

Abstract
The worldwide threat of tuberculosis to human health emphasizes the need to develop novel approaches to a global epidemiological surveillance. The current standard for Mycobacterium tuberculosis typing based on IS6110 restriction fragment length polymorphism (RFLP) suffers from the difficulty of comparing data between independent laboratories. Here, we propose a high-resolution typing method based on variable number tandem repeats (VNTRs) of genetic elements named mycobacterial interspersed repetitive units (MIRUs) in 12 human minisatellite-like regions of the M. tuberculosis genome. MIRU-VNTR profiles of 72 different M. tuberculosis isolates were established by PCR analysis of all 12 loci. From 2 to 8 MIRU-VNTR alleles were identified in the 12 regions in these strains, which corresponds to a potential of over 16 million different combinations, yielding a resolution power close to that of IS6110-RFLP. All epidemiologically related isolates tested were perfectly clustered by MIRU-VNTR typing, indicating that the stability of these MIRU-VNTRs is adequate to track outbreak episodes. The correlation between genetic relationships inferred from MIRU-VNTR and IS6110-RFLP typing was highly significant. Compared with IS6110-RFLP, high-resolution MIRU-VNTR typing has the considerable advantages of being fast, appropriate for all M. tuberculosis isolates, including strains that have a few IS6110 copies, and permitting easy and rapid comparison of results from independent laboratories. This typing method opens the way to the construction of digital global databases for molecular epidemiology studies of M. tuberculosis.
Tuberculosis is the leading cause of death in adults from a single infectious agent, killing about 3 million people every year. One-third of the human population is thought to be infected by the causative agent Mycobacterium tuberculosis, and about 200 million additional people are at risk for developing the disease in the next 20 years if current trends continue (1, 2). In several regions of the world, there is an alarming rise of incidence, linked to the increasing devastating impact of HIV epidemics and deficiencies of current tuberculosis control programs. The worldwide development of transport and migration contributes to globalize those threats (3, 4).
This context emphasizes the need for powerful approaches in the epidemiological tuberculosis surveillance, providing quantitative bases to assess or define new control strategies at a global level (5). To achieve this goal, effective methods for accurate identification and monitoring of large numbers of M. tuberculosis strains are required. However, all current typing markers suffer from significant drawbacks. IS6110-restriction fragment length polymorphism (RFLP) is the current gold standard method for M. tuberculosis typing (6) and is extensively used for epidemiological and population-based studies (e.g., refs. 7–11). This method is labor intensive and requires culturing of the slow-growing mycobacteria. Moreover, strains with low IS6110 copy numbers, frequently found in some parts of the world (12) or devoid of these sequences (6), cannot be adequately analyzed by this method. Perhaps the most important limitation of this method is that the data obtained in independent studies are difficult to compare because of the lack of reproducibility (6) and portability of the results between different laboratories. Spoligotyping, a PCR-based method targeted against repetitive sequences in a single locus (13), is frequently used as an alternative. However, this method does not sufficiently well discriminate different strains (6, 14). Multilocus sequence typing, appropriate for large multicenter studies of many bacteria (15), is not applicable for M. tuberculosis because the gene sequence polymorphism is very restricted among strains of this species (6, 16).
Variable number tandem repeat (VNTR) typing is an invaluable tool for genotyping in higher eukaryotes and provides data in a simple and nonambiguous format based on the number of repetitive sequences in so-called polymorphic micro- or minisatellite regions. Despite some attempts to develop equivalent approaches for typing bacterial pathogens, only limited applications for bacterial molecular epidemiology could be developed up to now (17, 18). Frothingham and Meeker-O'Connell (19) have identified five VNTR loci in M. tuberculosis that have recently been tested for epidemiological purposes. However, the polymorphism of these loci proved to be much too low to accurately discriminate different clones (6, 11). We have previously identified a type of VNTR sequences called mycobacterial interspersed repetitive units (MIRUs), which are scattered throughout the M. tuberculosis complex genomes (20–22). A recent genome scan revealed that 12 of the 41 MIRU loci present in the M. tuberculosis H37Rv genome correspond to human minisatellite-like VNTR regions among nonrelated isolates of different geographic origins (23). Here, we show that a PCR-based typing method by using these 12 loci provides a resolution comparable to that of IS6110-RFLP. Our results demonstrate the potential of the MIRU-VNTR method for tracking epidemiological key events, such as transmission or relapses. This 12-loci-based approach allows for direct and reliable comparison of results between laboratories and thus for construction and analysis of global databases via the Internet for large-scale epidemiological and population genetic studies.
Experimental Procedures
Bacterial Strains and Genomic DNA.
Isolates from transmissions, relapses, and laboratory crosscontaminations were selected from the collection of the Centre de Référence des Mycobactéries (Institut Pasteur, Paris, France). The mycobacteria were grown at 37°C and then kept on Löwenstein–Jensen medium (Sanofi Diagnostics Pasteur, Marnes la Coquette, France) at 4°C. The genomic DNA used for the PCR analyses was purified as described in ref. 24 or obtained by resuspending mycobacterial colonies into 100–200 μl 10 mM Tris-HCl/1 mM EDTA (pH 7.0) followed by incubation at 95°C for 45 min. After centrifugation of the suspension, the supernatant containing the DNA was harvested and stored at −20°C until further use. The M. tuberculosis isolates from Paris hospitals were described previously (14, 24). For these isolates, PCR analyses were carried out mostly on frozen stocks of heat-treated colonies, except in cases where purified DNA was still available. The set of isolates with no known epidemiological links from this collection is referred to as the Paris Collection. The set of isolates from transmissions, relapses, or laboratory crosscontaminations also included isolates 58, 59, 72, 73, 74, and 75 from ref. 24.
PCR and MIRU-VNTR Analysis.
PCRs were carried out by using the HotStartTaq DNA Polymerase kit (Qiagen, Hilden, Germany), as described in ref. 23. The primers and MgCl2 concentrations are available in Table 4 which is published as supplemental data on the PNAS web site, www.pnas.org. The PCR fragments were analyzed by agarose gel electrophoresis by using 2% NuSieve agarose (FMC). The sizes of the amplicons were estimated by comparison with 20- and 100-bp superladders-low from Eurogentec (Seraing, Belgium). The numbers of MIRUs per locus were calculated on the basis of conventions described in ref. 23.
Genetic Relationships Analysis.
The Jaccard distance (D ij) (25) was used for the MIRU-VNTR data of the isolates of the Paris collection, according to the formula D ij = 1 − [a/(a + b + c)], where a is the number of MIRU-VNTR alleles that are common to the i and j isolates, b the number of MIRU-VNTR alleles that are specific to the i isolate, and c the number of MIRU-VNTR alleles that are specific to the j isolate. The same formula was used for the IS6110-RFLP data from ref. 24 for the same isolates, by using the RFLP bands instead of the MIRU-VNTR alleles. RFLP analysis was done by using the taxotron software package (Institut Pasteur Taxolab, Paris) with a fragment length error tolerance set to 3.5–5% tolerance. Genetic relationships among isolates were estimated by the Unweighted Pair-Group Method with Arithmetic Averages (26). Agreement between the genetic relationships based on MIRU-VNTR and those based on IS6110-RFLP was tested by calculating the correlation between the genetic distances inferred from the two methods for any possible pair of isolates with a nonparametric Mantel test (27, 28). Briefly, this test uses a Monte Carlo simulation with 104 iterations, which randomly permutes the different cells of each distance matrix. In contrast to the classical correlation test, this randomization procedure requires no assumption about the number of degrees of freedom.
Analysis of the MIRU-VNTR Allelic Diversity.
The allelic diversity (h) at a locus was calculated as h = 1 − ∑ xi 2 [n/(n − 1)], where xi is the frequency of the ith allele at the locus, and n the number of isolates (29).
Results
Typing M. tuberculosis by Using MIRU-VNTR.
Twelve M. tuberculosis minisatellite-like loci contain variable copy numbers of 51- to 77-bp MIRUs. Only two of them are common to the VNTR loci used by Meeker-O'Connell and Frothingham (locus 4 and 31 identical to VNTR-D and VNTR-E, respectively) (19). All loci except loci 4 and 20 also display minor sequence variations between the repeat units, which consist of a few substitutions or insertions/deletions. The changes in these loci virtually always consist of simple sequential additions or deletions of identical size units (23). Therefore, for each locus, the number of variable units can be calculated by measuring the size of fragments amplified with specific primers hybridizing to flanking DNA regions. Thus, each strain is typed by a numerical MIRU-VNTR code corresponding to the numbers of variable MIRUs in each of the 12 loci (23).
Stability of the MIRU-VNTR Profiles.
Analysis of the 12 MIRU loci in genealogically distant BCG daughter strains indicated that 11 of them remained unchanged during hundreds of passages over more than 30 years in axenic culture conditions (23). Only locus 4 featured differences between some strains. These observations indicate the long-term stability of these minisatellite-like regions during in vitro cultivation. This stability is further documented by the analysis of three groups of M. tuberculosis strains from three distinct laboratory crosscontaminations. As shown in Table 1, identical MIRU-VNTR combinations were found within each of the three different groups containing a total of eight isolates. Interestingly, in one of the three groups, one isolate (951591) had an additional band in its IS6110-RFLP profile compared with the others (not shown), although its link to the two other strains by laboratory crosscontamination was clearly documented.
Table 1.
Stability of MIRU-VNTRs in epidemiologically related M. tuberculosis isolates
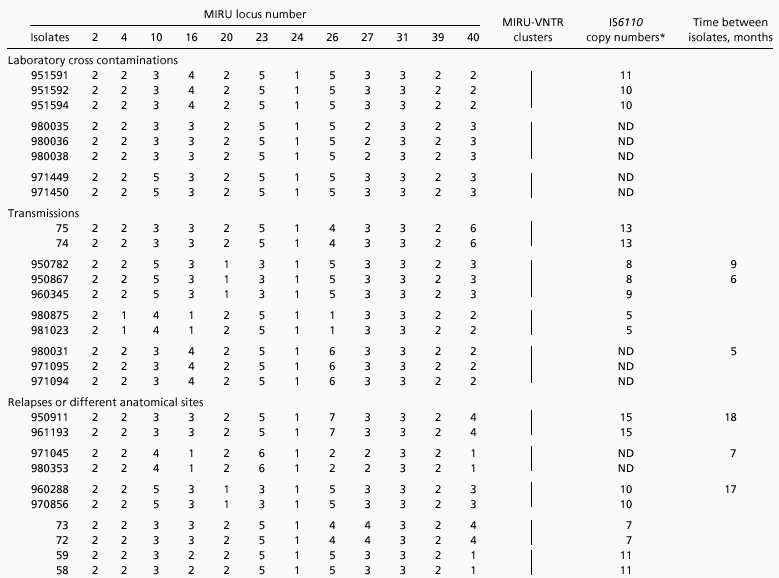 |
ND, not determined (corresponding isolates were found to have identical spoligotypes). Vertical bars correspond to MIRU-VNTR clusters.
IS6110-RFLP data for isolates 75, 74, 73, 72, 59, and 58 are from ref. 24.
In vivo stability was assessed by analyzing 20 isolates from transmissions, from relapses, or obtained from different anatomical sites of a same patient (Table 1). In all cases, the isolates could be properly grouped by MIRU-VNTR typing, although again, in one case, one isolate (960345) had an additional IS6110 copy compared with the two other strains within the same group. Taking into account the time elapsed between the first and subsequent isolations within the same patient or transmission group, the MIRU-VNTR profiles were estimated to be stable for at least 18 months.
Resolution of MIRU Typing.
To compare the discriminatory power of MIRU-VNTR typing with the current standard, we performed a retrospective MIRU-VNTR analysis on frozen isolates collected over 1 year from three different Paris hospitals. All isolates were previously typed by spoligotyping and IS6110-RFLP, and for each strain epidemiological records were available (14, 24).
Forty-four isolates with no known epidemiological links were chosen within 12 groups (Table 2). The first group contained eight isolates containing one or two IS6110 copies. Six of them were not distinguished by their IS6110-RFLP profiles (24), reflecting poor resolution obtained with this method for typing strains that contain low copy numbers of IS6110. In contrast, all eight isolates were discriminated by unique MIRU-VNTR allelic combinations. This discrimination was similar to that obtained by spoligotyping (14).
Table 2.
MIRU-VNTR analysis of epidemiogically unrelated M. tuberculosis isolates from Paris hospitals
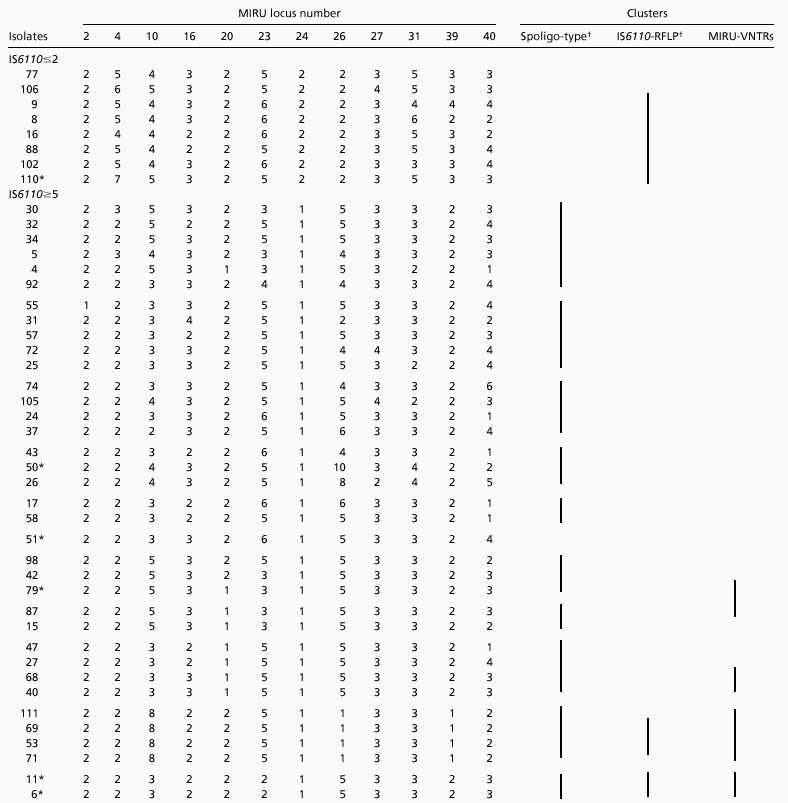 |
The other 36 isolates contained high (≥5) IS6110 copy numbers. On the basis of spoligotyping, these isolates were grouped into 11 different clusters. Almost all of the isolates could be distinguished from each other by IS6110-RFLP and MIRU typing. Only one group of two isolates (11 and 6) and one group of three isolates (69, 53, and 71) had identical IS6110-RFLP profiles within the groups (24). These strains also could not be distinguished by MIRU typing. A few isolates had identical MIRU-VNTR codes (isolate 40 was identical to 68, 79 to 87, and 111 to 69, 53, and 71), yet had distinct IS6110-RFLP profiles (see Table 2). Only two of them displayed also distinct spoligotypes (79 and 87). However, the isolates within these MIRU-VNTR clusters displayed very closely related IS6110 profiles (24). Taken together for all 44 isolates analyzed with no obvious epidemiological links, 19, 36, and 38 distinct patterns were obtained by spoligotyping, IS6110-RFLP and by MIRU-VNTR typing, respectively. When the results from this collection were combined with those obtained for the crosscontamination, transmission and relapse groups, only one additional MIRU-VNTR cluster was detected, which included Paris 34 and the crosscontamination cluster 971449/971450. The MIRU-VNTRs of isolates of one transmission group (950782/950867/960345) and one relapse group (960288/970856) also matched those of the Paris 79/87 MIRU-VNTR cluster.
When compared with each other, the 12 polymorphic MIRU loci did not display equal discrimination power. Some loci (20 and 24) contained only 2 alleles, whereas others contained up to 8 alleles (locus 26) among the strains analyzed. Moreover, within a given locus, some alleles were clearly much more frequent than others (for instance, the allele with two 77-bp MIRUs in locus 4 was encountered in 80% of the isolates, although 7 different alleles were identified in this locus). The allelic diversity detected by each locus was quantified, which allowed us to establish a hierarchy of the polymorphisms of the 12 loci (Table 3). The six loci with the highest diversity (loci 40, 10, 26, 23, 16, and 31) reached over 90% of the discriminatory power provided by all 12 loci. However, some strains could be distinguished only by a single locus with a very restricted allele distribution (for instance, isolates 47 and 58, and 42 and 79 by locus 20; see Table 2), which shows that the marginal effects of such loci can add significantly to the resolution power.
Table 3.
MIRU-VNTR allelic distribution among unrelated M. tuberculosis complex strains from Paris hospitals and from cross contamination, transmission, and relapse groups
| Locus | Number of isolates with the specified MIRU
copy number
|
Allelic diversity (h) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| 2 | 1 | 52 | 0.02 | ||||||||
| 4 | 1 | 42 | 2 | 1 | 5 | 1 | 1 | 0.35 | |||
| 10 | 1 | 22 | 12 | 14 | 4 | 0.69 | |||||
| 16 | 2 | 15 | 33 | 3 | 0.52 | ||||||
| 20 | 10 | 43 | 0.29 | ||||||||
| 23 | 2 | 9 | 1 | 32 | 9 | 0.58 | |||||
| 24 | 45 | 8 | 0.24 | ||||||||
| 26 | 5 | 10 | 5 | 27 | 3 | 1 | 1 | 1 | 0.67 | ||
| 27 | 3 | 47 | 3 | 0.19 | |||||||
| 31 | 3 | 41 | 3 | 5 | 1 | 0.37 | |||||
| 39 | 4 | 42 | 6 | 1 | 0.34 | ||||||
| 40 | 7 | 13 | 19 | 12 | 1 | 1 | 0.74 | ||||
MIRU-VNTR from one member of each cross contamination, transmission, and relapse group (see Table 1) was included in the analysis.
Correlation Between MIRU-VNTR Typing and IS6110-RFLP.
A dendrogram of the 44 isolates with no known epidemiological links of the Paris collection was built from Jaccard's genetic distance matrix calculated from the MIRU-VNTR data (Fig. 1). Interestingly, two main distant groups could be distinguished that correspond to low IS6110 copy number and high IS6110 copy number strains, respectively. When a dendrogram was built on the basis of IS6110-RFLP data from ref. 24 (not shown) and compared with the MIRU-VNTR dendrogram by using a nonparametric Mantel test (27, 28), the correlation between the two dendrograms (0.512) was found to be highly significant (P < 10−4).
Figure 1.

Dendrogram of genetic relationships among 44 M. tuberculosis isolates from Paris hospitals based on the 12 MIRU-VNTR loci. The dendrogram was built by using the Unweighted Pair-Group Method with Arithmetic Averages method and the Jaccard's genetic distance matrix, as described in Material and Methods. Linkage distance is indicated at the bottom.
Discussion
Micro- and minisatellites are powerful genetic tools for evolutionary as well as population genetic studies in higher eukaryotes (30–32). Although microsatellites also exist in many bacteria (17), their potential as genetic markers for molecular epidemiology has remained largely unexploited up to now. This fact is at least in part because of the hypervariability of many of these sequences, which blurs recognition between epidemiologically linked isolates (refs. 33 and 34; for review, see ref. 18). Bacterial minisatellites containing genetic elements named MIRUs (20) have recently been described (23). Here, we describe the use of a VNTR approach based on these minisatellites for high-resolution molecular epidemiology analyses of the major pathogen M. tuberculosis and the comparison of its performances with those of the current standard method based on IS6110-RFLP.
M. tuberculosis strains with identical IS6110 RFLP profiles isolated from different patients are generally considered to be clonal and to correspond to recent transmission events. Consistently, strains identified in outbreaks most often display the same IS6110-RFLP patterns, and population-based studies have revealed that clustering is associated with identified risk factors for transmission (e.g., refs. 9, 11, and 35). In this study, all epidemiologically linked isolates, resulting from either recent transmission or relapses, isolation from different anatomical sites of a same patient, or isolation coming from laboratory crosscontaminations, were perfectly matched by identical MIRU-VNTR alleles. Consistent with our recent analysis of the Mycobacterium bovis BCG vaccine phylogeny (23), these results indicate that the MIRU-VNTRs are remarkably stable and therefore adequate for tracking key events in epidemiological investigations. All strains with no known epidemiological links featuring identical IS6110-RFLP patterns also displayed identical MIRU-VNTR patterns, provided they contain at least five copies of IS6110. This concordance between these two markers with high resolving capacity suggests that the relevant patients were infected with the same strain, although no evidence of direct epidemiological links could be established from traditional investigations (24).
In contrast, a few isolates with identical MIRU-VNTR types had slightly distinct IS6110-RFLP patterns, although some of them were clearly epidemiologically linked. This observation suggests that the evolution rate of MIRU-VNTRs could be slightly slower than that of IS6110-RFLP. Gain or loss of one IS6110 copy in isolates from patients in case clusters has been reported in several studies (11, 35–38). More extensive changes in IS6110-RFLP were observed in strains that apparently underwent clonal expansion in the New York area within a few years (39). On the basis of these observations, it has been suggested that IS6110-RFLP may evolve too fast in certain strains to be reliably used to trace transmissions in outbreak investigations (38). The IS6110-RFLP evolution rate could be enhanced in such strains because of the effects of the local genomic context of specific IS6110 insertion sites on the transposition rate (40). Therefore, the equation “nonidentical IS6110-RFLP = unrelated case” is not always true and may lead to an underestimation of the epidemiological links between isolates (37). In contrast to the diversity of IS6110 insertion sites, the nature of the 12 loci containing the MIRU-VNTR is identical whichever strain is analyzed. Therefore, the rate of change of these VNTRs might be globally more stable among different strains, i.e., these markers might thus function as more regular molecular clocks than IS6110-RFLP. Together, these elements support that MIRU-VNTR typing may be particularly appropriate for long-term epidemiological analyses and may be used to reevaluate previously estimated degrees of transmission in populations (7, 9).
In addition to their stability, MIRU-VNTRs provide a high resolution essential to differentiate unrelated strains. In this study, the 12 minisatellite-like MIRU regions displayed each from 2 to 8 alleles, which provides a minimal theoretical potential to resolve over 16 million VNTR combinations. MIRU-VNTR typing performed considerably better than IS6110-RFLP when strains contained low copy numbers of IS6110, for which IS6110-RFLP is less useful (6, 14). Most of the unrelated strains distinguished by their high IS6110 copy number profiles were also discriminated by MIRU typing, with a few exceptions (see above). In a single case, two strains with distinct spoligotypes (79 and 87) were not resolved by MIRU-VNTRs. As our data indicate that even MIRU-loci with low resolving capacity can add significantly to the resolution power, the inclusion of some other identified VNTRs (19) might improve the resolution of these strains. For all 44 Paris strains analyzed, MIRU-VNTRs displayed an 86% discrimination level as compared with 82% for IS6110-RFLP and 85% when the results were combined with those of the crosscontamination, transmission, and relapse groups (this study) and 22 M. tuberculosis isolates of other geographic origins (23). Kremer et al. (6) recently reported a resolution level of 93% with IS6110-RFLP for 90 M. tuberculosis strains from different geographic origins. This set of strains was presumably more heterogeneous than the one used in our study, which contains many isolates collected in the same geographic area. Altogether, these results demonstrate that the discriminatory power of the 12 MIRU-VNTR regions is much higher than that of spoligotyping and close to that of IS6110-RFLP for typing of M. tuberculosis.
The correlation calculated between the genetic relationships among isolates inferred from MIRU-VNTR and IS6110-RFLP typing is statistically highly significant. The existence of IS6110 insertion hotspots, which could cause convergent evolution of IS6110-RFLP among unrelated isolates, has cast doubt on the appropriateness of IS6110 as a genetic marker for M. tuberculosis phylogenetic analyses (e.g., refs. 41–43). However, the correlation between these two independent methods supports the validity of phylogenetic analyses based on either marker alone. The MIRU-VNTR dendrogram also provides an independent indication that low IS6110 copy number strains constitute a group genetically distant from high copy number strains. This result is consistent with the observation that sites with IS6110 insertions in low copy number strains only rarely contain insertions in high copy number strains, suggesting separate lineages for these two groups of strains (44). Finally, the correlation between two independent sets of genetic markers is a striking case of linkage disequilibrium or nonrandom association of genotypes at different loci and provides therefore strong circumstantial evidence of clonal population structure within microbial populations (45). However, we cannot exclude a Wahlund effect bias of allopatry because of the cosmopolitan origin of many patients in the Paris Collection (24), which could generate artifactual linkage disequilibrium within the bacterial population considered (46). Future studies may address this important issue. MIRU-VNTR, unlike IS6110-RFLP, is well suited for such population genetic analyses, for it unambiguously reveals the variability of independent genetic loci, a requisite for linkage disequilibrium analysis.
MIRU-VNTR typing offers several other important advantages over IS6110-RFLP typing. Firstly, MIRU-VNTR typing is based on amplification by PCR and can be performed on mycobacterial colonies without extensive DNA purification. It can be applied to various biological materials, including nonviable material, permitting, as in this study, retrospective analyses of stocks of nonviable cells. Secondly, in addition to PCR amplification, it requires only sizing of the amplified fragments by gel electrophoresis with a resolution of about 50 bp (19–23) and should thus be broadly accessible for research and public health laboratories. The PCR conditions used in this study are very robust, as judged by the reproducible amplification of all 12 loci from all of the isolates analyzed to date (corresponding to more than 1,000 PCR amplifications). This approach is particularly suitable for the development of high-throughput automated analyses, by using multiplex PCR amplification combined with size analysis on a fluorescence-based sequencer. This will provide access to efficient large-scale genotyping, for which simple individual analyses of the 12 MIRU-VNTR loci are limiting. Thirdly, perhaps the most important advantage of MIRU-VNTR typing is the simple numerical format of the data generated (19, 23). As for multilocus sequence typing (15), this format is unambiguous and fully portable, which should allow laboratories in different parts of the world to compare their local isolates with those found elsewhere by submitting their VNTR data to a central database that can be created on a web site. On the basis of these advantages, we propose MIRU-VNTR typing as a new basis for global analysis of M. tuberculosis molecular epidemiology and population genetics.
Supplementary Material
Acknowledgments
We gratefully thank Yves Goguet de la Salmonière and Anne Varnerot for clinical isolates, Marc Lange for providing laboratory facilities, and Pablo Bifani for critical reading of the manuscript. This work was supported by Institut National de la Santé et de la Recherche Médicale, Institut Pasteur de Lille, Institut Pasteur à Paris, and by a grant from the Ministère de l'Education Nationale, de la Recherche et de la Technologie. P.S. is a Chercheur du Centre National de Recherche Scientifique.
Abbreviations
- RFLP
restriction fragment length polymorphism
- VNTR
variable number tandem repeats
- MIRUs
mycobacterial interspersed repetitive units
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Bloom B R, Murray C J L. Science. 1992;257:1055–1064. doi: 10.1126/science.257.5073.1055. [DOI] [PubMed] [Google Scholar]
- 2.World Health Organization. The World Health Report. Geneva: W.H.O.; 1998. [Google Scholar]
- 3.Grange J M, Zumla A. Lancet. 1999;353:996. doi: 10.1016/S0140-6736(99)01196-4. [DOI] [PubMed] [Google Scholar]
- 4.Gleissberg V. Lancet. 1999;353:998–999. doi: 10.1016/S0140-6736(99)01439-7. [DOI] [PubMed] [Google Scholar]
- 5.Van Helden P. In: Mycobacteria. Molecular Biology and Virulence. Ratledge C, Dale J, editors. Oxford: Blackwell; 1999. pp. 110–122. [Google Scholar]
- 6.Kremer K, van Soolingen D, Frothingham R, Haas W H, Hermans P W, Martin C, Palittapongarnpim P, Plikaytis B B, Riley L W, Yakrus M A, et al. J Clin Microbiol. 1999;37:2607–2618. doi: 10.1128/jcm.37.8.2607-2618.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Small P M, Hopewell P C, Singh S P, Paz A, Parsonnet J, Ruston D C, Schecter G F, Daley C L, Schoolnik G K. N Engl J Med. 1994;330:1703–1709. doi: 10.1056/NEJM199406163302402. [DOI] [PubMed] [Google Scholar]
- 8.van Soolingen D, Qian L, de Haas P E, Douglas J T, Traore H, Portaels F, Qing H Z, Enkhsaikan D, Nymadawa P, van Embden J D. J Clin Microbiol. 1995;33:3234–3238. doi: 10.1128/jcm.33.12.3234-3238.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Soolingen D, Borgdorff M W, de Haas P E, Sebek M M, Veen J, Dessens M, Kremer K, van Embden J D. J Infect Dis. 1999;180:726–736. doi: 10.1086/314930. [DOI] [PubMed] [Google Scholar]
- 10.Bifani P J, Mathema B, Liu Z, Moghazeh S L, Shopsin B, Tempalski B, Driscol J, Frothingham R, Musser J M, Alcabes P, Kreiswirth B N. J Am Med Assoc. 1999;282:2321–2327. doi: 10.1001/jama.282.24.2321. [DOI] [PubMed] [Google Scholar]
- 11.Yaganehdoost A, Graviss E A, Ross M W, Adams G J, Ramaswamy S, Wanger A, Frothingham R, Soini H, Musser J M. J Infect Dis. 1999;180:1245–1251. doi: 10.1086/314991. [DOI] [PubMed] [Google Scholar]
- 12.Dale J W, Nor R M, Ramayah S, Tang T H, Zainuddin Z F. J Clin Microbiol. 1999;37:1265–1268. doi: 10.1128/jcm.37.5.1265-1268.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kamerbeek J, Schouls L, Kolk A, van Agterveld M, van Soolingen D, Kuijper S, Bunschoten A, Molhuizen H, Shaw R, Goyal M, van Embden J. J Clin Microbiol. 1997;35:907–914. doi: 10.1128/jcm.35.4.907-914.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goguet de la Salmoniere Y O, Li H M, Torrea G, Bunschoten A, van Embden J, Gicquel B. J Clin Microbiol. 1997;35:2210–2214. doi: 10.1128/jcm.35.9.2210-2214.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maiden M C, Bygraves J A, Feil E, Morelli G, Russell J E, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant D A, et al. Proc Natl Acad Sci USA. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sreevatsan S, Pan X, Stockbauer K E, Connell N D, Kreiswirth B N, Whittam T S, Musser J M. Proc Natl Acad Sci USA. 1997;94:9869–9874. doi: 10.1073/pnas.94.18.9869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Belkum A, Scherer S, van Alphen L, Verbrugh H. Microbiol Mol Biol Rev. 1998;62:275–293. doi: 10.1128/mmbr.62.2.275-293.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van Belkum A. Curr Opin Microbiol. 1999;2:306–311. doi: 10.1016/S1369-5274(99)80053-8. [DOI] [PubMed] [Google Scholar]
- 19.Frothingham R, Meeker-O'Connell W A. Microbiology. 1998;144:1189–1196. doi: 10.1099/00221287-144-5-1189. [DOI] [PubMed] [Google Scholar]
- 20.Supply P, Magdalena J, Himpens S, Locht C. Mol Microbiol. 1997;26:991–1003. doi: 10.1046/j.1365-2958.1997.6361999.x. [DOI] [PubMed] [Google Scholar]
- 21.Magdalena J, Vachee A, Supply P, Locht C. J Clin Microbiol. 1998;36:937–943. doi: 10.1128/jcm.36.4.937-943.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Magdalena J, Supply P, Locht C. J Clin Microbiol. 1998;36:2471–2476. doi: 10.1128/jcm.36.9.2471-2476.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Supply P, Mazars E, Lesjean S, Vincent V, Gicquel B, Locht C. Mol Microbiol. 2000;36:762–771. doi: 10.1046/j.1365-2958.2000.01905.x. [DOI] [PubMed] [Google Scholar]
- 24.Torrea G, Offredo C, Simonet M, Gicquel B, Berche P, Pierre-Audigier C. J Clin Microbiol. 1996;34:1043–1049. doi: 10.1128/jcm.34.5.1043-1049.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jaccard P. Bull Soc Vaudoise Sci Nat. 1908;44:223–270. [Google Scholar]
- 26.Sneath P H A, Sokal R R. Numerical Taxonomy. San Francisco: Freeman; 1973. [Google Scholar]
- 27.Mantel N. Cancer Res. 1967;27:209–220. [PubMed] [Google Scholar]
- 28.Tibayrenc M. C R Acad Sci III. 1995;318:135–139. [PubMed] [Google Scholar]
- 29.Selander R K, Caugant D A, Ochman H, Musser J M, Gilmour M N, Whittam T S. Appl Environ Microbiol. 1986;51:873–884. doi: 10.1128/aem.51.5.873-884.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeffreys A J, MacLeod A, Tamaki K, Neil D L, Monckton D G. Nature (London) 1991;354:204–209. doi: 10.1038/354204a0. [DOI] [PubMed] [Google Scholar]
- 31.Sutherland G R, Richards R I. N Engl J Med. 1994;331:191–193. doi: 10.1056/NEJM199407213310310. [DOI] [PubMed] [Google Scholar]
- 32.Epplen C, Santos E J, Maueler W, van Helden P, Epplen J T. Electrophoresis. 1997;18:1577–1585. doi: 10.1002/elps.1150180916. [DOI] [PubMed] [Google Scholar]
- 33.van Belkum A, Scherer S, van Leeuwen W, Willemse D, van Alphen L, Verbrugh H. Infect Immun. 1997;65:5017–5027. doi: 10.1128/iai.65.12.5017-5027.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Renders N, Licciardello L, C, I J, Sijmons M, van Alphen L, Verbrugh H, van Belkum A. FEMS Microbiol Lett. 1999;173:95–102. doi: 10.1111/j.1574-6968.1999.tb13489.x. [DOI] [PubMed] [Google Scholar]
- 35.de Boer A S, Borgdorff M W, de Haas P E, Nagelkerke N J, van Embden J D, van Soolingen D. J Infect Dis. 1999;180:1238–1244. doi: 10.1086/314979. [DOI] [PubMed] [Google Scholar]
- 36.Cave M D, Eisenach K D, Templeton G, Salfinger M, Mazurek G, Bates J H, Crawford J T. J Clin Microbiol. 1994;32:262–266. doi: 10.1128/jcm.32.1.262-266.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yeh R W, Ponce de Leon A, Agasino C B, Hahn J A, Daley C L, Hopewell P C, Small P M. J Infect Dis. 1998;177:1107–1111. doi: 10.1086/517406. [DOI] [PubMed] [Google Scholar]
- 38.Alito A, Morcillo N, Scipioni S, Dolmann A, Romano M I, Cataldi A, van Soolingen D. J Clin Microbiol. 1999;37:788–791. doi: 10.1128/jcm.37.3.788-791.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bifani P J, Plikaytis B B, Kapur V, Stockbauer K, Pan X, Lutfey M L, Moghazeh S L, Eisner W, Daniel T M, Kaplan M H, et al. J Am Med Assoc. 1996;275:452–457. [PubMed] [Google Scholar]
- 40.Wall S, Ghanekar K, McFadden J, Dale J W. Microbiology. 1999;145:3169–3176. doi: 10.1099/00221287-145-11-3169. [DOI] [PubMed] [Google Scholar]
- 41.Fang Z, Forbes K J. J Clin Microbiol. 1997;35:479–481. doi: 10.1128/jcm.35.2.479-481.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McHugh T D, Gillespie S H. J Clin Microbiol. 1998;36:1410–1413. doi: 10.1128/jcm.36.5.1410-1413.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fang Z, Morrison N, Watt B, Doig C, Forbes K J. J Bacteriol. 1998;180:2102–2109. doi: 10.1128/jb.180.8.2102-2109.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fomukong N, Beggs M, el Hajj H, Templeton G, Eisenach K, Cave M D. Tuber Lung Dis. 1997;78:109–116. doi: 10.1016/s0962-8479(98)80003-8. [DOI] [PubMed] [Google Scholar]
- 45.Tibayrenc M, Kjellberg F, Ayala F J. Proc Natl Acad Sci USA. 1990;87:2414–2418. doi: 10.1073/pnas.87.7.2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tibayrenc M. Annu Rev Genet. 1999;33:449–477. doi: 10.1146/annurev.genet.33.1.449. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.