PubMed] [Google Scholar]
- 10.Wilden U, Hall SW, Kuhn H. Phosphodiesterase activation by photoexcited rhodopsin is quenched when rhodopsin is phosphorylated and binds the intrinsic 48-kDa protein of rod outer segments. Proc Natl Acad Sci USA. 1986;83:1174–1178. doi: 10.1073/pnas.83.5.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu J, Dodd RL, Makino CL, et al. Prolonged photoresponses in transgenic mouse rods lacking arrestin. Nature. 1997;389:505–509. doi: 10.1038/39068. [DOI] [PubMed] [Google Scholar]
- 12.Miller JL, Fox DA, Litman BJ. Amplification of phosphodiesterase activation is greatly reduced by rhodopsin phosphorylation. Biochemistry. 1986;25:4983–4988. doi: 10.1021/bi00366a002. [DOI] [PubMed] [Google Scholar]
- 13.Arshavsky VY, Dizhoor AM, Shestakova IK, et al. The effect of rhodopsin phosphorylation on the light-dependent activation of phosphodiesterase from bovine rod outer segments. FEBS Lett. 1985;181:264–266. doi: 10.1016/0014-5793(85)80272-6. [DOI] [PubMed] [Google Scholar]
- 14.Chen J, Simon MI, Matthes MT, et al. Increased susceptibility to light damage in an arrestin knockout mouse model of Oguchi disease (stationary night blindness) Invest Ophthalmol Vis Sci. 1999;40:2978–2982. [PubMed] [Google Scholar]
- 15.Zhu X, Brown B, Li A, et al. GRK1-dependent phosphorylation of S and M opsins and their binding to cone arrestin during cone phototransduction in the mouse retina. J Neurosci. 2003;23:6152–6160. doi: 10.1523/JNEUROSCI.23-14-06152.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carter-Dawson LD, LaVail MM. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J Comp Neurol. 1979;188:245–262. doi: 10.1002/cne.901880204. [DOI] [PubMed] [Google Scholar]
- 17.Lem J, Applebury ML, Falk JD, et al. Tissue-specific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron. 1991;6:201–210. doi: 10.1016/0896-6273(91)90356-5. [DOI] [PubMed] [Google Scholar]
- 18.Zhu X, Li A, Brown B, et al. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Mol Vis. 2002;8:462–471. [PubMed] [Google Scholar]
- 19.Gurevich VV, Benovic JL. Arrestin: mutagenesis, expression, purification, and functional characterization. Methods Enzymol. 2000;315:422–437. doi: 10.1016/s0076-6879(00)15859-8. [DOI] [PubMed] [Google Scholar]
- 20.Faktorovich EG, Steinberg RH, Yasumura D, et al. Basic fibroblast growth factor and local injury protect photoreceptors from light damage in the rat. J Neurosci. 1992;12:3554–3567. doi: 10.1523/JNEUROSCI.12-09-03554.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krispel CM, Chen CK, Simon MI, et al. Novel form of adaptation in mouse retinal rods speeds recovery of phototransduction. J Gen Physiol. 2003;122:703–712. doi: 10.1085/jgp.200308938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pugh EN, Jr, Nikonov S, Lamb TD. Molecular mechanisms of vertebrate photoreceptor light adaptation. Curr Opin Neurobiol. 1999;9:410–418. doi: 10.1016/S0959-4388(99)80062-2. [DOI] [PubMed] [Google Scholar]
- 23.Baylor DA, Hodgkin AL. Changes in time scale and sensitivity in turtle photoreceptors. J Physiol. 1974;242:729–758. doi: 10.1113/jphysiol.1974.sp010732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Strissel KJ, Sokolov M, Trieu LH, et al. Arrestin translocation is induced at a critical threshold of visual signaling and is superstoichiometric to bleached rhodopsin. J Neurosci 25. 2006;26:1146–1153. doi: 10.1523/JNEUROSCI.4289-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broekhuyse RM, Janssen AP, Tolhuizen EF. Effect of light-adaptation on the binding of 48-kDa protein (S-antigen) to photoreceptor cell membranes. Curr Eye Res. 1987;6:607–610. doi: 10.3109/02713688709025220. [DOI] [PubMed] [Google Scholar]
- 26.Broekhuyse RM, Tolhuizen EF, Janssen AP, et al. Light induced shift and binding of S-antigen in retinal rods. Curr Eye Res. 1985;4:613–618. doi: 10.3109/02713688508999993. [DOI] [PubMed] [Google Scholar]
- 27.Mangini NJ, Pepperberg DR. Immunolocalization of 48K in rod photoreceptors: light and ATP increase OS labeling. Invest Ophthalmol Vis Sci. 1988;29:1221–1234. [PubMed] [Google Scholar]
- 28.Philp NJ, Chang W, Long K. Light-stimulated protein movement in rod photoreceptor cells of the rat retina. FEBS Lett. 1987;225:127–132. doi: 10.1016/0014-5793(87)81144-4. [DOI] [PubMed] [Google Scholar]
- 29.Whelan JP, McGinnis JF. Light-dependent subcellular movement of photoreceptor proteins. J Neurosci Res. 1988;20:263–270. doi: 10.1002/jnr.490200216. [DOI] [PubMed] [Google Scholar]
- 30.Mendez A, Burns ME, Sokal I, et al. Role of guanylate cyclase-activating proteins (GCAPs) in setting the flash sensitivity of rod photoreceptors. Proc Natl Acad Sci USA. 2001;98:9948–9953. doi: 10.1073/pnas.171308998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.LaVail MM, Gorrin GM. Protection from light damage by ocular pigmentation: analysis using experimental chimeras and translocation mice. Exp Eye Res. 1987;44:877–889. doi: 10.1016/s0014-4835(87)80050-7. [DOI] [PubMed] [Google Scholar]
- 32.Sanyal S, Zeilmaker GH. Retinal damage by constant light in chimaeric mice: implications for the protective role of melanin. Exp Eye Res. 1988;46:731–743. doi: 10.1016/s0014-4835(88)80059-9. [DOI] [PubMed] [Google Scholar]
- 33.Roca A, Shin KJ, Liu X, et al. Comparative analysis of transcriptional profiles between two apoptotic pathways of light-induced retinal degeneration. Neuroscience. 2004;129:779–790. doi: 10.1016/j.neuroscience.2004.08.021. [DOI] [PubMed] [Google Scholar]
- 34.Gurevich VV, Gurevich EV. The molecular acrobatics of arrestin activation. Trends Pharmacol Sci. 2004;25:105–111. doi: 10.1016/j.tips.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 35.Hirsch JA, Schubert C, Gurevich VV, et al. The 2.8 A crystal structure of visual arrestin: a model for arrestin’s regulation. Cell. 1999;97:257–269. doi: 10.1016/s0092-8674(00)80735-7. [DOI] [PubMed] [Google Scholar]
- 36.Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268:11628–11638. [PubMed] [Google Scholar]
- 37.Kefalov V, Fu Y, Marsh-Armstrong N, et al. Role of visual pigment properties in rod and cone phototransduction. Nature. 2003;425:526–531. doi: 10.1038/nature01992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burns ME, Mendez A, Chen CK, et al. Deactivation of phosphorylated and nonphosphorylated rhodopsin by arrestin splice variants. J Neurosci. 2006;26:1036–1044. doi: 10.1523/JNEUROSCI.3301-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lyubarsky AL, Chen C, Simon MI, et al. Mice lacking G-protein receptor kinase 1 have profoundly slowed recovery of cone-driven retinal responses. J Neurosci. 2000;20:2209–2217. doi: 10.1523/JNEUROSCI.20-06-02209.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nikonov SS, Kholodenko R, Lem J, et al. Physiological features of the S- and M-cone photoreceptors of wild-type mice from single-cell recordings. J Gen Physiol. 2006;127:359–374. doi: 10.1085/jgp.200609490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mears AJ, Kondo M, Swain PK, et al. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- 42.Nir I, Ransom N. S-antigen in rods and cones of the primate retina: different labeling patterns are revealed with antibodies directed against specific domains in the molecule. J Histochem Cytochem. 1992;40:343–352. doi: 10.1177/40.3.1372630. [DOI] [PubMed] [Google Scholar]
- 43.Nork TM, Mangini NJ, Millecchia LL. Rods and cones contain antigenically distinctive S-antigens. Invest Ophthalmol Vis Sci. 1993;34:2918–2925. [PubMed] [Google Scholar]