

Abstract
Metal boryl complexes have received significant attention in the literature in recent years due to their role as key intermediates in a number of metal-catalyzed borylation reactions. The ligand scaffold is known to have a significant impact on the observed reactivity of these metal boryl complexes. A synthetic strategy to access ruthenium boryl analogues of the Shvo metal–ligand catalysts is described. Heating a precursor to Shvo’s catalyst (1) with bis(catecholato)diboron at 50 °C provided ruthenium boryl complex 3 [2,5-Ph2-3,4-Tol2(η5-C4COBcat)Ru(CO)2Bcat] (Bcat = catecholatoboryl). Addition of bis(catecholato)diboron to complex 1 in the presence of a phenol results in ruthenium boryl complex5 [2,5-Ph2-3,4-Tol2(η5-C4COH)Ru(CO)2Bcat] at 22 °C in 30% isolated yield. A single crystal X-ray analysis of complex 5 confirmed the assigned structure. An improved synthesis of ruthenium boryl complex 5 was developed by the in situ formation of complex 3 [2,5-Ph2-3,4-Tol2(η5-C4COBcat)Ru(CO)2Bcat] followed by addition of the phenol, resulting in a 51% yield.
Keywords: Metal, ligand Bifunctional Catalyst, Ruthenium Boryl
Introduction
Metal boryl complexes have received significant attention over the past two decades due to their known intermediacy in metal-catalyzed reactions that incorporate boron substituents into organic substrates.1–9 The design of metal boryl complexes with unique ligand frameworks has led to complexes with previously unknown reactivity. C–H borylation reactions, for example, utilize iridium, rhodium, rhenium, and ruthenium catalysts in the selective incorporation of boron substituents into aryl or alkyl organic substrates. 1,10–15 This process allows for the conversion of inexpensive starting materials into valuable fine chemicals based on the ability to convert C–B bonds into C–O, C–N, and C–C bonds.16–21
The continued development of metal boryl complexes with unique ligand frameworks has the potential to provide unexplored reactivity. Metal–ligand bifunctional complexes have led to highly active catalysts in hydrogenation and conjugate addition reactions in recent years.22,23 The Shvo and Noyori catalysts have served as prototypes of these metal–ligand bifunctional catalysts, which have been shown to have mechanistically unique catalytic cycles.24–27 Our group is interested in boron-substituted analogues of the Shvo complex with the ultimate goal of designing metal boryl complexes with unique catalytic activity based on metal–ligand bifunctional catalysis.28–30 We herein report the synthesis of two ruthenium boryl complexes [2,5-Ph2-3,4-Tol2(η5-C4COBcat)Ru(CO) 2Bcat] and [2,5-Ph2-3,4-Tol2(η5-C4COH)Ru(CO)2Bcat] (Bcat = catecholatoboronate) by the activation of bis(catecholato)diboron by a coordinatively unsaturated precursor to the Shvo catalyst.
Results and Discussion
The synthesis of [2,5-Ph2-3,4-Tol2(η5-C4COBcat)Ru(CO)2Bcat] 3 was achieved by reaction of bis(catecholato)diboron with ruthenium dimer 1. Previous studies on boron-substituted analogues of Shvo complexes revealed that the activation of pinacolborane could be achieved by ruthenium dimer 1 at 50 °C to provide ruthenium hydride 2 in 74% isolated yield.28 Direct replacement of pinacolborane with bis(catecholato)diboron resulted in the formation of 3 in 70% NMR yield at 50 °C after 6 hours. The structure of 3 was supported by 11B NMR spectroscopy with a broad resonance at 47.8 ppm (Ru–Bcat) and a sharp resonance at 22.3 ppm (Cp′OBcat).15,28,31,32 Repeated attempts to isolate 3 were unsuccessful due to the labile nature of the O–B bond. The greater kinetic stability of the O–B bonds of 2 compared with 3 is attributed to the known increased stability of pinacolato-substituted boron substituents over those of the catecholato-substituted.33 Analogous reactions of ruthenium dimer 1 with bis(pinacolato)diboron (or other diboron reagents), however, were unsuccessful in generating the desired [2,5-Ph2-3,4-Tol2(η5-C4COBpin)Ru(CO)2Bpin] in appreciable yields. These results are consistent with the known increased reactivity of B2cat2 over B2pin2 in metal-mediated reactions.34
The oxidative addition of diboron reagents in the presence of an alcohol was expected to provide [2,5-Ph2-3,4-Tol2(η5-C4COH)Ru(CO)2B(OR)2] complexes directly. Detailed mechanistic and computational studies by Casey and Cui on the activation of H2 have revealed a cyclic proton transfer transition state (TS-A, Scheme 2) involving ethanol for this oxidative addition.35 The activation of diboron reagents in the presence of an alcohol was anticipated to proceed through an analogous transition state (TS-B). Unlike the activation of H2, where the exchange of a hydrogen atom is degenerate, activation of a diboron reagent in the presence of an alcohol was expected to result in the net addition of (RO)2B–H, rather than (RO)2B–B(OR)2.
Scheme 2.

The Proposed Transition State for Diboron Activation Compared to Hydrogen Activation
Addition of B2cat2 to ruthenium dimer 1 in the presence of 4-methoxyphenol resulted in the formation of [2,5-Ph2-3,4-Tol2(η5-C4COH)Ru(CO)2Bcat] 5 in 74% NMR spectroscopic yield after 2 hours at 22 °C (eq 1).36 The reaction required that the alcohol additive did not possess an α-hydrogen. The presence of an α-hydrogen resulted in dehydrogenation of the alcohol to the carbonyl and formation of the corresponding ruthenium hydride.24 Upon screening numerous phenols (electron rich, electron poor, and 2,6-disubstituted) and tert-butyl alcohol, 4-methoxyphenol was found to provide the highest yield of 5. The formation of 5 is believed to proceed via TS-B (Scheme 2), rather than by initial activation of B2cat2 and protonolysis of the O–B bond by the phenol, based on the reduced reaction time and temperature as compared to the addition of B2cat2 in the absence of the phenol (Scheme 1). The increased reaction rate in the presence of the phenol is consistent with a lower kinetic barrier to diboron activation than the reaction in the absence of the phenol.37
Scheme 1.

Synthesis of Ruthenium Boryl Complex 3
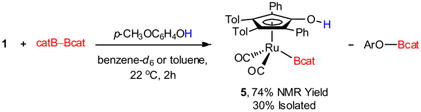 |
(1) |
In addition to the formation of complex 5, bridging hydride 6 (Figure 1) was identified by 1H NMR spectroscopic analysis (10–25% NMR yield).24 The quantity of complex 6 was highly dependent on both the identity and number of equivalents of the phenol that was used. Initially, the source of complex 6 was unclear since phenols lack α-hydrogens and therefore cannot act as a source of H2. The O–H borylation of the phenol by the in situ generated complex 5, however, could account for the generation of bridging hydride 6.38,39 To test this hypothesis, isolated complex 5 was treated with 4-methoxyphenol, resulting in 25% conversion into terminal hydride 4 after 4 h at 22 °C (eq 2). In the presence of ruthenium dimer 1, terminal hydride 4 is known to readily convert into bridging hydride 6.24,40 The formation of complex 6, therefore, is consistent with competitive reaction rates of the phenol in diboron activation (via TS-B, Scheme 2) and O–H borylation (eq 2).
Figure 1.
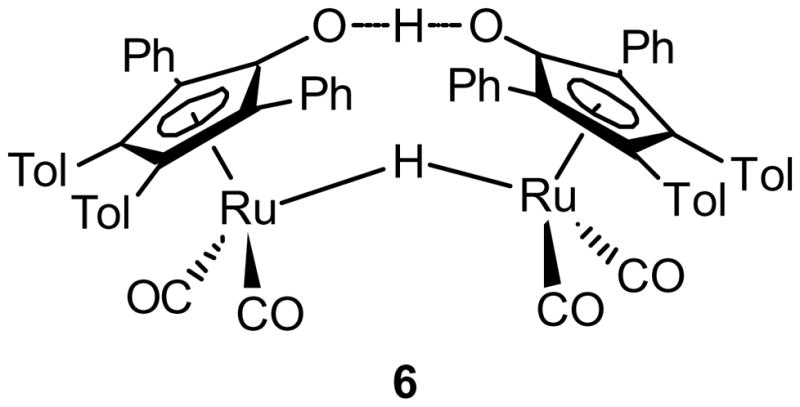
Bridging Hydride 6
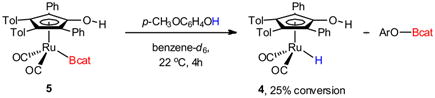 |
(2) |
Purification of ruthenium boryl complex 5 by trituration with diethyl ether and pentane resulted in clean isolation of 5 in 30% yield. Bridging hydride 6 was found to be soluble in a diethyl ether/pentane mixture, allowing its removal upon successive triturations. The characterization data obtained for complex 5 confirms the proposed ruthenium boryl structure. Notably, the 11B NMR spectrum revealed a broad resonance at 47.0 ppm.15,28 The IR spectrum also showed characteristic CO stretching frequencies at 2025 and 1968 cm-1.
Although the spectroscopic data was consistent with the assigned structure of 5, interactions between the metal-bound boron substituent and the ligand-bound hydrogen were considered. The presence of a α-borane complex was ruled out based on the 1H NMR chemical shifts. Known α-borane complexes have broad resonances (due to interactions with boron) of the boron-bound hydrogen and have similar chemical shifts to typical metal hydrides (−5 to −10 ppm),41,42 which is in stark contrast to the observed sharp singlet at +6.45 ppm. A transient α-borane complex was also ruled out based on the known regiochemistry of dialkoxyboranes with the coordinatively unsaturated complex derived from dimer 1 (Scheme 1), which provides the corresponding ruthenium hydride (such as 2, Scheme 1).
The regiochemistry of boron addition and the nature of the interactions between boron and hydrogen in complex 5 were probed in the solid state using X-ray crystallography. X-ray quality crystals were grown from CH2Cl2 and pentane at −30 °C. The crystal structure confirms the identity of 5 and revealed two unusual conformational features in the solid state arising from hydrogen bonding of the Cp′OH to an oxygen atom on the boryl group. First, the plane of the catecholatoboryl substituent is turned toward the hydroxycyclopentadienyl ligand in spite of the steric congestion imposed by the tetraaryl-substituted cyclopentadienyl ligand (19.9° torsion angle for Cp′ centroid–Ru–B–O(2)).43-45 Second, the hydroxy substituent on the cyclopentadienyl ligand and catecholatoboryl substituent are nearly eclipsed. X-ray crystal structures of the Shvo type complexes were found to have the metal–hydride bond rotated nearly anti to the hydroxy substituent of the cyclopentadienyl ligand in the solid state.46–48
Although the synthesis of complex 5 was achieved in the presence of 4-methoxyphenol, the isolated yields were difficult to obtain reproducibly. This resulted in the evaluation of other approaches to generate the ruthenium boryl complex. The labile nature of the O–B bond in 3 prompted attempts to selectively protonolyze the O–B bond in the presence of the Ru–B bond. A solution to the irreproducible yields was established with the initial formation of complex 3 (Scheme 3), followed by addition of 4-methoxyphenol, providing 5 as the major product (eq 3). Again, the addition of the phenol resulted in formation of bridging hydride 6, but complex 5 was formed more cleanly using this protocol, ultimately providing complex 5 in 51% isolated yield. Notably, this optimized protocol resulted in readily reproducible yields of 5.
Scheme 3.

Independent Synthesis of Ruthenium Hydride 7
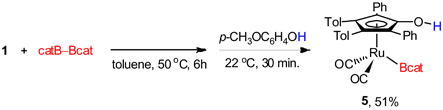 |
(3) |
Preliminary studies of the reactivity of 5 have been limited by the thermal instability of the complex. Heating 5 in benzene-d6 to 80 °C results in decomposition to the corresponding hydride, [2,5-Ph2-3,4-Tol2(η5-C4COBcat)Ru(CO)2H] 7 (eq 4). In addition to complex 7, bridging hydride 8 was observed (Scheme 3). Both of these complexes can be synthesized by treatment of ruthenium dimer 1 with catecholborane. The addition of 2 equivalents of catecholborane to ruthenium dimer 1 results in the predominant formation of bridging hydride 8 in 30 minutes at 22 °C (~10% terminal hydride 7 observed by 1H NMR spectroscopy). The generation of terminal hydride 7 could also be facilitated by heating 8 to 50 °C with a slight excess catB–H (3 equivalents). Unfortunately, only up to 75% conversion to 7 was possible under these conditions and higher temperatures led to decomposition of 7 and 8. The two complexes were readily distinguishable by 1H NMR spectroscopy based on the characteristic hydride resonances (7: δ = −9.3 ppm; 8: δ = −18.3 ppm), which are consistent with the analogous complexes from activation of dihydrogen (terminal hydride: δ = −9.8 ppm; bridging hydride 6: δ = −18.2 ppm).24 Again, the labile nature of the O–Bcat bond precluded the isolation of complexes 7 or 8.
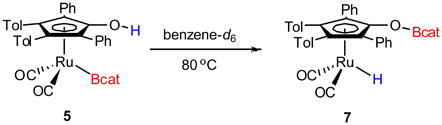 |
(4) |
Conclusions
The activation of bis(catecholato)diboron by ruthenium dimer 1 was explored as a means to prepare ruthenium boryl complexes possessing both acidic and Lewis acidic hydroxycyclopentadienyl ligands. The direct activation of bis(catecholato)diboron by 1 at 50 °C provided the corresponding ruthenium boryl complex (3) with a second boron substituent bound to oxygen on the hydroxycyclopentadienyl ligand. Conducting the same experiment in the presence of a phenol resulted in rapid formation of ruthenium boryl complex 5 at 22 °C (with an O–H bond on the hydroxycyclopentadienyl ligand). The concurrent formation of bridging hydride 6 decreased the isolated yield. Optimized yield of complex 5 was obtained by a two-step procedure in which ruthenium boryl complex 3 was first generated, followed by selective cleavage of the ligand O–B bond to provide 5 in 51% isolated yield. An X-ray crystal structure of 5 was obtained to confirm the identity of this ruthenium boryl complex.
Experimental Section
General Experimental Information
All materials were manipulated under dry nitrogen in an MBraun Inc. glovebox. NMR spectra were collected on UNITY-Inova spectrometer at 500 MHz for 1H NMR, 125 MHz for 13C NMR, and 160 MHz for 11B NMR. 1H NMR spectra are referenced to benzene-d6 at 7.16 ppm. The 1H NMR data are reported as follows: chemical shift in ppm, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, qn = quintet, sep = septet, m = multiplet), coupling constants (Hz), and integration. 13C NMR spectra are referenced to benzene-d6 at 125.39 ppm. 11B NMR spectra were referenced to an external BF3•Et2O sample in benzene-d6 (0.0 ppm). Elemental analyses were performed by Desert Analytics in Tucson, AZ. Elemental analysis data are reported as follows: empirical formula; calculated percent carbon, calculated percent hydrogen, calculated percent nitrogen; observed percent carbon, observed percent hydrogen, observed percent nitrogen. Deuterated solvents and pentane were dried, distilled, and degassed prior to use. Toluene and diethyl ether were deoxygenated and dried in a solvent purification system (Innovative Technology, Inc.) by passing through an activated alumina column and an oxygen scavenging column under nitrogen, followed by degassing by three freeze, pump, thaw cycles. Bis(catecholato)diboron and 4-methoxyphenol were purchased from Aldrich and used without further purification. Catecholborane was purchased from Aldrich and distilled under vacuum prior to use. [2,5-Ph2-3,4-Tol2(η4-C4CO)]Ru(CO)2]2 (1) was synthesized according to the literature procedure.49
[2,5-Ph2-3,4-Tol2(η5-C4COBcat)Ru(CO)2Bcat] 3
To a resealable NMR tube containing {[2,5-Ph2-3,4-Tol2(η4-C4CO)Ru(CO)2}2 (1) (0.016 g, 0.014 mmol) and 0.60 mL of benzene- d6 was added bis(catecholato)diboron (0.007 g, 0.029 mmol). The reaction mixture was agitated to disperse the solids and was heated to 50 °C for 6 h. A 1H NMR spectrum of the reaction mixture showed a 70% NMR spectroscopic yield of 3 relative to an internal PhSiMe3 standard. 1H NMR (benzene-d6, 300 MHz) δ 7.72 (m, 4H), 7.25 (d, J = 8.1, 4H), 6.92 (m, 6H), 6.72 (m, 2H), 6.60 (m, 8H), 6.45 (m, 2H), 1.85 (s, 6H). 11B NMR (benzene-d6, 160 MHz) δ 47.8, 22.3.
[2,5-Ph2-3,4-Tol2(η4-C4OH)]Ru(CO)2Bcat (5)
Method A
A slurry of {[2,5-Ph2-3,4-Tol2(η4-C4CO)Ru(CO)2}2 (1) (0.300 g, 0.263 mmol), bis(catecholato)diboron (0.126 g, 0.530 mmol), and 4-methoxyphenol (0.066 g, 0.532 mmol) in 11 mL of toluene was stirred for 2 h at 22° C. The solvent was evaporated in vacuo, providing a yellow solid. Et2O was added to the flask (12 mL) and the solution was stirred in the glove box for 2 h. Pentane (6 mL) was then added to the solution and the mixture was stirred for 10 min, during which a yellow precipitate formed. The precipitate was filtered and rinsed with addition pentane (~30 mL) and dried in vacuo to provide 5 as a pale yellow solid (0.110 g, 30%). Crystals suitable for X-ray diffraction were obtained by crystallization in a solution of CH2Cl2/pentane at −30 °C. 1H NMR (benzene-d6, 500 MHz) δ 7.58 (d, J = 6.5 Hz, 4H), 7.22 (d, J = 8.0 Hz, 4H), 6.93 (m, 6H), 6.76 (dd, J = 5.8, 3.2 Hz, 2H), 6.66 (d, J = 8.0 Hz, 4H), 6.61 (dd, J = 5.5, 3.5 Hz, 2H), 6.45 (s, 1H), 1.88 (s, 6H). 13C NMR (benzene-d6, 125 MHz) δ 201.7, 149.9, 138.0, 133.2, 132.6, 131.0, 129.4, 129.0, 128.9, 128.7, 122.5, 112.1, 107.1, 94.8, 21.3. 11B NMR (C6D6, 160 MHz) δ 47.0. IR (thin film) 3054, 2987, 2025, 1968, 1422 cm−1. Anal. Calcd for C39H29O5BRu: C, 67.74; H, 4.52. Found: C, 67.64; H, 4.38.
Method B
A slurry of {[2,5-Ph2 -3,4-Tol2(η4-C4CO)Ru(CO)2}2 (1) (0.500 g, 0.438 mmol) and bis(catecholato)diboron (0.313 g, 1.32 mmol) in 38 mL of toluene was heated to 50 °C for 5 h. The reaction mixture was removed from heat and 4-methoxyphenol (0.114 g, 0.918 mmol) was added. After 20 minutes, the solvent was evaporated in vacuo to provide an yellow solid. Et2O was added to the flask (20 mL) and the solution was stirred in the glove box for 2 h. Pentane (12 mL) was then added to the solution and the mixture was stirred for 10 min, during which a yellow precipitate formed. The precipitate was filtered and rinsed with addition pentane (~40 mL) and dried in vacuo to provide 5 as a pale yellow solid (0.308 g, 51%).
{[2,5-Ph2-3,4-Tol2(η5-C4CO)]2Bcat}Ru2(CO)4(μ-H) (8)
To a resealable NMR tube containing {[2,5-Ph2-3,4-Tol2(η4-C4CO)Ru(CO)2}2 (1) (0.033 g, 0.029 mmol) and 0.60 mL of benzene-d6 was added catecholborane (0.007 mL, 0.066 mmol). The reaction mixture was agitated to disperse the solid. After 25 min a 1H NMR of the reaction mixture showed complete conversion to 8 with about 10% 7. 1H NMR (benzene-d6, 300 MHz): δ 7.25 (d, J = 6.6 Hz, 6H), 7.13 (m, 6H), 6.99 (m, 18H), 6.71 (m, 2H), 6.49 (m, 8H), 1.80 (s, 12H), −18.36 (s, 1H). 11B NMR (benzene-d6, 160 MHz) δ 22.0.
Supplementary Material
Figure 2.
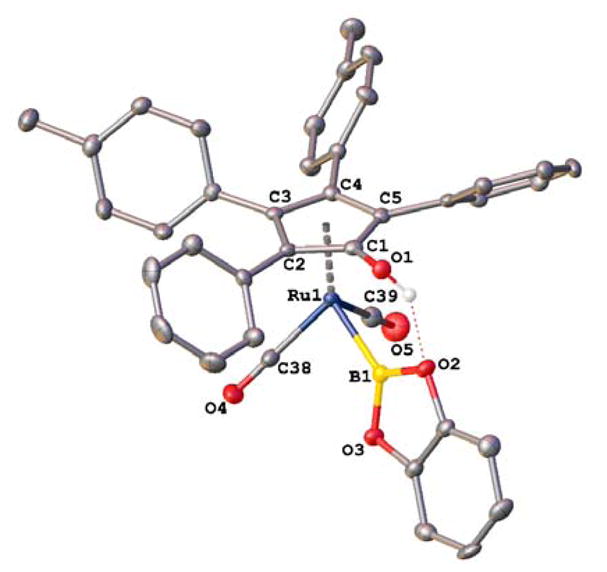
X-ray crystal structure of 5.
Acknowledgments
We thank Charles P. Casey for helpful discussions. Acknowledgement is made to the donors of the American Chemical Society Petroleum Research Fund for partial support of this research (47598-GB3), the M.J. Murdock Charitable Trust and Research Corporation for Science Advancement in the form of a department development grant, Western Washington University, and the National Institutes of Health in the form of a NRSA fellowship (GM077962-01).
Footnotes
Supporting Information Available. 1H, 13C, 11B NMR spectra and the X-ray crystallographic data of 5 are provided as supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem Rev. 2010;110:890–931. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- 2.Dang L, Lin Z, Marder TB. Chem Comm. 2009:3987–3995. doi: 10.1039/b903098k. [DOI] [PubMed] [Google Scholar]
- 3.Fritschi CB, Wernitz SM, Vogels CM, Shaver MP, Decken A, Bell A, Westcott SA. Eur J Inorg Chem. 2008:779–785. [Google Scholar]
- 4.Braunschweig H, Kollann C, Rais D. Angew Chem, Int Ed. 2006;45:5254–5274. doi: 10.1002/anie.200600506. [DOI] [PubMed] [Google Scholar]
- 5.Aldridge S, Coombs DL. Coord Chem Rev. 2004;248:535–559. [Google Scholar]
- 6.Irvine GJ, Lesley MJG, Marder TB, Norman NC, Rice CR, Robins EG, Roper WR, Whittell GR, Wright LJ. Chem Rev. 1998;98:2685–2722. doi: 10.1021/cr9500085. [DOI] [PubMed] [Google Scholar]
- 7.Braunschweig H. Angew Chem, Int Ed. 1998;37:1786–1801. [Google Scholar]
- 8.Wadepohl H. Angew Chem, Int Ed Engl. 1997;36:2441–2444. [Google Scholar]
- 9.Baker RT, Calabrese JC. J Am Chem Soc. 1993;115:4367–4368. [Google Scholar]
- 10.Murphy JM, Lawrence JD, Kawamura K, Incarvito C, Hartwig JF. J Am Chem Soc. 2006;128:13684–13685. doi: 10.1021/ja064092p. [DOI] [PubMed] [Google Scholar]
- 11.Chotana GA, Rak MA, Smith MR., III J Am Chem Soc. 2005;127:10539–10544. doi: 10.1021/ja0428309. [DOI] [PubMed] [Google Scholar]
- 12.Lawrence JD, Takahashi M, Bae C, Hartwig JF. J Am Chem Soc. 2004;126:15334–15335. doi: 10.1021/ja044933x. [DOI] [PubMed] [Google Scholar]
- 13.Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J Am Chem Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- 14.Cho JY, Tse MK, Holmes D, Maleczka RE, Jr, Smith MR., III Science (Washington D C) 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 15.Waltz KM, Hartwig JF. J Am Chem Soc. 2000;122:11358–11369. [Google Scholar]
- 16.Brown HC, Kramer GW, Levy AB, Middland MM. Organic Syntheses via Boranes. Vol. 1 Wiley-Interscience; New York: 1975. [Google Scholar]
- 17.Brown HC, Zaidlewicz M. Organic Syntheses via Boranes-Recent Developments. Vol. 2 Aldrich Chemical Company; Milwaukee: 2001. [Google Scholar]
- 18.Brown HC, Singaram B. Acc Chem Res. 1988;21:287–293. [Google Scholar]
- 19.Miyaura N, Suzuki A. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 20.Matteson DS. Tetrahedron. 1998;54:10555–10607. [Google Scholar]
- 21.Matteson DS. Stereodirected Synthesis with Organoboranes. In: Hafner K, Rees CW, Trost BM, Lehn J-M, von Ragué Schleyer P, Zahradník R, editors. Reactivity and Structure Concept in Organic Chemistry. Springer-Verlag; Berlin: 1995. p. 32. [Google Scholar]
- 22.For reviews of metal-ligand bifunctional catalysis, see: Noyori R, Ohkuma T. Angew Chem Int Ed. 2001;40:40–73.Noyori R, Kitamura M, Ohkuma T. Proc Natl Acad Sci USA. 2004;101:5356–5362. doi: 10.1073/pnas.0307928100.Clapham SE, Hadzovic A, Morris RH. Coord Chem Rev. 2004;248:2201–2237.Ikariya T, Murata K, Noyori R. Org Biomol Chem. 2006;4:393–406. doi: 10.1039/b513564h.Conley BL, Pennington-Boggio MK, Boz E, Williams TJ. Chem Rev. 2010;110:2294–2312. doi: 10.1021/cr9003133.
- 23.For examples of additional metal-ligand bifunctional catalysts that employ functional groups other than hydrogen, see: Watanabe M, Murata K, Ikariya T. J Am Chem Soc. 2003;125:7508–7509. doi: 10.1021/ja035435b.Watanabe M, Ikagawa A, Wang H, Murata K, Ikariya T. J Am Chem Soc. 2004;126:11148–11149. doi: 10.1021/ja046296g.Shibasaki M, Yoshikawa N. Chem Rev. 2002;102:2187–2209. doi: 10.1021/cr010297z.Shibasaki M, Matsunaga S. Chem Soc Rev. 2006;35:269–279. doi: 10.1039/b506346a.
- 24.Casey CP, Singer SW, Powell DR, Hayashi RK, Kavana M. J Am Chem Soc. 2001;123:1090–1100. doi: 10.1021/ja002177z. [DOI] [PubMed] [Google Scholar]
- 25.Casey CP, Clark TB, Guzei IA. J Am Chem Soc. 2007;129:11821–11827. doi: 10.1021/ja073370x. [DOI] [PubMed] [Google Scholar]
- 26.Abdur-Rashid K, Clapham SE, Hadzovic A, Harvey JN, Lough AJ, Morris RH. J Am Chem Soc. 2002;124:15104–15118. doi: 10.1021/ja016817p. [DOI] [PubMed] [Google Scholar]
- 27.Casey CP, Johnson JA. J Org Chem. 2003;68:1998–2001. doi: 10.1021/jo0205457. [DOI] [PubMed] [Google Scholar]
- 28.Koren-Selfridge L, Londino HN, Vellucci JK, Simmons BJ, Casey CP, Clark TB. Organometallics. 2009;28:2085–2090. [Google Scholar]
- 29.Conley BL, Williams TJ. J Am Chem Soc. 2010;132 doi: 10.1021/ja909858a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Conley BL, Williams TJ. Chem Commun. 2010:4815–4817. doi: 10.1039/c003157g. [DOI] [PubMed] [Google Scholar]
- 31.11B NMR spectra of borate esters typically have resonances in the range of 20–25 ppm; EtOBcat δ = 23.0 ppm. Nöth H, Wrackmeyer B. Nuclear Magnetic Resonance Spectroscopy of Boron Compounds. In: Diehl P, Fluck E, Kosfeld R, editors. NMR Basic Principles and Progress 14. Springer-Verlag; Berlin: 1978. pp. 136–138.
- 32.11B NMR chemical shift of B2cat2 is 30.7 ppm.
- 33.Tucker CE, Davidson J, Knochel P. J Org Chem. 1992;57:3482–3485. [Google Scholar]
- 34.Dang L, Zhao H, Lin Z, Marder TB. Organometallics. 2008;27:1178–1186. [Google Scholar]
- 35.Casey CP, Johnson JB, Singer SW, Cui Q. J Am Chem Soc. 2005;127:3100–3109. doi: 10.1021/ja043460r. [DOI] [PubMed] [Google Scholar]
- 36.NMR spectroscopic yields were determined by 1H NMR spectroscopy relative to a phenyltrimethylsilane as an internal standard and a ten second relaxation delay for integral integrity.
- 37.Casey CP, Johnson JB, Singer SW, Cui Q. J Am Chem Soc. 2005;127:3100–3109. doi: 10.1021/ja043460r. [DOI] [PubMed] [Google Scholar]
- 38.Schlecht S, Hartwig JF. J Am Chem Soc. 2000;122:9435–9443. [Google Scholar]
- 39.Marciniec B, Walkowiak J. Chem Commun. 2008:2695–2697. doi: 10.1039/b801013g. [DOI] [PubMed] [Google Scholar]
- 40.Casey CP, Beetner SE, Johnson JA. J Am Chem Soc. 2008;130:2285–2295. doi: 10.1021/ja077525c. [DOI] [PubMed] [Google Scholar]
- 41.Hartwig JF, Muhoro CN, He X. J Am Chem Soc. 1996;118:10936–10937. [Google Scholar]
- 42.Montiel-Palma V, Lumbierres M, Donnadieu B, Sabo-Etienne S, Chaudret B. J Am Chem Soc. 2002;124:5624–5625. doi: 10.1021/ja017429q. [DOI] [PubMed] [Google Scholar]
- 43.Rankin MA, MacLean DF, McDonald R, Ferguson MJ, Lumsden MD, Stradiotto M. Organometallics. 2009;28:74–83. [Google Scholar]
- 44.Lam WH, Shimada S, Batsanov AS, Lin Z, Marder TB, Cowan JA, Howard JAK, Mason SA, McIntyre GJ. Organometallics. 2003;22:4557–4568. [Google Scholar]
- 45.Hartwig JF, Huber S. J Am Chem Soc. 1993;115:4908–4909. [Google Scholar]
- 46.Casey CP, Strotman NA, Beetner SE, Johnson JB, Priebe DC, Guzei IA. Organometallics. 2006;25:1236–1244. [Google Scholar]
- 47.Knölker HJ, Baum E, Goesmann H, Klauss R. Angew Chem, Int Ed. 1999;38:2064–2066. doi: 10.1002/(SICI)1521-3773(19990712)38:13/14<2064::AID-ANIE2064>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 48.Casey CP, Guan H. J Am Chem Soc. 2007;129:5816–5817. doi: 10.1021/ja071159f. [DOI] [PubMed] [Google Scholar]
- 49.Casey CP, Singer SW, Powell DR, Hayashi RK, Kavana M. J Am Chem Soc. 2001;123:1090–1100. doi: 10.1021/ja002177z. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.