

Abstract
The vascular tumors of the retina and choroid comprise a diverse group of congenital and acquired lesions. The major vascular tumors of the retina include retinal capillary hemangioma, cavernous hemangioma of the retina, retinal vasoproliferative tumor, and racemose hemangiomatosis of the retina or Wyburn–Mason syndrome. Choroidal vascular tumors include circumscribed choroidal hemangioma and diffuse choroidal hemangioma. While classified as benign, visual symptoms secondary to exudative retinal detachment and a variety of other mechanisms are common and are a major source of long-term visual disability. While many therapeutic modalities exist, treatment of symptomatic cases can be challenging. Of particular importance, many of the vascular tumors of the retina and choroid have significant associations with systemic disease. As ocular symptoms are often the most common presenting disease manifestation, the ophthalmologist plays an important role in accurate and early diagnosis. The ability to initiate prompt screening and treatment in appropriate cases is critical. In the following article, the key clinical and diagnostic features of the major retinal and choroidal vascular tumors, their systemic associations, and the literature pertaining to the most currently available treatment strategies are reviewed.
Keywords: Cavernous Hemangioma, Choroidal Hemangioma, Retinal Capillary Hemangioma, Retinal Vasoproliferative Tumor, Wyburn–Mason Syndrome
INTRODUCTION
The vascular tumors of the retina and choroid comprise a diverse group of congenital and acquired lesions. The principal vascular tumors of the retina include retinal capillary hemangioma, cavernous hemangioma of the retina, retinal vasoproliferative tumor, and racemose hemangiomatosis of the retina, or Wyburn–Mason syndrome. The major vascular tumors of the choroid include circumscribed choroidal hemangioma and diffuse choroidal hemangioma. While the vascular tumors of the retina and choroid are classified as benign, visual symptoms secondary to exudative retinal detachment as well as a variety of other mechanisms are common and are a major source of long-term visual disability. Treatment can often be challenging. Of particular importance, many of the vascular tumors of the retina and choroid have significant systemic associations, and therefore the ophthalmologist plays a critical role in accurate and early diagnosis. The purpose of this article is to discuss the key clinical features, systemic associations, and relevant diagnostic studies including fluorescein angiography, indocyanine green angiography (ICG), ultrasonography, and optical coherence tomography (OCT) for each of the major retinal and choroidal vascular tumors. Additionally, the literature pertaining to the most currently available treatment strategies for each is reviewed.
RETINAL VASCULAR TUMORS
Retinal capillary hemangioma
Clinical features and systemic associations
Retinal capillary hemangiomas, also termed retinal hemangioblastomas, occur both in isolation and in association with the multisystem familial cancer syndrome, von Hippel–Lindau (VHL) disease. VHL is inherited in an autosomal dominant fashion with age-dependent penetrance. The incidence is approximately 1 in 40,000 to 1 in 54,000 live births.1 Mutations in the VHL gene that have been mapped to chromosome 3p25-26 and currently available genetic testing has detection rates as high as 99%.2 The pathogenesis of VHL is due to the inability of affected cells to degrade hypoxia-inducible factors in the presence of oxygen. This results in the overproduction of hypoxia-inducible factors including erythropoietin, vascular endothelial growth factor, and platelet-derived growth factor. As a result, tumors associated with VHL are often highly vascular owing to increased levels of angiogenic agents.
Retinal capillary hemangiomas are among the most common disease manifestations in VHL. The mean age of diagnosis of retinal capillary hemangioma in patients with VHL is 25 years with the majority of patients presenting between the ages of 10 and 40 years.3 Aside from retinal capillary hemangiomas, individuals with VHL also have a high incidence of renal cell carcinoma, central nervous system hemangiomas, pheochromocytomas, and other tumors. Renal cell carcinoma is the leading cause of mortality in patients with VHL, occurring in 5% of patients by age 30 and 40% by 60 years of age.4 Central nervous system hemangiomas occur in more than 50% of patients with VHL, and they are diagnosed at a younger age than in sporadic cases. The most commonly involved sites include the cerebellum (75%) and the spinal cord (15%).4 Pheochromocytomas are observed in <25% of patients with VHL. When associated with VHL, these tumors tend to be multiple and bilateral.5 As retinal capillary hemangiomas are often the earliest and most frequent disease manifestation seen in VHL, accurate diagnosis by the ophthalmologist plays an important role in the initiation of appropriate screening and treatment.
Clinically, retinal capillary hemangiomas appear as round, circumscribed, orange-to-red retinal lesions found either in a juxtapapillary location or in the mid-peripheral retina. Peripheral tumors occur most frequently in the superotemporal or inferotemporal quadrants and are associated with prominent feeder vessels extending from the optic disc.6 Lipid exudates and subretinal fluid surrounding the tumor are common and can affect visual function if the macula is involved. Although more commonly observed as a solitary tumor, approximately one-third of patients with VHL have multiple lesions, and one-half are affected bilaterally.4
Imaging studies
The clinical features of retinal capillary hemangioma are characteristic and diagnosis can generally be made based upon ophthalmoscopic appearance. Aside from ophthalmoscopy, fluorescein angiography is the most informative diagnostic study owing to the high degree of vascularity in these tumors. Fluorescein angiography typically demonstrates marked early hyperfluorescence with variable late leakage. Angiographic studies are also helpful in differentiating feeding arterioles from draining venules in cases that necessitate treatment. B-scan ultrasonography shows a round retinal mass lesion and can be useful in determining overall tumor dimensions as well as the presence or absence of subretinal fluid. A-scan ultrasound typically demonstrates medium-to-high internal reflectivity. OCT of the macula is also useful in detecting subretinal fluid and in accurately measuring foveal thickness particularly as in cases where treatment response is being assessed [Figure 1].
Figure 1.

(A) Fundus photograph of a retinal capillary hemangioma. (B) Note prominent feeder vessels that show hyperfluorescence on fluorescein angiography. (C) B-scan ultrasonography showing a circumscribed retinal mass with a localized retinal detachment
Treatment and prognosis
Treatment of retinal capillary hemangioma is based upon tumor size, location, presence of subretinal fluid or retinal traction, and visual acuity. Observation alone is warranted for smaller-sized lesions (<500 μm), tumors with the absence of exudate and subretinal fluid, and in cases in which visual acuity is not threatened.7 In particular, juxtapapillary retinal capillary hemangiomas have a natural history of stability over time and in many cases may be appropriately managed with observation.8 For larger tumors and those cases in which visual acuity is affected, other treatment modalities including laser photocoagulation, cryotherapy, photodynamic therapy, radiotherapy, and various other vitreoretinal procedures can be employed. Photocoagulation, targeted either directly at the tumor, to feeder vessels, or both can be applied over many sessions and has been shown to be 91–100% effective in preserving visual acuity at a mean follow-up time of 5.1 years.9,10 Photocoagulation has been successful in treating retinal capillary hemangiomas that are up to 4.5 mm in size, but is particularly effective for tumors that are 1.5 mm or smaller.10 Cryotherapy may be preferable to photocoagulation in cases where tumors are more anteriorly located, have a moderate degree of subretinal fluid, or are larger than 3.0 mm.11 Photodynamic therapy has recently been reported to induce the occlusion of both juxtapapillary and peripheral retinal capillary hemangiomas.8,12–14 For larger tumors (greater than 4.0 mm) or those that have been resistant to either prior photocoagulation or cryotherapy, plaque radiotherapy or low-dose external beam radiotherapy have been used successfully.15,16 Vitreoretinal surgery is generally reserved for larger retinal capillary hemangiomas complicated by rhegmatogenous or tractional retinal detachment. Vitreoretinal surgery has no role and is not appropriate for retinal capillary hemangiomas that have no tractional retinal detachment or related complications. Most recently, systemic and intravitreal administration of inhibitors of vascular endothelial growth factor (VEG-F) have demonstrated mixed treatment outcomes, suggesting that the general efficacy of anti-angiogenic agents in VHL is uncertain.17 Prospective clinical trials are still needed to further evaluate the efficacy of these agents.
Visual prognosis in patients with retinal capillary hemangioma is dependent upon the number of tumors, size, location, and degree of exudative or tractional retinal detachment. In general, prognosis is guarded, even in cases that have been adequately treated. More than 25% of eyes develop permanent visual loss and approximately 20% have a visual acuity worse than 20/100 in at least one eye.6 Patients who present with multiple retinal capillary hemangiomas are predisposed to developing new lesions, and therefore need to be examined more frequently.9 Early diagnosis and treatment of retinal capillary hemangiomas prior to onset of symptoms results in improved visual outcomes.6
Cavernous hemangioma of the retina
Clinical features
Cavernous hemangiomas of the retina are believed to be rare congenital vascular hamartomas. In some patients, these tumors have been observed to coexist with intracranial cavernous hemangiomas and angiomatous skin hamartomas, suggesting that the disease may exist in both a sporadic and syndromic form; however, the data regarding these associations are inconsistent.18 Patients may be asymptomatic, or they may present with decreased visual acuity depending upon the location of the tumor, presence of macular fibrosis, or vitreous hemorrhage. In a small series of nine patients, the mean age of presentation ranged from 1 to 55 years.19
Ophthalmoscopically, retinal cavernous hemangiomas appear as a grouping of blood-filled saccules within the inner retinal layers or on the surface of the optic disc.18 These tumors are often described as having a “cluster of grapes” appearance. The lesions are variable in size and location, and frequently follow the course of a major retinal vein. Epiretinal membranes are a common feature. In contrast to retinal capillary hemangiomas, cavernous hemangiomas lack prominent feeder vessels, intraretinal exudate, and surrounding subretinal fluid. Because the fundoscopic features are characteristic, diagnosis can generally be made based upon clinical appearance.
Imaging studies
Fluorescein angiography can be helpful in confirming the diagnosis and shows delayed filling during the venous phase owing to the low flow nature of the tumor. The composition of multiple dilated vascular channels results in hyperfluorescent saccular caps secondary to staining of the plasma, which overlies the focal collections of stagnant erythrocytes. Angiography can also be helpful in differentiating cavernous hemangioma of the retina from retinal capillary hemangioma as the former generally has absence of feeder vessels and late leakage. While ultrasonography and OCT are not routinely performed, features such as surface gliosis and epiretinal membrane formation can be further characterized using OCT [Figure 2].
Figure 2.
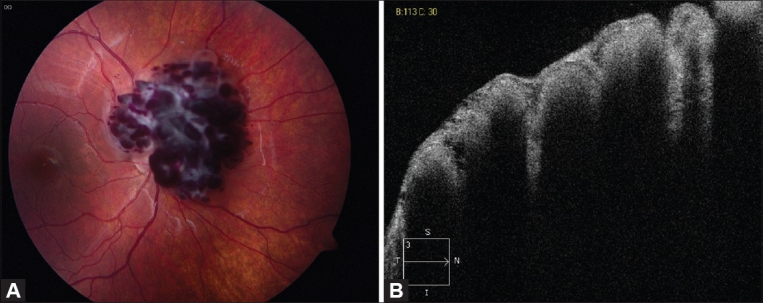
(A) Fundus photograph of a peripapillary cavernous hemangioma of the retina. (B) Note the absence of retinal exudation. OCT confirms multilobulated consistency
Treatment and prognosis
As the majority of cavernous hemangiomas of the retina remains stable over time, most can simply be followed with periodic observation. Some tumors may undergo spontaneous thrombosis and subsequently develop increased surface gliosis. A small number of reported cases have been associated with self-limiting vitreous hemorrhage.18 No effective treatment has been determined for these tumors; however, the use of laser photocoagulation has been reported in a few cases.18
Retinal vasoproliferative tumor
Clinical features and systemic associations
Retinal vasoproliferative tumors are very rare and only recently obtained status as a distinct clinical entity in the early 1980s.20 They have been noted to be either primary (74%) or secondary to a variety of pre-existing ocular conditions (26%).21 When their occurrence is secondary, the underlying cause has been attributed to inflammatory, vascular, traumatic, dystrophic, and degenerative retinal disease.21 Other possible associations that have been reported include bilateral occurrence in monozygotic twins,22 Waardenburg’s syndrome,23 neurofibromatosis type I,24 systemic hypertension, and hyperlipidemia.21 Males and females appear to be equally affected. The mean age of presentation is in the third to fourth decade of life.21
On ocular examination, retinal vasoproliferative tumors appear as a pinkish-yellow raised vascular mass in the pre-equitorial retina, classically located inferiorly. The majority of tumors are solitary (87%); however, secondary tumors have been observed to be multiple in 42% of cases.21 Normal caliber retinal vessels may be observed entering the posterior aspect of the tumor, however in contrast to retinal capillary hemangioma, vasoproliferative tumors do not generally have dilated tortuous feeder vessels. Extensive subretinal exudation is common and occurs in approximately 80% of cases.21 Additional clinical findings include exudative retinal detachment, retinal or vitreous hemorrhage, or both, and presence of vitreous cells. Vasoproliferative tumors may be surrounded by retinal pigment epithelium (RPE) hyperplasia, particularly in cases of secondary tumors.21 Diagnosis is usually based upon clinical appearance.
Imaging studies
Ancillary studies such as fluorescein angiography are often of minimal value because of the tumor’s predilection for the peripheral retina. In cases where angiography is able to capture the tumor, rapid filling in early phases followed by increasing hyperfluorescence and diffuse leakage in the later phase are seen in the angiogram. Telangiectatic and sometimes dilated vessels can be seen within the tumor. A-scan and B-scan ultrasonography demonstrate a raised, solid mass with high internal reflectivity. OCT is not routinely performed, but may be helpful in further characterizing tumor features including the degree of subretinal exudation, retinal detachment, or RPE hyperplasia [Figure 3]. In more difficult cases, tumor biopsy may be required to establish a definitive diagnosis.25
Figure 3.
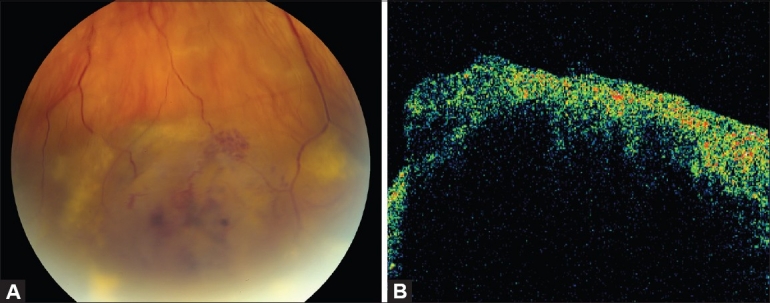
(A) Fundus appearance of a vasoproliferative retinal tumor. (B) OCT reveals irregularly thickened retina on the surface of the tumor. Due to limited penetration, deeper aspects of the tumor are not visualized
Treatment and prognosis
Because retinal vasoproliferative tumors are most commonly found in the periphery, they may be asymptomatic and it is reasonable to periodically observe small tumors that lack significant exudate. Patients who are symptomatic generally present with reduced vision, photopsia, and metamorphopsia. Aside from exudative retinal detachment and vitreous hemorrhage, visual impairment from macular fibrosis and macular edema are seen in approximately 31% and 15% of cases, respectively.21 Multiple treatment options exist for retinal vasoproliferative tumor including cryotherapy, plaque brachytherapy, laser photocoagulation, and photodynamic therapy. Triple freeze-thaw transconjunctival cryotherapy has been reported to be successful in a small number of cases; however, repeated treatment sessions may be required.21 Both iodine I-125 and ruthenium-106 brachytherapy have been used to treat vasoproliferative tumors.26,27 In a series of 30 eyes with retinal vasoproliferative tumor treated with iodine I-125 brachytherapy, tumor regression was observed in 97%, exudative retinal detachment resolved in 65%, and visual acuity improved or remained stable in 73%.26 Photocoagulation and photodynamic therapy have been used in limited number of cases with some success; however, larger number of cases and longer follow-up time are required to further assess these treatment modalities.21,28,29 More recently, bevacizumab (Avastin) which has been approved by the FDA for the treatment of metastatic colorectal cancer, has been used as an off-label therapy for several ocular diseases with underlying vascular etiology. A single case of successfully treated vasoproliferative tumor using bevacizumab was reported in 2007.30 Vitrectomy is generally reserved for cases of vasoproliferative tumor resulting in tractional retinal detachment and macular pucker.31 In the largest reported series of 103 patients, prognosis was variable. Approximately one-third of patients were managed with observation alone; however, even small peripheral tumors were associated with significant visual loss. Advanced cases may eventually develop neovascular glaucoma requiring enucleation.21
Wyburn-Mason syndrome
Clinical features and systemic associations
Wyburn-Mason syndrome was first described in 1943 when the association was made between racemose hemangiomatosis of the retina and arteriovenous malformations (AVMs) of the brain.32 This non-hereditary, sporadic disorder is also referred to by some authors as Bonnet–Dechaumme–Blanc syndrome.33 Aside from the retina and brain, other affected tissues include the skin, bones, kidneys, muscles, and gastrointestinal tract.32,34 Wyburn–Mason syndrome is also considered to be one of the phakomatoses, or neuro-oculocutaneous syndromes. In contrast to the other clinical entities in the group of phakomatoses, Wyburn–Mason syndrome does not commonly have cutaneous involvement. The clinical findings are believed to be congenital in origin; however, diagnosis is typically delayed until later in childhood as there are not typically apparent external features of the disease. The incidence of coexisting intracranial AVMs in patients with retinal AVMs is approximately 30%; however, only 8% of patients with intracranial AVMs harbor retinal AVMs.34
The ophthalmoscopic appearance of the retinal AVM of Wyburn–Mason syndrome is characteristic, with dilated and tortuous retinal vessels extending from the optic disc to the retinal periphery. They have been classified into three distinct groups depending upon the severity of the vascular malformation. Group I AVMs possess an abnormal capillary plexus between the major vessels of the AVM. In group II, there is a lack of an intervening capillary network between the artery and the vein. Group III encompasses the most extensive AVMs, with dilated and tortuous vessels and loss of the ability to distinguish between arteries and veins.35 Diagnosis of Wyburn–Mason syndrome is generally based upon clinical findings.
Imaging studies
Fluorescein angiography can be helpful in supporting the diagnosis and further classifying the disease. Angiography demonstrates anomalous arteriovenous communications and the presence or absence of intervening capillary networks. In the most severe cases seen in grade III disease, arteries and veins cannot usually be differentiated even with fluorescein angiography. Unlike several of the previously described vascular hamartomas, retinal AVMs do not leak in late phases of the angiogram. Ultrasonography and OCT are generally necessary for diagnostic confirmation; however, structural changes in the nerve fiber layer over time may be evident with OCT imaging [Figure 4]. Intracranial AVMs are the best diagnosed by magnetic resonance imaging (MRI) or by arteriography.
Figure 4.
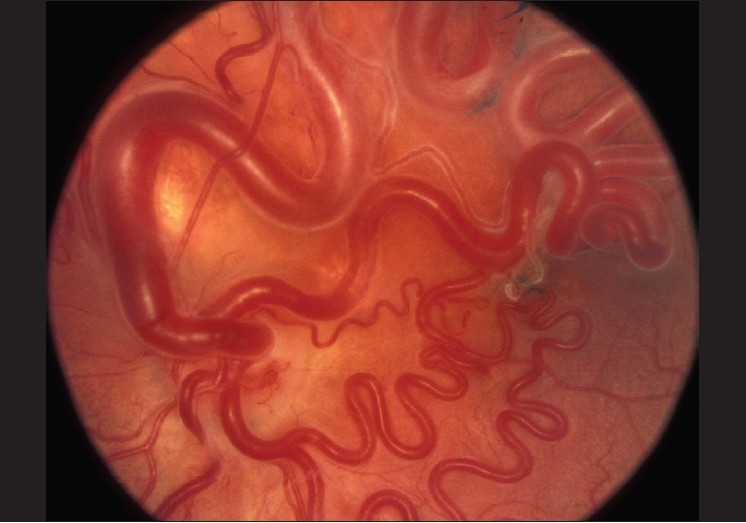
Fundus appearance of a typical retinal arteriovenous malformation. Reproduced with permission from: Singh AD, Rundle PA, Rennie IG. Retinal vascular tumors. In: Singh AD, Damato BE, Pe'er J, Murphree AL, Perry JD, editors. Clinical Ophthalmic Oncology. Philadelphia: Saunders-Elsevier, 2007:341-7
Treatment and prognosis
Patients with Wyburn–Mason syndrome are at high risk for loss of visual function, and unfortunately the disease is not amenable to currently available therapies. Unlike intracranial AVMs, retinal AVMs typically do not hemorrhage; however, vision loss occurs through a variety of mechanisms. Over many years, retinal vascular malformations may exhibit increased tortuosity36 which can lead to vascular occlusions,37 retinal ischemia, and eventually neovascular glaucoma. If neovascular glaucoma occurs, symptomatic treatment can be offered. Patients with group III disease are at the highest risk for vision loss either secondary to retinal decompensation or due to direct compression of the optic nerve or retinal nerve fiber layer.38,39 Similarly, the intracranial AVMs seen in Wyburn–Mason syndrome are usually inoperable because of their predilection for location in the midbrain. Embolization may be effective for intracranial AVMs in some cases.
CHOROIDAL VASCULAR TUMORS
Circumscribed choroidal hemangioma
Clinical features
Circumscribed choroidal hemangiomas are benign hamartomas that typically present from second to fourth decade of life.40 They usually occur sporadically in the absence of systemic disease. Although commonly asymptomatic, choroidal hemangiomas can be associated with exudative retinal detachment resulting in reduced visual function, metamorphopsia, and photopsia.
On ophthalmoscopic examination, a circumscribed choroidal hemangioma appears as an orange choroidal mass with indistinct margins that blend with the surrounding choroid. They are frequently located in the macular region of the posterior pole, and are not usually thicker than 6 mm.41 Although these tumors are highly vascular, dilated and tortuous feeder vessels are not typically observed. Surrounding subretinal fluid leading to exudative retinal detachment with macular involvement is common in symptomatic cases. Retinal hard exudates are minimal or absent.
Imaging studies
While the fundoscopic features are characteristic, angiographic studies such as fluorescein and ICG can be helpful in establishing the diagnosis and differentiating these benign lesions from other tumors in the differential diagnosis including amelanotic malignant melanoma and choroidal metastases. Fluorescein angiography demonstrates a hyperfluorescent mass with a fine lacy vascular network of intrinsic vessels in the early choroidal filling phase. The hyperfluorescence increases throughout the angiogram, and there is variable leakage in late views.42 ICG is the most useful study for demonstrating the intrinsic vascular pattern of circumscribed choroidal hemangioma.43 Within 30 s of injection of the ICG dye, the tumor’s intrinsic tumor vascular pattern becomes apparent. Following, is a rapid increase in hyperfluorescence, which peaks around 3 to 4 min. In the late phases of the ICG angiogram, a “washout” effect with reduction of the initial hyperfluorescence is observed secondary to the outflow of dye from the hemangioma.43 Ultrasonography with A-scan and B-scan can also be helpful in making the diagnosis of choroidal hemangioma and differentiating these benign lesions from other malignant tumors in the differential diagnosis. B-scan ultrasound shows a dome-shaped choroidal mass with smooth contours and A-scan reveals high internal reflectivity. In contrast, malignant melanoma classically demonstrates low-to-medium reflectivity on A-scan. Optical coherence tomography can also be helpful in evaluating secondary changes in the overlying retina such as the presence of shallow subretinal fluid or cystoid macular edema [Figure 5]. Fundus autofluorescence (AF) has recently been used to further assess the state of the overlying RPE and to detect the presence of subtle subretinal fluid in circumscribed choroidal hemangiomas.44 In a series of 27 circumscribed choroidal hemangiomas, AF of the tumor (intrinsic AF) was iso-AF to hypo-AF in comparison to surrounding tissue in the majority of cases. AF of the surrounding tissue (extrinsic AF) primarily represented the status of the overlying RPE. Overlying orange pigment demonstrated hyper-AF, RPE fibrous metaplasia was hypo-AF, and RPE atrophy was hypo-AF. Tumors with fresh subretinal fluid demonstrated extrinsic hyper-AF.44 While biopsy is not typically indicated to confirm the diagnosis, histopathology reveals that the tumor is composed vascular channels lined with endothelium. The tumor involves the full thickness of the choroid with secondary changes of the overlying RPE and the retina.45
Figure 5.

(A) Fundus photograph of the right eye showing a circumscribed choroidal haemangioma. (B) On B-scan ultrasonography, there is a smooth-contoured, dome-shaped choroidal mass (C) that demonstrates high internal reflectivity on A-scan
Treatment and prognosis
Treatment of circumscribed choroidal hemangioma is based upon tumor location, the presence of subretinal fluid, extent of symptoms, and potential for visual recovery. Periodic observation alone may be warranted for asymptomatic cases where tumors lack subretinal fluid threatening the macula. When vision loss occurs, therapy is aimed at inducing tumor atrophy with resolution of subretinal fluid and foveal distortion. Current treatment options include laser photocoagulation, radiotherapy, transpupillary thermotherapy (TTT), and photodynamic therapy. Laser photocoagulation does little to induce tumor regression; however, it has been reported to be successful in inducing resolution of subretinal fluid and exudative retinal detachment.44 Unfortunately, in these cases the subretinal fluid has a tendency to recur and as many as 40% of patients require additional treatment.40 Radiotherapy may offer an advantage to laser photocoagulation in that the former has been reported to cause tumor regression in addition to resolution of exudative retinal detachment and improvement in visual acuity in the majority of cases. Radiotherapy can be administered in several forms including lens-sparing external beam radiotherapy,45–48 stereotactic radiotherapy,49 plaque radiotherapy (iodine-125,45,46 cobalt-60,50 ruthenium-10646,48 ), and proton beam radiotherapy.51–53 Complications of circumscribed choroidal hemangioma treated with radiotherapy include development of cataract,54 radiation optic neuropathy,53 and radiation retinopathy50 particularly in cases where hemangiomas occur in a juxtapapillary or macular location. TTT may avoid some of the limitations of laser photocoagulation (i.e., failure to induce tumor regression and need for repeated treatments) while avoiding complications of radiotherapy. In a series of 36 patients with circumscribed choroidal hemangioma, treated with TTT, over 90% showed complete or partial tumor regression with corresponding improvement in visual acuity.55 Reported complications of TTT in these cases included cystoid macular edema, preretinal fibrosis, and retinal vascular occlusion; therefore, TTT may not be suited for tumors that are subfoveal or located in a juxtapapillary region.55 For sufficiently extrafoveal tumors, TTT offers several advantages to photocoagulation as previously mentioned, and may be an appropriate treatment option. Photodynamic therapy may be the most ideally suited treatment modality for circumscribed choroidal hemangioma as it offers the capability for site-specific tumor destruction, sparing of the overlying retina, and the ease of performing the treatment in the outpatient setting. Photodynamic therapy is the preferred method of treatment in subfoveal or juxtafoveal circumscribed choroidal hemangiomas as other treatments are associated with significant treatment morbidity for tumors at this location. One challenge of photodynamic therapy is that the various treatment variables including verteporfin injection parameters (bolus verses injection over 10 min), total number of treatment sessions (1–5), laser power settings (50–100 J/cm2), duration of exposure (83–186 s), and number of laser spots (1 or more) have differed between investigators.56 In general, tumor regression is most dramatic following the first session of photodynamic therapy and is usually evident within 3 months.57 Additional treatment sessions may be offered after a 3-month period if the tumor or subretinal fluid persist; however, repeated treatments and over-treatment may result in delayed choroidal atrophy adversely affecting final visual acuity outcomes.58 Long-term visual prognosis for patients with circumscribed choroidal hemangioma is guarded secondary to vision loss induced by subretinal fluid, cystoid macular edema, foveal distortion, and treatment side effects. Even in adequately treated patients, complications from longstanding chronic exudative retinal detachment including neovascular glaucoma can occur.
Diffuse choroidal hemangioma
Clinical features and systemic associations
Diffuse choroidal hemangiomas are usually evident at birth and typically occur as part of neuro-oculocutaneous hemangiomatosis, or Sturge–Weber syndrome. Unlike other phakomatoses, Sturge–Weber syndrome is not inherited and occurs sporadically. Approximately 50% of patients with Sturge–Weber syndrome have a diffuse choroidal hemangioma.59 The diffuse choroidal hemangioma is most commonly unilateral and ipsilateral to the nevus flammeus, or port-wine stain.
On ophthalmic examination, these tumors appear as orange, diffuse choroidal thickening [Figure 6]. This has been likened to the classically described “tomato-catsup fundus.” Focal regions of excessively thickened choroid within the diffuse hemangioma may simulate circumscribed choroidal hemangioma. As with circumscribed hemangiomas, there may be associated exudative retinal detachment that often does not become manifest until adolescence.
Figure 6.

(A) Fundus photograph showing diffuse choroidal thickening with shallow subretinal fluid in the right eye. (B) Note diffuse orange color of the fundus as compared to the normal left eye. (C) On B-scan ultrasonography, there is a smooth-contoured, diffuse thickening of the choroid
Imaging studies
In the setting of Sturge–Weber syndrome with typical cutaneous features, the diagnosis of diffuse choroidal hemangioma is usually straightforward. In some cases, additional studies may be useful in confirming the diagnosis. Fluorescein angiography demonstrates early hyperfluorescence with persistence of hyperfluorescence through the late phases of the angiogram. The hyperfluorescent pattern is diffuse and corresponds with the tumor margins. Leakage of fluorescein in the late phase of the angiogram is less common than is seen with circumscribed choroidal hemangioma. ICG has a similar appearing pattern with rapid diffuse filling in early phases of the angiogram with intense persistence of hyperfluorescence into the late phases. A fine lacy intrinsic vascular pattern with a diffuse distribution is characteristic. The “washout” phenomenon observed in circumscribed choroidal hemangiomas is generally absent. B-scan ultrasonography demonstrates a dome-shaped choroidal mass and diffusely thickened choroid. A-scan ultrasound shows high internal reflectivity. OCT can be used to identify the presence of and to assess the degree of subretinal fluid in appropriate cases [Figure 6]. Fundus AF has also been used in cases of diffuse choroidal hemangioma. Similar to circumscribed choroidal hemangioma, the majority of these tumors demonstrated intrinsic hypo to iso-AF. The extrinsic AF patterns showed that overlying orange pigment was hyper-AF, RPE hyperplasia was hypo-AF, RPE fibrous metaplasia was hypo-AF, and RPE atrophy was hypo-AF.44
Treatment and prognosis
Diffuse choroidal hemangioma may be asymptomatic; however, visual loss can occur secondary to refractive error, foveal distortion, and exudative retinal detachment. Observation may be appropriate for asymptomatic cases that lack subretinal fluid exudation. Decision to treat should be based upon the extent of visual symptoms and potential for visual recovery. When visual loss occurs, therapy is aimed at inducing tumor atrophy, resolving exudative retinal detachment, and minimizing foveal distortion. Radiotherapy and photodyndamic therapy are currently the preferred methods of treatment. Low-dose lens-sparing radiotherapy or proton beam irradiation can induce tumor regression and resolution of subretinal fluid. In a series of 15 eyes treated with low-dose lens-sparing radiotherapy, complete resolution of exudative retinal detachment was seen in all cases, tumor regression was present in five cases, and improved visual acuity was observed in seven cases at a mean follow-up time of 5 years.47 Short-term treatment success using photodynamic therapy has also been reported.60–62 Photodynamic therapy offers potential advantages over radiotherapy including avoidance of radiation and associated complications, ease of delivery, and few side effects. Larger studies with long-term follow-up are required to fully assess the efficacy of photodynamic therapy in the treatment of diffuse choroidal hemangioma. In addition to visual loss from exudative retinal detachment, congenital glaucoma is seen in approximately 70% of cases of Sturge–Weber syndrome.59 The mechanism of elevated intraocular pressure is believed to be secondary to developmental anomalies of the anterior chamber angle and increased episcleral venous pressure.63 Medical therapy for glaucoma is effective in a minority of cases; however, most require multiple surgical procedures including trabeculectomies, combined trabeculotomy with trabeculectomy or insertion of drainage implant devices.64
CONCLUSION
Each of the major vascular tumors of the retina and choroid possess unique clinical features which are discernable on a thorough ophthalmic examination. The distinct fundoscopic appearance of each entity is generally enough to establish the diagnosis. In more challenging cases, ancillary studies including fluorescein and ICG, A-scan and B-scan ultrasonography, and OCT can be helpful in confirming the diagnosis. While considered to be benign, many cases are associated with significant visual impairment. Treatment for symptomatic cases can be difficult and although many therapeutic modalities exist, large-scale studies with long-term follow-up results are still required in order to fully understand the efficacy of each therapy. Newer treatment strategies including the systemic and intravitreal use of novel anti-angiogenic agents are still undergoing investigation but may provide promising therapies in the future. Early and accurate diagnosis of the retinal and choroidal vascular tumors is important because of the associations with systemic disease and the ability to initiate prompt screening and treatment in appropriate cases.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Maher ER, Kaelin WG. Von Hippel-Lindau disease. Medicine. 1997;76:381–91. doi: 10.1097/00005792-199711000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Stolle C, Glenn G, Zbar B, Humphrey JS, Choyke P, Walther M, et al. Improved detection of germline mutation in the von Hippel-Lindau disease tumor suppression gene. Hum Mutat. 1998;12:417–23. doi: 10.1002/(SICI)1098-1004(1998)12:6<417::AID-HUMU8>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 3.Chang JH, Spraul CW, Lynn ML, Drack A, Grossniklaus HE. The two-stage mutation model in retinal hemangioblastoma. Ophthalmic Genet. 1998;19:123–30. doi: 10.1076/opge.19.3.123.2185. [DOI] [PubMed] [Google Scholar]
- 4.Maher ER, Yates JR, Harries R, Benjamin C, Harris R, Moore AT, et al. Clinical features and natural history of von Hippel-Lindau disease. Q J Med. 1990;77:1151–63. doi: 10.1093/qjmed/77.2.1151. [DOI] [PubMed] [Google Scholar]
- 5.Richard S, Chauveau D, Chrétien Y, Beigelman C, Denys A, Fendler JP, et al. Renal lesions and pheochromocytomas in von Hippel-Lindau disease. Adv Nephrol Necker Hosp. 1994;23:1–27. [PubMed] [Google Scholar]
- 6.Webster AR, Maher ER, Moore AT. Clinical characteristics of ocular angiomatosis in von Hippel-Lindau disease and correlation with germline mutation. Arch Ophthalmol. 1999;117:371–8. doi: 10.1001/archopht.117.3.371. [DOI] [PubMed] [Google Scholar]
- 7.Singh AD, Nouri M, Shields CL, Shields JA, Perez N. Treatment of retinal capillary hemangioma. Ophthalmology. 2002;109:1799–806. doi: 10.1016/s0161-6420(02)01177-6. [DOI] [PubMed] [Google Scholar]
- 8.Schmidt-Erfurth UM, Kusserow C, Barbazetto IA, Laqua H. Benefits and complications of photodynamic therapy of papillary capillary hemangiomas. Ophthalmology. 2002;109:1256–66. doi: 10.1016/s0161-6420(02)01059-x. [DOI] [PubMed] [Google Scholar]
- 9.Schmidt D, Natt E, Neumann HP. Long-term results of laser treatment for retinal angiomatosis in von Hippel-Lindau disease. Eur J Med Res. 2000;5:47–58. [PubMed] [Google Scholar]
- 10.Blodi CF, Russell SR, Pulido JS, Folk JC. Direct and feeder vessel photocoagulation of retinal angiomas with dye yellow laser. Ophthalmology. 1990;97:791–7. doi: 10.1016/s0161-6420(90)32509-5. [DOI] [PubMed] [Google Scholar]
- 11.Welch RB. The recognition and treatment of early angiomatosis retinae and use of cryosurgery as an adjunct to therapy. Trans Am Ophthalmol Soc. 1970;68:367–424. [PMC free article] [PubMed] [Google Scholar]
- 12.Atebara NH. Retinal capillary hemangioma treated with verteporfin photodynamic therapy. Am J Ophthalmol. 2002;134:788–90. doi: 10.1016/s0002-9394(02)01648-3. [DOI] [PubMed] [Google Scholar]
- 13.Bakri SJ, Sears JE, Singh AD. Transient closure of a retinal capillary hemangioma with verteporfin photodynamic therapy. Retina. 2005;25:1103–34. doi: 10.1097/00006982-200512000-00024. [DOI] [PubMed] [Google Scholar]
- 14.Bhattacharjee H, Deka H, Deka S, Barman MJ, Mazumdar M, Medhi J. Verteporfin photodynamic therapy of retinal capillary hemangioblastoma in von Hippel-Lindau disease. Indian J Ophthalmol. 2010;58:73–5. doi: 10.4103/0301-4738.58479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kreusel KM, Bornfeld N, Lommatzsch A, Wessing A, Foerster MH. Ruthenium-106 brachytherapy for peripheral retinal capillary hemangioma. Ophthalmology. 1998;105:1386–92. doi: 10.1016/S0161-6420(98)98017-4. [DOI] [PubMed] [Google Scholar]
- 16.Raja D, Benz MS, Murray TG, Escalona-Benz EM, Markoe A. Salvage external beam radiotherapy of retinal capillary hemangiomas secondary to von Hippel-Lindau disease: Visual and anatomic outcomes. Ophthalmology. 2004;111:150–3. doi: 10.1016/j.ophtha.2003.04.003. [DOI] [PubMed] [Google Scholar]
- 17.Wong WT, Chew EY. Ocular von Hippel-Lindau disease: Clinical update and emerging treatments. Curr Opin Ophthalmol. 2008;19:213–7. doi: 10.1097/ICU.0b013e3282fb7c04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gass JD. Cavernous hemangioma of the retina. A neuro-oculo-cutaneous syndrome. Am J Ophthalmol. 1971;71:799–814. doi: 10.1016/0002-9394(71)90245-5. [DOI] [PubMed] [Google Scholar]
- 19.Messmer E, Laqua H, Wessing A, Spitznas M, Weidle E, Ruprecht K, et al. Nine cases of cavernous hemangioma of the retina. Am J Ophthalmol. 1983;95:383–90. doi: 10.1016/s0002-9394(14)78309-6. [DOI] [PubMed] [Google Scholar]
- 20.Baines PS, Hiscott PS, McLeod D. Posterior non-vascularized proliferative extraretinopathy and peripheral nodular retinal telangiectasis. Trans Ophthalmol Soc UK. 1982;102:487–91. [PubMed] [Google Scholar]
- 21.Shields CL, Shields JA, Barrett J, De Potter P. Vasoproliferative tumors of the ocular fundus. Classification and clinical manifestations in 103 patients. Arch Ophthalmol. 1995;113:615–23. doi: 10.1001/archopht.1995.01100050083035. [DOI] [PubMed] [Google Scholar]
- 22.Wachtlin J, Heimann H, Jandeck C, Kreusel KM, Bechrakis NE, Kellner U, et al. Bilateral vasoproliferative retinal tumors with identical localization in a pair of monozygotic twins. Arch Ophthalmol. 2002;120:860–2. [PubMed] [Google Scholar]
- 23.Rundle P, Shields JA, Shields CL, Singh AD, Peairs R. Vasoproliferative tumour of the ocular fundus associated with Waardenburg’s syndrome. Eye. 2000;14:105–6. doi: 10.1038/eye.2000.27. [DOI] [PubMed] [Google Scholar]
- 24.Hood CT, Janku L, Lowder CY, Singh AD. Retinal vasoproliferative tumor in association with neurofibromatosis type 1. J Pediatr Ophthalmol Strabismus. 2009;25:1–3. doi: 10.3928/01913913-20090616-05. [DOI] [PubMed] [Google Scholar]
- 25.Bechrakis NE, Foerster MH, Bornfeld N. Biopsy in indeterminate intraocular tumors. Ophthalmology. 2002;109:235–42. doi: 10.1016/s0161-6420(01)00931-9. [DOI] [PubMed] [Google Scholar]
- 26.Cohen VM, Shields CL, Demirci H, Shields JA. Iodine I 125 plaque radiotherapy for vasoproliferative tumors of the retina in 30 eyes. Arch Ophthalmol. 2008;126:1245–51. doi: 10.1001/archopht.126.9.1245. [DOI] [PubMed] [Google Scholar]
- 27.Anastassiou G, Bornfeld N, Schueler AO, Schilling H, Weber S, Fluehs D, et al. Ruthenium-106 plaque brachytherapy for symptomatic vasoproliferative tumours of the retina. Br J Ophthalmol. 2006;90:447–50. doi: 10.1136/bjo.2005.081422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blasi MA, Scupola A, Tiberti AC, Sasso P, Balestrazzi E. Photodynamic therapy for vasoproliferative retinal tumors. Retina. 2006;26:404–9. doi: 10.1097/01.iae.0000238554.61165.fd. [DOI] [PubMed] [Google Scholar]
- 29.Saldanha MJ, Edrich C. Treatment of vasoproliferative tumors with photodynamic therapy. Ophthalmic Surg Lasers Imaging. 2008;39:143–5. doi: 10.3928/15428877-20080301-13. [DOI] [PubMed] [Google Scholar]
- 30.Kenawy N, Groenwald C, Damato B. Treatment of a vasoproliferative tumour with intravitreal bevacizumab (Avastin) Eye. 2007;21:893–4. doi: 10.1038/sj.eye.6702782. [DOI] [PubMed] [Google Scholar]
- 31.Heimann H, Bornfeld N, Vij O, Coupland SE, Bechrakis NE, Kellner U, et al. Vasoproliferative tumours of the retina. Br J Ophthalmol. 2000;84:1162–9. doi: 10.1136/bjo.84.10.1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wyburn-Mason R. Arteriovenous aneurysm of midbrain and retina, facial nevi and mental changes. Brain Dev. 1943;66:163–203. [Google Scholar]
- 33.Muthukumar N, Sundaralingam MP. Retinocephalic vascular malformation: Case report. J Neurosurg. 1998;12:458–60. doi: 10.1080/02688699844718. [DOI] [PubMed] [Google Scholar]
- 34.Theron J, Newton TH, Hoyt WF. Unilateral retinocephalic vascular malformations. Neuroradiology. 1974;7:185–96. doi: 10.1007/BF00342696. [DOI] [PubMed] [Google Scholar]
- 35.Archer DB, Deutman A, Ernest JT, Krill AE. Arteriovenous communications of the retina. Am J Ophthalmol. 1973;75:224–41. doi: 10.1016/0002-9394(73)91018-0. [DOI] [PubMed] [Google Scholar]
- 36.Augsburger JJ, Goldberg RE, Shields JA, Mulberger RD, Magargal LE. Changing appearance of retinal arteriovenous malformation. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1980;215:65–70. doi: 10.1007/BF00413398. [DOI] [PubMed] [Google Scholar]
- 37.Bech K, Jensen OA. On the frequency of coexisting racemose hemangiomata of the retina and brain. Acta Psychiatr Scand. 1961;36:47–56. doi: 10.1111/j.1600-0447.1961.tb01756.x. [DOI] [PubMed] [Google Scholar]
- 38.Shah GK, Shields JA, Lanning RC. Branch retinal vein obstruction secondary to retinal arteriovenous communication. Am J Ophthalmol. 1998;126:446–8. doi: 10.1016/s0002-9394(98)00103-2. [DOI] [PubMed] [Google Scholar]
- 39.Effron L, Zakov ZN, Tomsak RL. Neovascular glaucoma as a complication of the Wyburn-Mason syndrome. J Clin Neuroophthalmol. 1985;5:95–8. [PubMed] [Google Scholar]
- 40.Anand R, Augsburger JJ, Shields JA. Circumscribed choroidal hemangiomas. Arch Ophthalmol. 1989;107:1338–42. doi: 10.1001/archopht.1989.01070020408045. [DOI] [PubMed] [Google Scholar]
- 41.Witschel H, Font R. Hemangioma of the choroid. A clinicopathologic study of 71 cases and a review of the literature. Surv Ophthalmol. 1976;20:415–31. doi: 10.1016/0039-6257(76)90067-9. [DOI] [PubMed] [Google Scholar]
- 42.Singh A, Kaiser P, Sears J. Choroidal hemangioma. Ophthalmol Clin North Am. 2005;18:151–61. doi: 10.1016/j.ohc.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 43.Arevalo JF, Shields CL, Shields JA, Hykin PG, De Potter P. Circumscribed choroidal hemangioma: Characteristic features with indocyanine green videoangiography. Ophthalmology. 2000;107:344–50. doi: 10.1016/s0161-6420(99)00051-2. [DOI] [PubMed] [Google Scholar]
- 44.Ramasubramanian A, Shields CL, Harmon SA, Shields JA. Autofluorescence of choroidal hemangioma in 34 consecutive eyes. Retina. 2010;30:16–22. doi: 10.1097/IAE.0b013e3181bceedb. [DOI] [PubMed] [Google Scholar]
- 45.Shields CL, Honavar SG, Shields JA, Cater J, Demirci H. Circumscribed choroidal hemangioma: Clinical manifestations and factors predictive of visual outcome in 200 consecutive cases. Ophthalmology. 2001;108:2237–48. doi: 10.1016/s0161-6420(01)00812-0. [DOI] [PubMed] [Google Scholar]
- 46.Madreperla SA, Hungerford JL, Plowman PN, Laganowski HC, Gregory PT. Choroidal hemangiomas: Visual and anatomic results of treatment by photocoagulation or radiation therapy. Ophthalmology. 1997;104:1773–8. doi: 10.1016/s0161-6420(97)30027-x. discussion 1779. [DOI] [PubMed] [Google Scholar]
- 47.Schilling H, Sauerwein W, Lommatzsch A. Long term results after low dose ocular irradiation for choroidal hemangioma. Br J Ophthalmol. 1997;81:267–73. doi: 10.1136/bjo.81.4.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ritland JS, Eide N, Tasujo J. External beam irradiation therapy for choroidal hemangiomas: Visual and anatomical results after a dose of 20-25 Gy. Acta Ophthalmol Scand. 2001;79:184–6. doi: 10.1034/j.1600-0420.2001.079002184.x. [DOI] [PubMed] [Google Scholar]
- 49.Kivelä T, Tenhunen M, Joensuu T, Tommila P, Joensuu H, Kouri M. Stereotactic radiotherapy of symptomatic circumscribed choroidal hemangiomas. Ophthalmology. 2003;110:1977–82. doi: 10.1016/S0161-6420(03)00483-4. [DOI] [PubMed] [Google Scholar]
- 50.Zografos L, Bercher L, Chamot L, Gailloud C, Raimondi S, Egger E. Cobalt-60 treatment of choroidal hemangiomas. Am J Ophthalmol. 1996;121:190–9. doi: 10.1016/s0002-9394(14)70584-7. [DOI] [PubMed] [Google Scholar]
- 51.Hannouche D, Frau E, Desjardins L, Cassoux N, Habrand JL, Offret H. Efficacy of proton therapy in circumscribed choroidal hemangiomas associated with serous retinal detachment. Ophthalmology. 1997;104:1780–4. doi: 10.1016/s0161-6420(97)30026-8. [DOI] [PubMed] [Google Scholar]
- 52.Lee V, Hungerford JL. Proton beam therapy for posterior pole circumscribed choroidal haemangioma. Eye. 1998;12:925–8. doi: 10.1038/eye.1998.240. [DOI] [PubMed] [Google Scholar]
- 53.Zografos L, Egger E, Bercher L, Chamot L, Munkel G. Proton beam irradiation of choroidal hemangiomas. Am J Ophthalmol. 1998;126:261–8. doi: 10.1016/s0002-9394(98)00150-0. [DOI] [PubMed] [Google Scholar]
- 54.Scott TA, Augsburger JJ, Brady LW, Hernandez C, Woodleigh R. Low dose ocular irradiation for diffuse choroidal hemangiomas associated with bullous nonrhegmatogenous retinal detachment. Retina. 1991;11:389–93. doi: 10.1097/00006982-199111040-00004. [DOI] [PubMed] [Google Scholar]
- 55.Gunduz K. Transpupillary thermotherapy in the management of circumscribed choroidal hemangioma. Surv Ophthalmol. 2004;49:316–27. doi: 10.1016/j.survophthal.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 56.Singh AD, Kaiser PK. Uveal vascular tumors. In: Singh AD, Damato BE, Pe’er J, editors. Clinical Ophthalmic Oncology. Philadelphia (PA): Saunders-Elsevier; 2007. pp. 289–99. [Google Scholar]
- 57.Madreperla SA. Choroidal hemangioma treated with photodynamic therapy using verteporfin. Arch Ophthalmol. 2001;119:1606–10. doi: 10.1001/archopht.119.11.1606. [DOI] [PubMed] [Google Scholar]
- 58.Jurklies B, Anastassiou G, Ortmans S, Schüler A, Schilling H, Schmidt-Erfurth U, et al. Photodynamic therapy using verteporfin in circumscribed choroidal haemangioma. Br J Ophthalmol. 2003;87:84–9. doi: 10.1136/bjo.87.1.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sullivan TJ, Clarke MP, Morin JD. The ocular manifestations of the Sturge-Weber syndrome. J Pediatr Ophthalmol Strabismus. 1992;29:349–56. doi: 10.3928/0191-3913-19921101-05. [DOI] [PubMed] [Google Scholar]
- 60.Anand R. Photodynamic therapy for diffuse choroidal hemangioma associated with Sturge Weber syndrome. Am J Ophthalmol. 2003;136:758–60. doi: 10.1016/s0002-9394(03)00423-9. [DOI] [PubMed] [Google Scholar]
- 61.Singh AD, Rundle PA, Vardy SJ, Rennie IG. Photodynamic therapy of choroidal hemangioma associated with Sturge-Weber syndrome. Eye. 2004;19:365–7. doi: 10.1038/sj.eye.6701474. [DOI] [PubMed] [Google Scholar]
- 62.Bains HS, Cirino AC, Ticho BH, Jampol LM. Photodynamic therapy using verteporfin for a diffuse choroidal hemangioma in Sturge-Weber syndrome. Retina. 2004;24:152–5. doi: 10.1097/00006982-200402000-00022. [DOI] [PubMed] [Google Scholar]
- 63.Phelps CD. The pathogenesis of glaucoma in Sturge-Weber syndrome. Ophthalmology. 1978;85:276–86. doi: 10.1016/s0161-6420(78)35667-0. [DOI] [PubMed] [Google Scholar]
- 64.Mandal AK. Primary combined trabeculotomy-trabeculectomy for early-onset glaucoma in Sturge-Weber syndrome. Ophthalmology. 1999;106:1621–7. doi: 10.1016/S0161-6420(99)90462-1. [DOI] [PubMed] [Google Scholar]