

Abstract
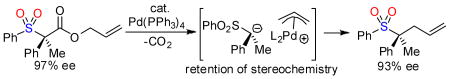
Allyl sulfonyl acetic esters undergo highly stereospecific, palladium-catalyzed decarboxylative allylation. The reaction allows the stereospecific formation of tertiary homoallylic sulfones in high yield. In contrast to related reactions that proceed at -100 °C and require highly basic preformed organometallics, the decarboxylative coupling described herein occurs under mild non-basic conditions and requires no stoichiometric additives. Allylation of the intermediate α-sulfonyl anion is more rapid than racemization, leading to a highly enantiospecific process. DFT calculations indicate that the barrier for racemization is 9.9 kcal/mol and thus the barrier of allylation must be <9.9 kcal/mol.
Catalytic decarboxylative coupling is a powerful method for C–C bond formation that avoids the use of strong bases and/or preformed organometallics that are typically required for cross-coupling reactions. In 2004, we reported the first asymmetric decarboxylative allylation of enolates.1 This was followed by several reports of decarboxylative enolate allylations that allow enantioselective formation of quaternary carbon centers α-to ketones.2 Since these reactions proceed through achiral enolate intermediates the reactions are stereoconvergent, allowing racemic starting materials to form highly enantioenriched products (eq. 1). Stereoconvergence is a common reaction profile in carbanion chemistry, where resonance-stabilized carbanions often exist as planar achiral intermediates and unstabilized carbanions rapidly racemize by inversion.3 Herein we report that optically active sulfonyl acetic esters undergo enantiospecific decarboxylative coupling, where the original stereochemistry is maintained in the product (eq. 2). Furthermore, DFT calculations are used to examine the origin of the enantiospecificity.
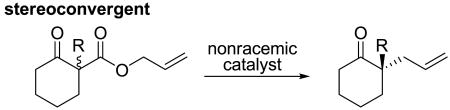 |
(1) |
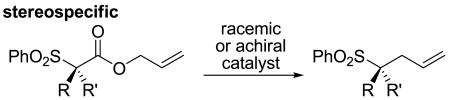 |
(2) |
From the elegant work of Corey and Cram in the early 1960's, it was known that the decarboxylative protonation of sulfonyl acetic esters is stereospecific.4 While there are several examples of stereospecific decarboxylative protonations,4,5 none has been extended to C–C bond forming reactions. With this in mind, we set out to develop a stereospecific decarboxylative C–C bond forming reaction.
To begin, sulfone 1a was synthesized and treated with 2 mol% Pd(PPh3)4 in toluene at room temperature (eq. 3).6 It was gratifying to find that the decarboxylative allylation was highly stereospecific, with a conservation of enantiomeric excess (cee) of 96 % (cee = 100 × (product ee)/(reactant ee)).
 |
(3) |
Next, a variety of different enantioenriched sulfones were similarly subjected to conditions for palladium-catalyzed decarboxylative coupling. As can be seen from the data in table 1, a variety of α-aryl and α,α-dialkyl sulfones all undergo decarboxylative allylation with a high degree of stereospecificity.
Table 1.
Stereospecificities of decarboxylation allylation
| entry | reactant ee(%)c | product | procedurea | yield (%)b | ee(%)c | cee (%)c |
|---|---|---|---|---|---|---|
| 1 | 46 | 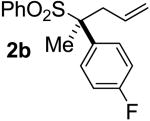 |
A | 96 | 43 | 91 |
| 2 | 89 | 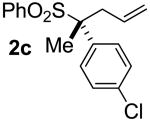 |
A | 99 | 87 | 98 |
| 3 | 97 | 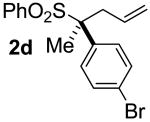 |
A | 99 | 96 | 99 |
| 4 | 61 | 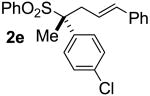 |
A | 99 | 61 | 99d |
| 5 | 73 | 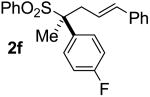 |
A | 99 | 69 | 96d |
| 6 | 97 | 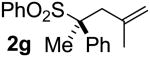 |
A | 99 | 93 | 96 |
| 7 | 80 | 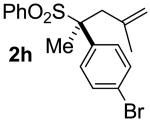 |
A | 99 | 80 | 99 |
| 8 | 94 | 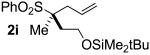 |
B | 82 | 92 | 99 |
| 9 | 94 | 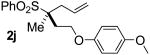 |
B | 95 | 92 | 98 |
| 10 | >99 | 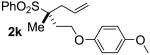 |
B | 97 | >99 | >99 |
| 11 | 98 | 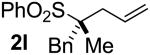 |
B | 85 | 95 | 97 |
| 12 | 98 | 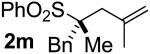 |
B | 93 | 95 | 97 |
Conditions A: 2 mol % Pd(PPh3)4, 0.2 M toluene, 23 °C, 0.25-2 h. Conditions B: 5 mol % Pd2dba3, 10 mol % (±)-binap, 0.2 M toluene, 95 °C, 11-15 h.
isolated yields.
determined via chiral stationary phase HPLC analysis
8:1 linear:branched
It is noteworthy that high cee's are obtained even at elevated temperatures (95 oC). Moreover, our reaction proceeds under formally neutral conditions, in contrast to more classical alkylations of sulfones which typically require stoichiometric, highly basic, organolithium reagents.7 Lastly, an X-ray crystal structure of a derivative of 2k confirms that the allylation proceeds with overall retention of configuration.8
The overall retention of stereochemistry in the decarboxylative allylation can be explained if decarboxylation directly produces either a configurationally stable alkyl palladium complex that undergoes reductive elimination (path 1, Scheme 1) or if decarboxylation produces a configurationally stable α-sulfonyl anion that attacks the palladium π-allyl complex (path 2, Scheme 1).9
Scheme 1.

To begin addressing whether path 1 or 2 (Scheme 1) is operative, we investigated the effect of palladium on the rates of decarboxylation of several α-sulfonyl acetates. Control studies show that the rate of decarboxylation is relatively unaffected by the addition of a variety of palladium sources (Table 2).8 For example, the cesium or triethylammonium salts of α-sulfonyl acetic acids readily decarboxylate under our reaction conditions. Addition of Pd(OAc)2 or (Ph3P)2Pd(allyl)Cl to these reaction mixtures does not substantially affect the rate of decarboxylation.10 When palladium is directly involved in the decarboxylation, such experiments usually show a dramatic acceleration of the decarboxylation.11 Ultimately, Corey and Cram have detailed thermal decarboxylations of α-sulfonyl acetates under similar conditions to those used by us,4 and we have failed to observe catalysis of decarboxylation by palladium. Thus, the simplest conclusion is that decarboxylation of the α-sulfonyl carboxylates is a thermal process that proceeds via path 2.
Table 2.
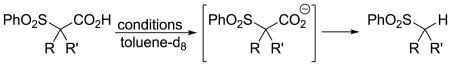 | |||||
|---|---|---|---|---|---|
| R | R' | conditions | Pd source | time | % conv. |
| H | H | 95 °C, Et3N | none | 5 min. | 17% |
| H | H | 95 °C, Et3N | Pd(allyl)Cl(PPh3)2 100 mol% | 5 min. | 23% |
| Me | Bn | 95 °C, Et3N | none | 45 min. | 25% |
| Me | Bn | 95 °C, Et3N | Pd(OAc)2 10 mol% | 45 min. | 27% |
| Me | H | 23 °C, Cs2CO3 | none | 36 h | 59% |
| Me | H | 23 °C, Cs2CO3 | Pd(OAc)2 10 mol% | 36 h | 59% |
Reaction via path 1 and path 2 can also be distinguished via the observed diastereoselectivity in nucleophilic attack of a stereochemically labeled allyl ester substrate (Scheme 2).10 In such a reaction, nucleophiles that are bound to palladium prior to reductive elimination, as in path 1, react with overall inversion of configuration. Alternatively, if the free nucleophile directly attacks the allyl ligand, as in path 2, then one observes retention of stereochemistry by way of the standard double-inversion mechanism.12 Toward this end, substrate 1n was prepared and allowed to react under our standard reaction conditions (Scheme 2).13 The reaction produced the 1,3-cis diastereomers as the only C–C bond forming product in 49% yield,14 suggesting that the decarboxylative coupling proceeds via attack of an α-sulfonyl anion on the backside of palladium π-allyl complex (path 2, Scheme 2). While we could not generate the α-chloro-α-sulfonyl ester in enantioenriched form in order to test the enantiospecificity of the coupling, the product is formed as a 1:1 mixture of diastereomers at the sulfonyl stereocenter. Thus, the stereochemistry of the α-position did not change during the reaction, which is consistent with a stereospecific process.
Scheme 2.

Our control studies and stereochemical studies both suggest that α-sulfonyl anions are formed, and react, outside the coordination sphere of palladium. In order to form highly enantioenriched products, these intermediate α-sulfonyl anions must be configurationally stable. While it is known that α-sulfonyl anions are more configurationally stable than typical carbanions, Gais has reported the rapid racemization of anion 3a at -80 °C.15 Thus, we became curious why our reaction was highly stereospecific, even at elevated temperatures.
To address the reasons for the apparently slower rate of racemization than coupling of the α-sulfonyl anion and Pd-π-allyl, DFT calculations were performed to determine the inversion barrier for the free α-sulfonyl carbanion. The calculations revealed that the most stable conformation is 3a, which has the major lobe of the lone-pair oriented anti-periplanar to the S–Ph bond (eq. 4). A definitive number for the inversion barrier could not be determined because all attempts to minimize the energy of conformation 3b led to convergence to the minimum energy structure (3a). While we could not obtain a specific energy for structure 3b, this result does support the conclusion that the barrier to inversion is small (< 2 kcal/mol). The small inversion barrier is supported by other studies, as is the slightly pyramidal structure of the carbanion.15-17
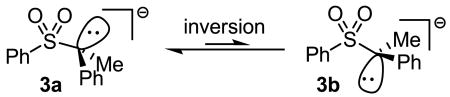 |
(4) |
Taking into account the small barrier to inversion, our mechanistic hypothesis is as follows. Ionization and decarboxylation of 1a generates sulfonyl anion, 3a which is likely ion-paired with the Pd-π-allyl (Scheme 3). The C–C formation proceeds via path A, where the carbanion can react through the low-energy, staggered, transition state to form the product with retention of stereochemistry. Coupling through the inverted conformer, 3b (path B) to form the enantiomeric product not only requires the anion to react through a higher energy conformer, but also requires a higher energy transition state to form the eclipsed product.4 Alternatively, the enantiomer, ent-2a, could be formed by allylation of the anion that has undergone inversion and rotation (path C). Thus, we hypothesized that the reaction is stereospecific because rotation about the C–S bond of the sulfone (path C) is slower than reaction of the α-sulfonyl anion, 3a, with the palladium π-allyl complex through path A.
Scheme 3.

DFT calculations carried out at the B3LYP/6-31+G* level of theory reveal that the lowest barrier to rotation about the αC–S bond is 9.9 kcal/mol, which is consistent with experimental measurements in related systems (Scheme 4).15-17 Thus, the barrier for allylation of the α-sulfonyl anion must be < 9.9 kcal/mol or rotation would lead to racemization. Such a low barrier is consistent with the reaction between an ion-pair of a highly nucleophilic α-sulfonyl anion and a palladium π-allyl complex.8,18 Thus, the stark contrast in our observations and that of Gais can be attributed to the fact that we are generating the reactive α-sulfonyl anion intermediates in the presence of highly electrophilic π-allyl palladium electrophiles, allowing us to effect the C–C bond formation faster than rotation.
Scheme 4.

In conclusion, we have developed a stereospecific decarboxylative coupling reaction that provides access to enantioenriched homoallylic sulfones. The enantiospecific nature of this reaction is unique among, yet complimentary to, existing decarboxylative allylation methodologies. The stereospecificity is made possible by the relatively high barrier for racemization of axially chiral α-sulfonyl anions and the low barrier for their reaction with palladium π-allyl complexes.
Supplementary Material
Acknowledgments
We thank the National Institute of General Medical Sciences (1R01GM079644) for financial support. We thank Dr. Victor Day for X-ray crystallographic analysis.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Burger EC, Tunge JA. Org Lett. 2004;6:4113. doi: 10.1021/ol048149t. [DOI] [PubMed] [Google Scholar]
- 2.(a) Trost BM, Xu J. J Am Chem Soc. 2005;127:17180. doi: 10.1021/ja055968f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Burger EC, Barron BR, Tunge JA. Synlett. 2006:2824. [Google Scholar]; (c) Trost BM, Bream RN, Xu J. Angew Chem, Int Ed. 2006;45:3109. doi: 10.1002/anie.200504421. [DOI] [PubMed] [Google Scholar]; (d) You SL, Dai LX. Angew Chem, Int Ed. 2006;45:5246. doi: 10.1002/anie.200601889. [DOI] [PubMed] [Google Scholar]; (e) Trost BM, Xu J, Schmidt T. J Am Chem Soc. 2009;131:18343. doi: 10.1021/ja9053948. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Behenna DC, Stoltz BM. J Am Chem Soc. 2004;126:15044. doi: 10.1021/ja044812x. [DOI] [PubMed] [Google Scholar]; (g) Nakamura M, Hajra A, Endo K, Nakamura E. Angew Chem, Int Ed. 2005;44:7248. doi: 10.1002/anie.200502703. [DOI] [PubMed] [Google Scholar]; (h) Mohr JT, Behenna DC, Harned AM, Stoltz BM. Angew Chem, Int Ed. 2005;44:6924. doi: 10.1002/anie.200502018. [DOI] [PubMed] [Google Scholar]
- 3.Baechler RD, Andose JD, Stackhouse J, Mislow K. J Am Chem Soc. 1972;94:8060. [Google Scholar]
- 4.(a) Corey EJ, Kaiser ET. J Am Chem Soc. 1961;83:490. [Google Scholar]; (b) Corey EJ, Koenig H, Lowry TH. Tetrahedron Lett. 1962:515. [Google Scholar]; (c) Corey EJ, Lowry TH. Tetrahedron Lett. 1965:803. [Google Scholar]; (d) Corey EJ, Lowry TH. Tetrahedron Lett. 1965:793. [Google Scholar]; (e) Cram DJ, Nielsen WD, Rickborn B. J Am Chem Soc. 1960;82:6415. [Google Scholar]; (f) Cram DJ, Rickborn B, Knox GR. J Am Chem Soc. 1960;82:6412. [Google Scholar]; (g) Cram DJ, Wingrove AS. J Am Chem Soc. 1963;85:1100. [Google Scholar]
- 5.Rozov LA, Rafalko PW, Evans SM, Brockunier L, Ramig K. J Org Chem. 1995;60:1319. [Google Scholar]
- 6.(a) Weaver JD, Morris DK, Tunge JA. Synlett. 2010:470. doi: 10.1055/s-0029-1219186. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Weaver JD, Tunge JA. Org Lett. 2008;10:4657. doi: 10.1021/ol801951e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gais HJ, Hellmann G. J Am Chem Soc. 1992;114:4439. [Google Scholar]
- 8.See supporting information for more details.
- 9.Complete crossover is observed when two sulfonyl esters are allowed to undergo coupling in the same flask. The interpretation of complete crossover is ambiguous since crossover can likely occur via either the intermediate π-allyl palladium carboxylates or the π-allyl palladium α-sulfonyl anion complexes.
- 10.The presence of acidic protons from the carboxylic acid results in the formation of the protonation product rather than the allylation product that may be expected from (allyl)PdCl(PPh3)2
- 11.Recio A, Tunge JA. Org Lett. 2009;11:5630. doi: 10.1021/ol902065p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) Trost BM, Verhoeven TR. J Am Chem Soc. 1980;102:4730. doi: 10.1021/ja00418a063. [DOI] [PubMed] [Google Scholar]; (b) Keinan E, Roth Z. J Org Chem. 1983;48:1770. [Google Scholar]
- 13.Unfortunately, the α-methyl,α-phenyl sulfonyl acetic ester of cyclohexen-3-ol gives 78% elimination product. Thus, it could not be used for the stereochemical test. Elimination is less problematic with an α-chloro substitutent see footnote 6b.
- 14.Analysis of the crude reaction mixture by 1H NMR spectroscopy indicates that the mass balance of the decarboxylative allylation of 1n is made up of elimination products.
- 15.Gais HJ, Hellmann G, Gunther H, Lopez F, Lindner HJ, Braun S. Angew Chem Int Ed. 1989;28:1025. [Google Scholar]
- 16.(a) Raabe G, Gais HJ, Fleischhauer J. J Am Chem Soc. 1996;118:4622. [Google Scholar]; (b) Reetz MT, Hutte S, Goddard R. Eur J Org Chem. 1999:2475. [Google Scholar]
- 17.This barrier to rotation is, in part, due to loss of negative hyperconjugation with the Ph-S σ* orbital.; Koch R, Anders E. J Org Chem. 1994;59:4529. [Google Scholar]
- 18.(a) Seeliger F, Mayr H. Org Biomol Chem. 2008;6:3052. doi: 10.1039/b805604h. [DOI] [PubMed] [Google Scholar]; (b) Kuhn O, Mayr H. Angew Chem Int Ed. 1999;38:343. doi: 10.1002/(SICI)1521-3773(19990201)38:3<343::AID-ANIE343>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.