

Abstract
Adenosine deaminases acting on RNA (ADARs) catalyse hyper-editing of long dsRNAs, whereby up to 50% of adenosines are converted to inosine (I). While hyper-edited dsRNAs (IU-dsRNAs) have been implicated in various cellular functions, we now provide evidence suggesting a novel role. We show that IU-dsRNA suppresses induction of interferon-stimulated genes (ISGs) and apoptosis in response to poly(IC). Moreover, we demonstrate that IU-dsRNA inhibits activation of IRF3, which is essential for induction of ISGs and apoptosis. Finally, we speculate that IRF3 inhibition results from specific binding of IU-dsRNA to MDA-5 or RIG-I, cytosolic sensors for poly(IC). While our data are consistent with a previous study in which ADAR1 deletion resulted in increased expression of ISGs and apoptosis, we show that IU-dsRNA per se suppresses ISGs and apoptosis. We therefore propose that any IU-dsRNA generated by ADAR1 can inhibit both pathways.
Keywords: ADAR, dsRNA, Editing, Interferon, Apoptosis, IRF3, MDA-5, RIG-I
Adenosine deaminases acting on RNA (ADARs) catalyze the deamination of adenosine (A) to inosine (I) within double-stranded RNA (dsRNA)1,2. ADARs have been characterized throughout the metazoa, although the number of ADAR genes expressed in each organism differs. Three ADARs have been described in mammals (ADAR1–3), although only ADAR1 and ADAR2 appear to be catalytically active. The importance of ADARs in post-transcriptional gene regulation has been demonstrated by analysis of ADAR-null mutants1,2.
While A-to-I editing can occur selectively within mRNA, hyper-editing of long dsRNA can result in up to 50% of A residues being changed to I. As I is decoded as guanosine by ribosomes, A-to-I editing can result in codon changes. Localized changes in RNA structure are also likely within hyper-edited inosine-containing dsRNAs (IU-dsRNAs), as IU pairs are weaker than conventional base pairs3. Most mammalian editing occurs within non-coding regions of RNA, including repetitive elements such as inverted Alus4-7. However, while countless RNAs may be extensively edited, the possible functions of IU-dsRNA in cells are not fully understood. Various studies have suggested diverse fates for IU-dsRNA8-13.
Two major isoforms of ADAR1 have been described in mammalian cells. A constitutively expressed truncated ADAR1 protein (p110) localizes to the nucleus, while full-length interferon-inducible ADAR1 (p150)14 shuttles between the nucleus and cytoplasm15. ADAR1 is essential, as demonstrated by death of ADAR1-null mice at ~E11.5 due to defective hematopoiesis and widespread apoptosis16,17. Recent findings can account for these observations. ADAR1 is indispensable for maintenance of hematopoietic stem cells in fetal liver and adult bone marrow17. In addition, ADAR1 suppressed activation of interferon-stimulated genes (ISGs), thereby protecting against premature apoptosis17. Several ideas were put forward to explain how ADAR1 might regulate interferon (IFN) signaling. Editing of crucial, as yet unidentified transcripts by ADAR1 may be important. A complex needed for IFN regulation may be disrupted in the absence of ADAR1. Alternatively, the lack of editing in ADAR1-deficient cells may give rise to immunoreactive dsRNA.
Here we present experimental data that supports an alternative explanation for how ADAR1 regulates IFN signaling. We provide evidence that IU-dsRNA is sufficient per se to suppress activation of ISGs. When human cells (HeLa) were transfected with short dsRNAs containing multiple IU pairs, induction of ISGs by long dsRNA was suppressed. Microarrays confirmed that suppression of gene expression by IU-dsRNA was largely restricted to genes involved in immunity and defense. We also showed that IU-dsRNA inhibits apoptosis induced by long dsRNA. Both suppressive effects mediated by IU-dsRNA could be accounted for by our observation that IU-dsRNA inhibits activation of IRF3 (IFN regulatory factor 3), a key component in the pathway by which long dsRNA induces ISGs and apoptosis18. Moreover, our data suggests that IU-dsRNA acts at an early step in the pathway by specifically inhibiting MDA-5 (melanoma differentiation-associated protein 5) or RIG-I (retinoic acid-inducible gene I), the cytosolic sensors for dsRNA19. These observations together lead us to propose that any IU-dsRNA generated by editing can directly inhibit IFN induction and apoptosis.
Results
IU-dsRNA does not induce an IFN response
We previously used short model dsRNAs to show that IU-dsRNA in HeLa cells downregulated both endogenous and reporter gene expression13. In addition, we showed that IU-dsRNA binds a complex that comprises stress-granule (SG) components13. SGs function during cellular stress to allow selective synthesis of proteins needed for survival20. In considering how IU-dsRNA downregulates gene expression, we speculated that IU-dsRNA might elicit an IFN response. Although IFN is typically induced by long dsRNAs, it is possible that IU-dsRNA in cells signifies stress and induces IFN. Induction of IFN would activate a signaling cascade, which culminates in transcription of hundreds of ISGs that function in cellular stress response pathways21. We therefore tested whether IU-dsRNA in HeLa cells triggered an IFN response.
HeLa cells were transfected with control (C) or IU-dsRNA (C-IU) duplexes (Table 1), with or without Firefly luciferase (Fluc) mRNA. Inclusion of Fluc mRNA enabled the effect of IU-dsRNA on reporter gene expression to be monitored (data not shown). C and C-IU were identical except for the four central base pairs; the control dsRNA (C) consisted of Watson-Crick base pairs, while C-IU contained IU pairs. Cells were harvested 6 or 12h post-transfection, and reverse transcription (RT) and quantitative PCR (qPCR) were used to quantify expression of various ISGs (Fig. 1a). The ISGs tested corresponded to a subset of those upregulated by IFN treatment or ADAR1 deficiency17. Expression of β-actin was also analyzed. Fold-change in mRNA levels at 12h were calculated relative to those at 6h with control dsRNA, and normalized to GapDH. At 6h post-transfection no induction of the ISGs was observed (data not shown). Unless stated otherwise, fold-change was calculated in the same way for subsequent experiments. For all genes tested, expression did not change when C or C-IU were used to transfect cells in the absence of Fluc mRNA (Fig. 1a). In contrast, expression of all ISGs tested was substantially higher in the presence of Fluc mRNA and C dsRNA (Fig. 1a). A significantly smaller increase was seen with C-IU and Fluc mRNA. β-actin remained constant. These data suggested that Fluc mRNA caused induction of the ISGs, and that IU-dsRNA suppressed the response.
Table 1. dsRNA sequences.
| Substrate | Sequence |
|---|---|
| GP | ACUGGACAGGUGCUCCGAGG |
| UGACCUGUCCACGAGGCUCC | |
| IIUI | ACUGGACAIIUICUCCGAGG |
| UGACCUGUUUIUGAGGCUCC | |
| IUI | ACUGGACAAIUICUCCGAGG |
| UGACCUGUUUIUGAGGCUCC | |
| UI | ACUGGACAAAUICUCCGAGG |
| UGACCUGUUUIUGAGGCUCC | |
| GGUG | ACUGGACAGGUGCUCCGAGG |
| UGACCUGUUUGUGAGGCUCC | |
| IC | CUGGACAIIUIUUCUCCGAG |
| UGACCUGUCCACGAGGCUCC | |
| 6I | CUGGACAIIUIUUCUCCGAG |
| GACCUGUUUIUIIGAGGCUC | |
| C | GGUCCGGCUCCCCCAAAUGdTdT |
| dTdTCCAGGCCGAGGGGGUUUAC | |
| C-IU | GGUCCGGCIIUICCAAAUGdTdT |
| dTdTCCAGGCCGUUIUGGUUUAC | |
|
miR-142
(142) |
A |
| CAUAAAGUAG AAGCACUAC | |
| GGUAUUUCAUC UUUGUGAUGU | |
| C | |
|
miR-142-IU
(142-IU) |
A |
| CAUIIIGUIG IAGCACUIC | |
| GGUIUUUCIUC UUUGUGIUGU | |
| C | |
| Bio-C | Bio-GGUCCGGCUCCCCCAAAUGdTdT |
| dTdTCCAGGCCGAGGGGGUUUAC | |
| Bio-C-IU | Bio-GGUCCGGCIIUICCAAAUGdTdT |
| dTdTCCAGGCCGUUIUGGUUUAC |
Base pairs that differ between the pairs of dsRNAs are in bold. Biotinylated (Bio) dsRNAs are indicated.
Figure 1. IU-dsRNA suppressed induction of ISGs.
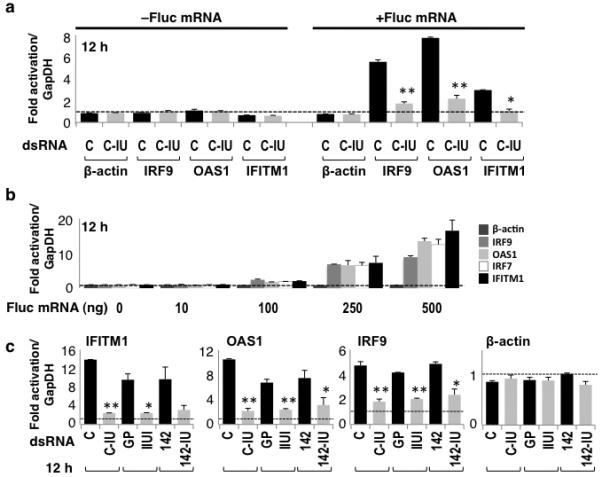
(a) HeLa cells were co-transfected with C or C-IU dsRNAs, ±Fluc mRNA. RT/qPCR was used to quantify expression of β-actin or ISGs (IRF9, OAS1, IFITM1; Supplementary Table 1) after 12h. Fold-change in mRNA was relative to that at 6h with C, and normalized to GapDH (n=4; P values = 0.001 (*) or <5×10−4 (**)). (b) HeLa cells were transfected with 0–500 ng Fluc mRNA. RT/qPCR was used to quantify expression of β-actin or ISGs (IRF9, OAS1, IRF7, IFITM1; Supplementary Table 1) (n=4). Fold-change in mRNA was relative to that without mRNA, and normalized to GapDH. (c) HeLa cells were co-transfected with Fluc mRNA and either control dsRNAs (C, GP, or 142) or IU-dsRNAs (C-IU, IIUI, or 142-IU), respectively. RT/qPCR was used to quantify expression of β-actin or ISGs (IFITM1, OAS1, IRF9; Supplementary Table 1) after 12h (n=4). Fold-change in mRNA levels were calculated relative to those at 6h with GP, and normalized to GapDH. P values were <5×10−3 (*) or <1×10−3 (**). All error bars are mean ± s.d.
To verify that upregulation of ISGs was due to Fluc mRNA, HeLa cells were transfected with Fluc mRNA alone. Fold-change in gene expression was analyzed after 12h using RT/qPCR, relative to that seen without Fluc mRNA (Fig. 1b). With increasing concentrations of Fluc mRNA, a corresponding increase in ISG expression was observed. β-actin was unchanged. These data confirmed that Fluc mRNA in HeLa cells induced ISGs. It was possible that this was due to contamination of the Fluc mRNA with a small amount of dsRNA, as reported previously22. Alternatively, any uncapped mRNA present in the in vitro transcribed preparation of ‘capped’ Fluc mRNA could activate an IFN response via interaction with RIG-I, which responds to 5′-triphosphate ssRNA19. Analysis of the Fluc mRNA 5′-end confirmed that a proportion of the RNA was uncapped, consistent with inefficient in vitro capping23 (Supplementary Fig. 1a). Moreover, RT/qPCR confirmed that RIG-I expression was induced by either ‘capped’ or uncapped Fluc mRNA (Supplementary Figs. 1b, 1c). Importantly, these data also showed that C-IU suppressed induction of ISGs when uncapped RNA was used to trigger the immune response.
These experiments conclusively demonstrated that IU-dsRNA in HeLa cells did not induce an IFN response. On the contrary, they unexpectedly showed that IU-dsRNA suppressed induction of ISGs.
Different IU-dsRNAs suppressed the IFN response
To ensure that the observed effect was unrelated to the sequence of the short dsRNAs, two additional pairs of duplexes, GP and IIUI, and miR-142 (142) and miR-142-IU (142-IU) (Table 1) were used to co-transfect HeLa cells with Fluc mRNA. For each pair, the dsRNAs were identical except for the presence or absence of IU pairs. Again, expression of β-actin and various ISGs was analyzed after 12h using RT/qPCR (Fig. 1c). This showed that expression of all ISGs tested was considerably higher with control dsRNAs (C, GP, 142) but not with the corresponding IU-dsRNA (C-IU, IIUI, 142-IU). β-actin was constant. These data therefore supported the idea that IU-dsRNA suppressed induction of ISGs, and showed that suppression occurred in a sequence-independent manner.
IU-dsRNA suppressed ISG induction by poly(IC)
IU-dsRNA inhibited induction of ISGs by Fluc mRNA. We next tested whether IU-dsRNA similarly suppressed induction of ISGs in response to poly(IC), which effectively mimics long dsRNA.
We initially analyzed induction of ISGs in HeLa cells transfected with increasing amounts of poly(IC) (Fig. 2a). RT/qPCR was used to quantify expression of various ISGs after 12h, relative to that without poly(IC). As the concentration of poly(IC) increased, a corresponding increase in ISG expression was observed (Fig. 2a). β-actin was constant. These data confirmed that transfection of HeLa cells with poly(IC) induced ISG expression, as expected24.
Figure 2. IU-dsRNA suppressed induction of ISGs by poly(IC).
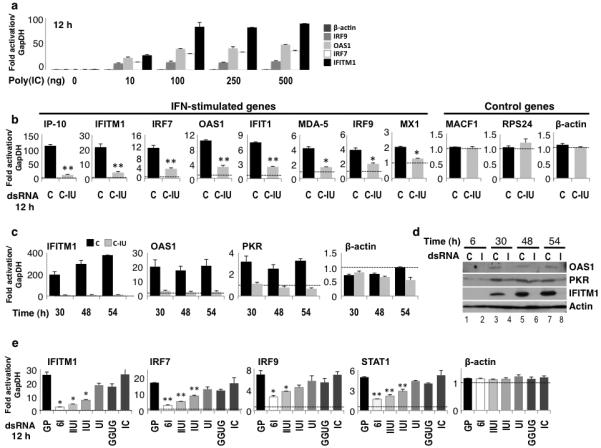
(a) HeLa cells were transfected with 0–500 ng poly(IC). RT/qPCR was used to quantify expression of β-actin or ISGs (IRF9, OAS1, IRF7, IFITM1; Supplementary Table 1) after 12h (n=4). Fold-change in mRNA levels were calculated relative to those without poly(IC), and normalized to GapDH. (b) HeLa cells were co-transfected with poly(IC) and C or C-IU dsRNAs. RT/qPCR was used to quantify expression of ISGs (IP-10, IFITM1, IRF7, OAS1, IFIT1, MDA-5, IRF9, MX1; Supplementary Table 1) or control genes (MACF1, RPS24, β-actin; Supplementary Table 1) after 12h (n=4). Fold-change in mRNA levels was relative to those at 6h with C, and normalized to GapDH. P values were <1×10−3 (*) or <5×10−4 (**). (c) HeLa cells were co-transfected with poly(IC) and C or C-IU. RT/qPCR was used to quantify expression of β-actin or ISGs (IFITM1, PKR, OAS1; Supplementary Table 1) after 30, 48 and 54h. Fold-change in mRNA levels were calculated relative to those at 6h with C, and normalized to GapDH. (d) HeLa cells were co-transfected with poly(IC) and C or C-IU (I). Immunoblotting was used to analyze expression of ISGs (IFITM1, PKR, OAS1; Supplementary Table 1) after 6, 30, 48 and 54h. Actin was a loading control. (e) HeLa cells were co-transfected with poly(IC) and either control dsRNA (GP) or IU-dsRNAs with 2, 3, 4 or 6 IU pairs (UI, IUI, IIUI, 6I, respectively). dsRNAs with GU or IC pairs (GGUG or IC, respectively) were also tested. RT/qPCR was used to quantify expression of β-actin or ISGs (IFITM1, IRF7, IRF9, STAT1; Supplementary Table 1) after 12h (n=4). Fold-change in mRNA was relative to those at 6h with GP, and normalized to GapDH. P values were <1×10−3 (*) or <1×10−4 (**). All error bars are mean ± s.d.
We next tested the effect of C-IU on induction of ISGs by poly(IC). HeLa cells were transfected with either C or C-IU, with or without poly(IC). Expression of various ISGs was quantified after 12h using RT/qPCR. Several control genes not induced by IFN were also tested. In the absence of poly(IC), expression of all genes was constant with either C or C-IU (data not shown). However, when HeLa cells were co-transfected with poly(IC) and C dsRNA, expression of all ISGs tested was increased (Fig. 2b). In contrast, induction of ISGs was suppressed by C-IU (Fig. 2b). Expression of control genes was constant (Fig. 2b). These data therefore reinforced the previous observations in that they clearly demonstrated that IU-dsRNA suppressed induction of ISGs by poly(IC). Moreover, the effect of IU-dsRNA on gene expression was specific for ISGs.
IU-dsRNA has a prolonged effect on ISGs
In the previous experiments, expression of ISGs was evaluated 12h after co-transfection of HeLa cells with short dsRNAs and poly(IC). We next asked whether suppression of ISGs by IU-dsRNA persisted over a longer time course.
HeLa cells were co-transfected with poly(IC) and C or C-IU. Cells were harvested after 6–54h, and expression of various ISGs analyzed by RT/qPCR. This showed that each ISG tested was upregulated with poly(IC) and C after 30–54h (Fig. 2c). In contrast, when C-IU dsRNA was co-transfected with poly(IC), induction of ISGs was suppressed (Fig. 2c). The presence of C-IU in cells therefore suppressed induction of ISGs during an extended time course.
Immunoblots were additionally used to analyze expression of the ISGs following co-transfection of HeLa cells with poly(IC) and C or C-IU (Fig. 2d). With poly(IC) and C, expression of all ISGs was increased 30–54h post-transfection (lanes 3, 5, 7), relative to 6h (lane 1). In contrast, increased expression was not seen with poly(IC) and C-IU (I) (lanes 4, 6, 8). These data again showed that IU-dsRNA suppressed induction of ISGs, therefore corroborating the findings described above where gene expression was analyzed by RT/qPCR.
Multiple IU pairs are needed to suppress ISGs
Several short duplexes (C-IU, IIUI, 142-IU) were used to show that IU-dsRNA suppressed induction of ISGs by poly(IC). We next investigated how many IU pairs were required for suppression.
HeLa cells were co-transfected with poly(IC) and either a perfect Watson-Crick duplex (GP; Table 1) or an IU-dsRNA containing 2, 3, 4 or 6 IU pairs (UI, IUI, IIUI or 6I, respectively). In addition, a duplex containing IC pairs (IC) was tested to determine whether inosine was sufficient per se. A duplex containing four isosteric GU pairs (GGUG) was also tested. Again, with the exception of the central 4–6 base pairs, the sequences of the dsRNAs tested were identical. Expression of β-actin and various ISGs was analyzed 12h post-transfection by RT/qPCR (Fig. 2e). In the presence of GP dsRNA and poly(IC), expression of all ISGs increased. β-actin was unchanged. In contrast, induction of ISGs was significantly reduced with 6I IU-dsRNA and poly(IC) (Fig. 2e; compare expression relative to that with GP). Moreover, as the number of IU pairs in the IU-dsRNAs decreased, a corresponding decrease in suppression of ISGs was seen. When the GGUG duplex was tested, a small amount of repression was observed (Fig. 2e). However, this was substantially less than that seen with the equivalent IU-dsRNA (IIUI). In contrast, IC dsRNA did not suppress induction of ISGs by poly(IC) (Fig. 2e). This observation confirmed that IU pairs were essential for suppression. These data together therefore demonstrated that multiple IU pairs were required to efficiently suppress induction of ISGs.
Previously we have shown that IU-dsRNAs undergo specific cleavage in various cell lysates11. Using various synthetic duplexes that correspond to potential cleavage products of IIUI IU-dsRNA11, we additionally demonstrated that relatively short IU-dsRNAs containing multiple inosine residues also suppressed induction of ISGs (Supplementary Fig. 2).
Inhibition of gene expression is specific for ISGs
Multiple duplexes were used to demonstrate that IU-dsRNA represses induction of various ISGs in response to poly(IC). We next used microarrays to look more globally at the effects of IU-dsRNA on gene expression.
HeLa cells were co-transfected with poly(IC) and C or C-IU, and RNA harvested after 12h. Three independent experiments were performed for microarray analyses. Microarrays were subsequently used to analyze gene expression in the presence of C or C-IU, where fold-change in mRNA with C was calculated relative to that with C-IU. This revealed that expression of 59 genes was ≥1.5-fold greater with C than with C-IU after 12h (Supplementary Table 2). Moreover, this gene set included most of the ISGs analyzed in Figures 1 and 2. Using gene ontology analyses (PANTHER)25, the set of 59 genes was found to be significantly enriched for genes involved in biological processes such as interferon-mediated immunity and immunity and defense (Fig. 3, Supplementary Table 3). This observation was consistent with the idea that IU-dsRNA suppressed induction of genes involved in the IFN response. Interestingly, a number of genes involved in apoptosis were also overrepresented. Analysis of pathways and molecular functions related to the 59 genes were also significantly enriched for terms that strongly supported roles in immunity and defense (Supplementary Table 3). These data confirmed that IU-dsRNA specifically suppressed induction of genes involved in immune responses.
Figure 3. Genes involved in immunity and defense were enriched.
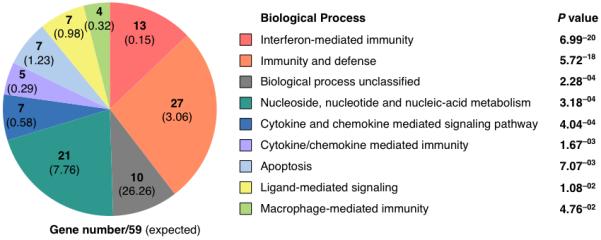
The 59 genes enriched ≥1.5-fold with poly(IC) and C relative to C-IU on the microarray were subject to Gene ontology analyses (PANTHER), and classified according to Biological Process (P≤0.05). The numbers of genes within the set of 59 that belong to each class are given; the number expected with reference to the control database (NCBI: H. sapiens genes (25431)) are shown in parentheses.
To validate the microarray data, expression of approximately half of the 59 genes identified was analyzed. HeLa cells were co-transfected with poly(IC) and C or C-IU, and harvested after 2 or 12h. RT/qPCR was used to analyze gene expression. Fold-change in mRNA levels at 12h were calculated relative to those at 2h with C, and normalized to GapDH. In the presence of poly(IC) and C, expression of all genes tested was upregulated 1.4–400-fold (Fig. 4a). In contrast, C-IU suppressed induction of gene expression (Fig. 4a). When seven genes that were unchanged on the array with poly(IC) and C or C-IU were additionally analyzed, no induction by poly(IC) or suppression by C-IU was observed, as expected (Fig. 4b). Together these results validated the data obtained using microarrays, where induction of ISGs by poly(IC) was specifically repressed by C-IU dsRNA.
Figure 4. Microarray validation by RT/qPCR.
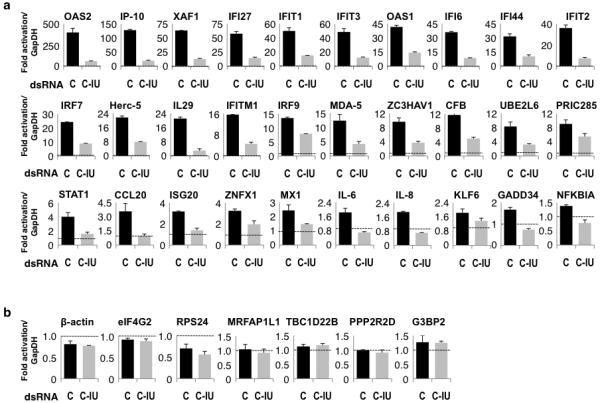
HeLa cells were co-transfected with poly(IC) and C or C-IU, and harvested after 2 and 12h. (a) Expression of numerous genes enriched ≥1.5-fold at 12h with C relative to C-IU on the array was validated using RT/qPCR (n=4). Fold-change in mRNA levels was relative to those at 2h with C dsRNA, and normalized to GapDH. (b) Expression of selected genes that are unchanged at 12h with C relative to C-IU was validated using RT/qPCR. Expression at 12h is relative to that at 2h with C (n=4). All error bars are mean ± s.d.
IU-dsRNA inhibited poly(IC)-induced apoptosis
Transfection of HeLa cells with poly(IC) triggered an IFN response characterized by induction of ISGs. Moreover, previous studies have shown that transfection of cells with poly(IC) also induced apoptosis26,27. Throughout the experiments described above, we consistently observed more cell death following transfection with poly(IC) and C than with poly(IC) and C-IU. We therefore investigated whether cell death was due to apoptosis, and to verify the effect of C or C-IU.
To quantify the amount of cell death following co-transfection with poly(IC) and C or C-IU, the number of dead cells in the supernatant were counted after 12–24h (Fig. 5a). This confirmed that the amount of cell death increased over time with C, but not C-IU. These data therefore suggested that C-IU protected against cell death.
Figure 5. poly(IC) induced apoptosis is inhibited by C-IU.
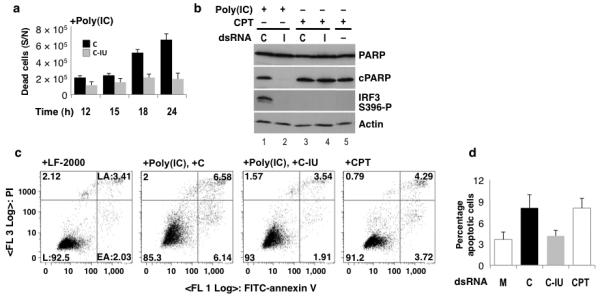
(a) Cells were co-transfected with poly(IC) and C or C-IU (I). Dead cells (supernatant) were counted after 12–24h (n=3). (b) HeLa cells were co-transfected with poly(IC) and C or C-IU, or treated with CPT ± C or C-IU. Immunoblotting was used to detect apoptosis by analyzing PARP cleavage (cPARP). Uncleaved PARP is also shown (PARP). IRF3 activation was analyzed (IRF3 S396-P), and actin was a loading control. (c) HeLa cells were mock transfected (LF-2000), co-transfected with poly(IC) and C or C-IU, or treated with CPT. Cells were labeled after 24h with Annexin V FITC and PI, and analyzed by FACS. The percentage of living (L; Annexin V−, PI−), early apoptotic (EA; Annexin V+, PI−) and late apoptotic (LA; Annexin V+, PI+) cells are given. (d) The proportion of apoptotic (EA+LA) cells observed in samples described in (c) were quantified by FACS, and summarized graphically (n≥3). All error bars are mean ± s.d.
To investigate whether apoptosis was responsible for cell death, HeLa cells were co-transfected with poly(IC) and C or C-IU, or treated with camptothecin (CPT), with or without C or C-IU. Treatment of cells with CPT induces apoptosis28. Cell lysates were prepared from adherent cells after 12h, and immunoblotting was used to analyze cleavage of poly(ADP-ribose) polymerase (PARP), a hallmark of apoptosis29. Under all conditions tested, full-length PARP (PARP) was detected (Fig. 5b, lanes 1–5). When cells were treated with CPT, with or without C or C-IU, a band corresponding to cleaved PARP (cPARP) was detected (lanes 3–5). Similarly, when cells were transfected with poly(IC) and C, cPARP was seen (lane 1). In contrast, cPARP was not detected in cells treated with poly(IC) and C-IU (lane 2). These data confirmed that cell death in response to poly(IC) was due to apoptosis. Moreover, these data strongly suggested that C-IU inhibited apoptosis.
Fluorescence-activated cell sorting (FACS) was used to confirm that cell death in response to poly(IC) resulted from apoptosis. HeLa cells were co-transfected with poly(IC) and C or C-IU, or treated with CPT as a control for apoptosis. Mock transfections were also performed. After 12–24h adherent cells were labeled with Annexin V and propidium iodide (PI), which stains for apoptotic or dead (apoptotic/necrotic) cells, respectively. FACS was then used to determine what proportion of cell death resulted from apoptosis. Representative FACS data from the 24h time point is shown (Fig. 5c), and data obtained from multiple experiments is summarized graphically (Fig. 5d; n=4). As expected, CPT treatment increased apoptosis (Figs. 5c, 5d). Similarly, HeLa cells co-transfected with poly(IC) and C underwent substantially more apoptosis than mock-transfected cells (+LF-2000). In contrast, no significant increase in apoptosis was observed with poly(IC) and C-IU, relative to the mock. These data thus support the observation that C-IU inhibited poly(IC)-induced apoptosis.
IU-dsRNA inhibits activation of IRF3
When poly(IC) is used to transfect mammalian cells, it is recognised by the cytosolic sensors MDA-5 and RIG-I (Supplementary Fig. 3)19. This in turn leads to activation of the transcription factor IRF3, which triggers production of type I IFNs and ultimately a transcriptional cascade18. Activation of IRF3 requires phosphorylation of multiple residues, followed by homodimerization and nuclear translocation30,31. IRF3 is also required for virus- or dsRNA-induced apoptosis32-35. We therefore investigated whether IU-dsRNA inhibited activation of IRF3, which would provide a plausible explanation for how IU-dsRNA suppressed both the IFN and apoptotic pathways.
HeLa cells were co-transfected with poly(IC) and C or C-IU, and harvested after 2–36h. RT/qPCR was used to verify that ISG expression was upregulated with poly(IC) and C at each time point, but suppressed by C-IU (Fig. 6a).
Figure 6. C-IU dsRNA inhibits IRF3 activation.
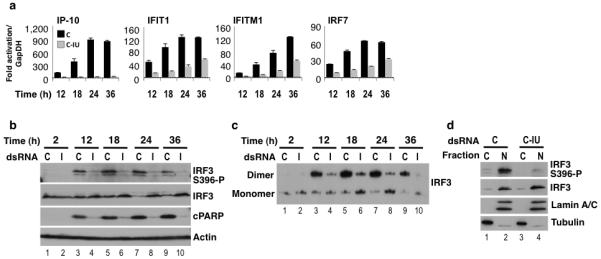
HeLa cells were co-transfected with poly(IC) and C or C-IU (I), and lysates prepared after 2-36h. (a) RT/qPCR was used to quantify expression of ISGs (IP-10, IFIT1, IFITM1, IRF-7; Supplementary Table 1) after 12-36h (n=4). Fold-change in mRNA was relative to that at 2h with C, and normalized to GapDH. Error bars are mean ± s.d. (b) Immunoblots were used to detect IRF3 activation (IRF3 S396-P) and total IRF3 (IRF3). Detection of cPARP indicated apoptosis. Actin was a loading control. (c) Dimerization of IRF3 was analyzed using native gels and immunoblotting (IRF3). (d) HeLa cells were co-transfected with poly(IC) and C or C-IU, and nuclear (N) and cytoplasmic (C) lysates prepared. Lamin A/C and Tubulin were markers for N and C, respectively. Immunoblots were used to detect total IRF3 (IRF3) and IRF3 activation (IRF3 S396-P).
Immunoblotting was first used to test whether IRF3 was activated by phosphorylation with poly(IC) and C or C-IU. Using a specific IRF3-phospho antibody (IRF3 S396-P), we showed that phosphorylation of IRF3 occurred with poly(IC) and C after 12–36h (Fig. 6b; lanes 3, 5, 7, 9). In contrast, C-IU suppressed IRF3 phosphorylation (lanes 4, 6, 8, 10). In the absence of poly(IC), IRF3 was not phosphorylated with either C or C-IU (Supplementary Fig. 4a). Equivalent results were obtained using another unrelated pair of duplexes (GP and 6I; Supplementary Fig. 4b). These data therefore demonstrated that IU-dsRNA inhibited activation of IRF3. When the immunoblot was additionally probed with anti-IRF3, which detects total IRF3, reduced levels of IRF3 were seen at 24 and 36h with C but not C-IU (Fig. 6b; compare lanes 7 and 8 or 9 and 10). This is consistent with previous studies showing proteolysis of IRF3 following activation30. Finally the immunoblot was probed with anti-PARP, which only detected cPARP with poly(IC) and C, but not C-IU (Fig. 6b; compare lanes 3, 5, 7, 9 with 4, 6, 8, 10). Thus C-IU inhibited IRF-3 activation and apoptosis. This observation was also made using other cell lines (Supplementary Fig. 4c) and in the previous experiment, where both IRF3 phosphorylation and cPARP was observed with poly(IC) and C, but not C-IU (Fig. 5b; lanes 1, 2). Together these data confirmed that C-IU prevented IRF-3 activation.
Dimerization of IRF3 was next analyzed using native gels and immunoblotting. When HeLa cells were co-transfected with poly(IC) and C, IRF3 dimers were observed after 12–36h (Fig. 6c; lanes 3, 5, 7, 9). In contrast, substantially less dimerization of IRF3 occurred with poly(IC) and C-IU (lanes 4, 6, 8, 10). These data were therefore consistent with the idea that IU-dsRNA inhibits activation, hence dimerization, of IRF3.
IRF3 translocates to the nucleus following phosphorylation and dimerization30,31. To investigate the sub-cellular localization of active IRF3, nuclear and cytoplasmic fractions were prepared 24h after co-transfection of cells with poly(IC) and C or C-IU, and analyzed using immunoblots (Fig. 6d). Lamin A/C and Tubulin were nuclear and cytoplasmic markers, respectively. With poly(IC) and C or C-IU, localization of total IRF3 (IRF3) was predominantly nuclear, consistent with the observation that inactive IRF3 shuttles into and out of the nucleus18. In contrast, phosphorylated-IRF3 (IRF3 S396-P) was strongly enriched in the nuclear fraction with poly(IC) and C (Fig. 6d) while very little phosphorylated-IRF3 was detected with C-IU. These data confirmed that activation of IRF3 via phosphorylation and nuclear translocation only occurred with poly(IC) and C but not C-IU.
By analyzing IRF3 phosphorylation, dimerization and nuclear localization we conclusively demonstrated that IU-dsRNA inhibits activation of IRF3. In so doing, we have provided an explanation for how IU-dsRNA inhibits both the IFN and apoptotic pathways.
IU-dsRNA interacts specifically with MDA-5 and RIG-I
We have shown that IU-dsRNA inhibits activation of IRF3. We next asked whether IU-dsRNA inhibits an earlier step in the pathway preceding IRF3 activation (Supplementary Fig. 3). We thus focused on whether IU-dsRNA interacts with MDA-5 or RIG-I, the cytosolic sensors for poly(IC).
We first tested whether IU-dsRNA interacts with MDA-5. MDA-5 was overexpressed in HeLa cells for 24h, followed by transfection with specific (Bio-C-IU; Table 1) or non-specific (C or Bio-C) dsRNAs. Lysates were prepared after 12h, and RNA–protein complexes captured using streptavidin beads. Immunoblotting showed that MDA-5 bound specifically to Bio-C-IU (bI), but not Bio-C (bC) or C dsRNAs (Fig. 7a; compare lane 4 with lanes 5 and 6). These data therefore demonstrated that IU-dsRNA specifically interacts with MDA-5.
Figure 7. IU-dsRNA binds specifically to MDA-5 and RIG-I.
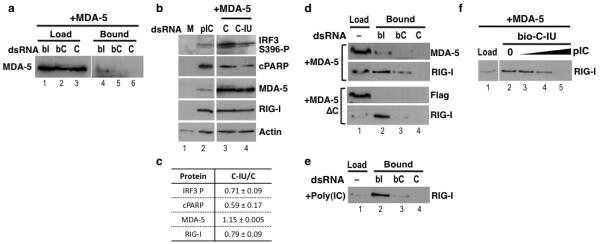
(a) HeLa cells were co-transfected with pUNO1-hMDA5 and C or Bio-C (bC) or Bio-C-IU (bI) dsRNAs. Protein complexes were captured using streptavidin beads, and immunoblotting was used to detect MDA-5 bound to C, bC or bI (n=3). Total protein is shown. (b) HeLa cells were mock transfected (M), transfected with poly(IC) (pIC), or co-transfected with pUNO1-hMDA5 and C or C-IU; n=3. Immunoblots were used to detect IRF3 activation (IRF3 S396-P), apoptosis (cPARP), MDA-5 expression (MDA-5), and RIG-I (RIG-I) (n=3). (c) Levels of each protein detected in (b) in the presence of overexpressed MDA-5 and C or C-IU were quantified (C-IU/C), relative to actin (n=3). (d) Lysates were prepared from HeLa cells overexpressing either MDA-5 or MDA-5ΔC, and then used to bind C, Bio-C (bC) or Bio-C-IU (bI) coupled streptavidin beads. Immunoblots were used to detect full length MDA-5, MDA-5ΔC (Flag) or RIG-I (n≥3). (e) HeLa cell lysates were prepared 12h after transfection with poly(IC), and then used to bind C, Bio-C (bC) or Bio-C-IU (bI) coupled streptavidin beads. Immunoblots were used to detect RIG-I. (f) Lysates were prepared from HeLa cells expressing MDA-5, and incubated with Bio-C-IU-coupled streptavidin beads in the presence of 0, 50, 200 or 300 ng poly(IC) (pIC). RIG-I binding was detected by immunoblotting (n=3).
We next investigated whether C-IU interfered with activation of IRF3 by MDA-5. MDA-5 was overexpressed in HeLa cells for 24h, followed by transfection with C or C-IU, and lysates prepared after 12h. Control transfections (mock, poly(IC)) were also performed. RT/qPCR confirmed that overexpression of MDA-5 induced ISGs36 (data not shown). Moreover, immunoblots showed that RIG-I was substantially upregulated by overexpressed MDA-5 (Fig. 7b (RIG-I)). However, as it was likely to be inactive in the absence of poly(IC)36,37, RIG-I was unlikely to contribute to IRF3 activation. Activation of IRF3 was analyzed using immunoblots. When MDA-5 was overexpressed, considerably more phosphorylated-IRF3 was detected with C than C-IU (Fig. 7b; compare lanes 3 and 4 (IRF3 S396-P), Fig. 7c). This was not due to differential expression of MDA-5 (Fig. 7b, lanes 3 and 4 (MDA-5), Fig. 7c). Furthermore, RT/qPCR confirmed that induction of ISGs by MDA-5 was substantially higher with C than C-IU (data not shown). Apoptosis was additionally analyzed using anti-PARP. In keeping with the differential levels of phosphorylated-IRF3, more cPARP was detected with C than C-IU (Fig. 7b; compare lanes 3 and 4 (cPARP), Fig. 7c). Together these data demonstrated that C-IU inhibited pathways initiated by MDA-5. This inhibition may result from specific binding of IU-dsRNA to MDA-5.
We next tested whether C-IU interacted with a truncated version of MDA-5 (MDA-5ΔC) lacking the C-terminal region implicated in ligand binding38. MDA-5 or MDA-5ΔC was expressed in HeLa cells (Fig. 7d; MDA-5 or Flag antibodies, respectively; lane 1), and lysates incubated with streptavidin beads coupled to specific (Bio-C-IU) or non-specific (C or Bio-C) dsRNAs. Analysis of the dsRNA–protein complexes showed that MDA-5 bound specifically to Bio-C-IU, as expected (lane 2; MDA-5). In contrast, no binding was seen with MDA-5ΔC (lane 2; Flag). These data suggest that C-IU binds MDA-5 via the C-terminal half of the protein, so may interfere with ligand binding and subsequent activation.
The immunoblot was additionally probed for RIG-I, which was induced by expression of either MDA-5 or MDA-5ΔC. This showed that RIG-I also bound preferentially to Bio-C-IU compared to Bio-C or C (Fig. 7d). Moreover, with reference to the load, RIG-I appeared to bind more efficiently to C-IU than MDA-5 (Fig. 7d; compare lanes 1 and 2). When RIG-I was induced by transfection of HeLa cells with poly(IC), specific binding of RIG-I to C-IU was similarly observed (Fig. 7e). These data thus demonstrated that C-IU dsRNA bound specifically to RIG-I as well as MDA-5.
Finally we investigated whether C-IU binding to RIG-I could potentially interfere with ligand binding. Lysates prepared from HeLa cells overexpressing MDA-5, thus containing increased levels of RIG-I, were incubated with Bio-C-IU-coupled streptavidin beads in the presence of increasing concentrations of poly(IC). In the absence of poly(IC), RIG-I bound C-IU (Fig. 7f, lane 2). As the concentration of poly(IC) increased, a corresponding decrease in RIG-I binding to C-IU was observed (Fig. 7f, lanes 3–5). In contrast, a non-specific RNA did not reduce binding (data not shown). These data suggest that binding of C-IU and poly(IC) to RIG-I are mutually exclusive, thus leading to the speculation that C-IU may inhibit RIG-I by interfering with ligand binding.
We have clearly shown that C-IU inhibits MDA-5 and binds specifically to both MDA-5 and RIG-I. We therefore propose that IU-dsRNA exerts its inhibitory effects on the IFN and apoptotic pathways by specifically interacting with RIG-I or MDA5, which in turn prevents IRF3 activation and subsequent signalling events.
Discussion
The data presented here shed interesting light upon the role of IU-dsRNA in mammalian cells. We have conclusively demonstrated that IU-dsRNA selectively suppresses induction of ISGs in response to long dsRNA. Previously we showed that IU-dsRNA downregulated expression of both endogenous and reporter genes in trans 13. While the identity of the endogenous genes was previously unknown, we now speculate that they correspond to ISGs. It will now be interesting to discover how downregulation of reporter genes relates to our current findings.
We have also shown that IU-dsRNA inhibits poly(IC)-induced apoptosis (Figs. 5-7). One mechanism by which poly(IC) induces apoptosis is via upregulation of the proapoptotic TNF-related apoptosis-inducing ligand (TRAIL), a direct transcriptional target of IRF339. We speculate that TRAIL may be important for the apoptosis we observed, as its expression was upregulated by poly(IC) and C, but repressed by C-IU (Supplementary Fig. 5). Furthermore, XAF1 (X-linked inhibitor of apoptosis-associated factor 1), an IFN-dependent factor that sensitizes cells to the proapoptotic effects of TRAIL40, was strongly activated by poly(IC) and C but not C-IU (Supplementary Fig. 5). However, regardless of the mechanism by which poly(IC)-induced apoptosis occurs, we conclusively demonstrated that apoptosis was suppressed by IU-dsRNA.
We have shown that IU-dsRNA inhibits phosphorylation of IRF3 (Fig. 6), which is essential for induction of ISGs and apoptosis31-35. Moreover, this finding was supported by the observation that IU-dsRNA was unable to inhibit ISG induction by a phosphomimetic IRF3 mutant (IRF3 5D30; Supplementary Fig. 6). Consistent with our observations, a recent study showed activation of IRF3 in ADAR1-deficient cells41. We further investigated the effect of IU-dsRNA at earlier steps in the pathway leading to IRF3 activation, and thus speculate that IU-dsRNA may interfere with initial recognition of poly(IC) by either RIG-I or MDA-5 (Fig. 7).
Our findings are in keeping with previous studies that suggest ADAR1 is essential for survival. Increased apoptosis was seen in ADAR1-deficient HeLa cells following viral infection41. Early embryonic death in Adar1−/− mice was the result of widespread apoptosis and defective hematopoiesis16. Mouse embryonic fibroblasts derived from Adar−/− embryos were prone to apoptosis during serum starvation16. Increased apoptosis was observed in ADAR1-deficient hematopoietic progenitor cells42. Loss of ADAR1 in hematopoietic stem cells led to global upregulation of ISGs and rapid apoptosis17. Based on these observations, ADAR1 was thought to promote cell survival by suppressing the potentially deleterious effects of IFN activation. Our findings now provide an explanation for how ADAR1 regulates IFN signaling.
Various targets for hyper-editing by ADARs have been described in mammalian cells. Analysis of RNA from human brain showed editing within non-coding regions5. Moreover, bioinformatic analyses predicted that >5% of human mRNAs contain editing sites within non-coding sequences, such as Alus4,6,7. miRNA precursors may also be edited43. dsRNA arising from viral infection is another likely editing target. We speculate that editing of any of these putative targets by ADAR1 will give rise to IU-dsRNA that has the potential to suppress the immune response.
Restricted expression of ADAR1 p150 may regulate IU-dsRNA production. p150 expression occurs in response to IFN14 and also due to serum starvation16. Induction of p150 by IFN may represent a negative feedback mechanism whereby its upregulation during stress acts to dampen down the IFN response, thus enabling cell survival. Expression of p150 was higher than the constitutive (p110) isoform in hematopoietic stem cells, where ADAR1 is essential for survival. In contrast, p110 was expressed almost exclusively in embryonic stem cells, where ADAR1 is dispensable17. Increased expression of p150 may be critical in maintaining the delicate balance between cell survival and death.
We have provided compelling evidence that IU-dsRNA per se suppresses ISG induction and apoptosis in response to long dsRNA. We have thus uncovered a novel role for IU-dsRNA, hence editing by ADARs, in mammalian cells.
Supplementary Material
Acknowledgements
We thank Chris Smith for helpful comments, John Hiscott (McGill University), Joari De Miranda (Columbia University) and Shizuo Akira (Osaka University) for reagents, Rachael Walker for FACS (Wellcome Trust Centre for Stem Cell Research), and Emily Clemente and Julien Bauer for microarrays (Cambridge Genomic Services). Funding was from the Wellcome Trust.
Appendix
Methods
Transfections
2 × 105 HeLa cells per well (6 well plate) were co-transfected with 120 pmol dsRNA (80 pmol specific and 40 pmol C) and typically either 250 ng Fluc mRNA or 10 ng poly(IC) (Sigma) using Lipofectamine-2000 (LF-2000; Invitrogen). Mock transfections used LF-2000 alone. Transfections were scaled up 5-fold for large-scale (10 cm plates). For expression of MDA-5 or MDA-5ΔC (amino acids 1–575), HeLa cells were transfected with 0.5 μg pUNO1-hMDA5 (Invivogen) or pFLAG-MDA-5ΔC44, respectively. RNA was isolated with TRIzol®, and lysates prepared using NET or RIPA buffer. NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) were also used. Protein concentrations were determined by Bradford assays.
RNAs
Short dsRNAs were from Dharmacon. Capped Fluc mRNA was transcribed in vitro from pLuc-MCS45.
RT/qPCR
2 μg DNase treated RNA was reverse transcribed (RT) using oligo(dT) and AMV-RT (Promega). –RT controls were performed. qPCR was performed using a Rotor-Gene 6000 (Corbett) and SensiMixPlus SYBR (Quantace). Fold-change in mRNA was relative to control, and normalized to GapDH. T-tests (2 tailed, unequal variance) were used. Primer sequences are in Supplementary Table 4.
Immunoblots
Proteins (30–50 μg) were separated by SDS-PAGE, transferred to PVDF, and detected by ECL. Antibodies were OAS1, PKR, IFITM1, Lamin A/C, IRF3 (Santa Cruz), Phospho-IRF3 (Ser396), MDA-5, RIG-I (Cell Signaling), PARP, tubulin (Abcam) and Flag, actin (Sigma). Secondary antibodies were HRP coupled (Jackson ImmunoResearch). Quantitation was performed by densitometry using ImageQuant™ TL (GE Healthcare).
Affinity purification
HeLa cells were transfected with pUNO1-hMDA5 for expression of MDA-5, then re-transfected after 24h with C, or biotinylated C or C-IU dsRNAs, as above. NET lysates were prepared after a further 12h, and dsRNA–protein complexes immediately recovered by incubation on ice with Streptavidin-coupled Dynabeads®. Alternatively, NET lysates were prepared 24h after transfection of HeLa cells with pUNO1-hMDA5 or pFlag-MDA-5ΔC, and incubated 60 min at room temperature with 200 μl Streptavidin-Dynabeads® coupled to C or Bio-C or Bio-C-IU dsRNAs (as per manufacturers instructions). For competition studies, 50, 200 or 300 ng poly(IC) was added to lysate for 30 min prior to incubation with beads. Beads were washed extensively, and proteins eluted with SDS buffer.
Native Gels
Proteins (50 μg) were electrophoresed at 25 mA (4 °C, 60 min) using Novex® 8% (w/v) Tris-Glycine Gels (Invitrogen). Gels were soaked in SDS buffer before immunoblotting.
Quantifying dead cells
Dead cells in the supernatant were counted after being harvested by centrifugation, washed and resuspended in 2 ml PBS (n=3).
FACS
Large-scale transfections were with poly(IC) and C or C-IU, and analyzed after 2-24h. 10 μM CPT was added to the media as an apoptosis control. Cells were washed with PBS, trypsinized and resuspended in binding buffer (Invitrogen). Cells were labeled with Annexin V FITC and PI (Invitrogen), and analyzed on a CyAn analyzer (Beckman Coulter) using 530/40 or 613/20 filters, respectively. Cells were gated on forward scatter vs. side scatter to eliminate debris and doublet discrimination was carried out to analyze only single cells.
Microarrays
HeLa cells were co-transfected with poly(IC) and C or C-IU, and RNA harvested after 12h. Samples were from three independent experiments. RNA was analyzed (Nanodrop ND1000, Bioanalyzer (Agilent)), and amplified (Illumina® TotalPrep™-96; Ambion). Samples were spotted on a Human-6 v2 Expression BeadChip (simultaneously assays 6 samples with >48,000 probes per sample, targeting genes and known alternative splice variants from the RefSeq database release 17, UniGene build 188), followed by Illumina Direct HYb Assay. BeadChips were scanned (Illumina Beadstation 500) and data analyzed by lumi 46. Genes enriched >1.5-fold with C relative to C-IU were subject to Gene Ontology analyses (PANTHER)25.
References
- 1.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annual Review of Biochemistry. 2002;71:817–846. doi: 10.1146/annurev.biochem.71.110601.135501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valente L, Nishikura K, Kivie M. ADAR Gene Family and A-to-I RNA Editing: Diverse Roles in Posttranscriptional Gene Regulation. Prog. Nucleic Acid Res. Mol. Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. Academic Press. [DOI] [PubMed] [Google Scholar]
- 3.Serra MJ, Smolter PE, Westhof E. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Research. 2004;32:1824–1828. doi: 10.1093/nar/gkh501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blow M, Futreal PA, Wooster R, Stratton MR. A survey of RNA editing in human brain. Genome Research. 2004;14:2379–2387. doi: 10.1101/gr.2951204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morse DP, Aruscavage PJ, Bass BL. RNA hairpins in noncoding regions of human brain and Caenorhabditis elegans mRNA are edited by adenosine deaminases that act on RNA. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:7906–7911. doi: 10.1073/pnas.112704299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Levanon EY, et al. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nature Biotechnology. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 7.Barak M, et al. Evidence for large diversity in the human transcriptome created by Alu RNA editing. Nucl. Acids Res. 2009;37:6905–6915. doi: 10.1093/nar/gkp729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L-L, DeCerbo JN, Carmichael GG. Alu element-mediated gene silencing. EMBO Journal. 2008;27:1694–1705. doi: 10.1038/emboj.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L-L, Carmichael GG. Altered Nuclear Retention of mRNAs Containing Inverted Repeats in Human Embryonic Stem Cells: Functional Role of a Nuclear Noncoding RNA. 2009;35:467–478. doi: 10.1016/j.molcel.2009.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hundley HA, Krauchuk AA, Bass BL. C. elegans and H. sapiens mRNAs with edited 3′UTRs are present on polysomes. RNA. 2008;14:2050–2060. doi: 10.1261/rna.1165008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scadden ADJ, Smith CWJ. Specific cleavage of hyper-edited dsRNAs. EMBO Journal. 2001;20:4243–4252. doi: 10.1093/emboj/20.15.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scadden ADJ. The RISC subunit Tudor-SN binds to hyper-edited double-stranded RNA and promotes its cleavage. Nature Structural & Molecular Biology. 2005;12:489–496. doi: 10.1038/nsmb936. [DOI] [PubMed] [Google Scholar]
- 13.Scadden ADJ. Inosine-Containing dsRNA Binds a Stress-Granule-like Complex and Downregulates Gene Expression In trans. Molecular Cell. 2007;28:491–500. doi: 10.1016/j.molcel.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.George CX, Samuel CE. Characterization of the 5′-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- 15.Eckmann CR, Neunteufl A, Pfaffstetter L, Jantsch MF. The Human But Not the Xenopus RNA-editing Enzyme ADAR1 Has an Atypical Nuclear Localization Signal and Displays the Characteristics of a Shuttling Protein. Mol. Biol. Cell. 2001;12:1911–1924. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang QD, et al. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. Journal of Biological Chemistry. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 17.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiscott J. Triggering the Innate Antiviral Response through IRF-3 Activation. J. Biol. Chem. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 19.Kato H, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008;205:1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anderson P, Kedersha N. RNA granules. Journal of Cell Biology. 2006;172:803–808. doi: 10.1083/jcb.200512082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stark GR, Kerr IM, Williams BRG, Silverman RH, Schreiber RD. How cells respond to interferons. Annual Review of Biochemistry. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- 22.Fire A, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 23.Grudzien EWA, et al. Novel cap analogs for in vitro synthesis of mRNAs with high translational efficiency. RNA. 2004;10:1479–1487. doi: 10.1261/rna.7380904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Field AK, et al. Poly I:C, an inducer of interferon and interference against virus infections. Medicine. 1972;51:169–174. doi: 10.1097/00005792-197205000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Thomas PD, et al. PANTHER: A Library of Protein Families and Subfamilies Indexed by Function. Genome Research. 2003;13:2129–2141. doi: 10.1101/gr.772403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dogusan Z, et al. Double-Stranded RNA Induces Pancreatic β-Cell Apoptosis by Activation of the Toll-Like Receptor 3 and Interferon Regulatory Factor 3 Pathways. Diabetes. 2008;57:1236–1245. doi: 10.2337/db07-0844. [DOI] [PubMed] [Google Scholar]
- 27.Besch R, et al. Proapoptotic signaling induced by RIG-I and MDA-5 results in type I interferon-independent apoptosis in human melanoma cells. J. Clin. Invest. 2009;119:2399–2411. doi: 10.1172/JCI37155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gorczyca W, Gong J, Ardelt B, Traganos F, Darzynkiewicz Z. The Cell Cycle Related Differences in Susceptibility of HL-60 Cells to Apoptosis Induced by Various Antitumor Agents. Cancer Res. 1993;53:3186–3192. [PubMed] [Google Scholar]
- 29.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 30.Lin R, Heylbroeck C, Pitha PM, Hiscott J. Virus-Dependent Phosphorylation of the IRF-3 Transcription Factor Regulates Nuclear Translocation, Transactivation Potential, and Proteasome-Mediated Degradation. Mol. Cell. Biol. 1998;18:2986–2996. doi: 10.1128/mcb.18.5.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoneyama M, et al. Direct triggering of the type I interferon system by virus infection: activation of a transcription factor complex containing IRF-3 and CBP/p300. EMBO Journal. 1998;17:1087–1095. doi: 10.1093/emboj/17.4.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weaver BK, Ando O, Kumar KP, Reich NC. Apoptosis is promoted by the dsRNA-activated factor (DRAF1) during viral infection independent of the action of interferon or p53. FASEB J. 2001;15:501–515. doi: 10.1096/fj.00-0222com. [DOI] [PubMed] [Google Scholar]
- 33.Peters K, Chattopadhyay S, Sen GC. IRF-3 Activation by Sendai Virus Infection Is Required for Cellular Apoptosis and Avoidance of Persistence. J. Virol. 2008;82:3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holm GH, et al. Retinoic Acid-inducible Gene-I and Interferon-beta Promoter Stimulator-1 Augment Proapoptotic Responses Following Mammalian Reovirus Infection via Interferon Regulatory Factor-3. J. Biol. Chem. 2007;282:21953–21961. doi: 10.1074/jbc.M702112200. [DOI] [PubMed] [Google Scholar]
- 35.Heylbroeck C, et al. The IRF-3 Transcription Factor Mediates Sendai Virus-Induced Apoptosis. J. Virol. 2000;74:3781–3792. doi: 10.1128/jvi.74.8.3781-3792.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saito T, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proceedings of the National Academy of Sciences. 2007;104:582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yoneyama M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5:730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- 38.Takahasi K, et al. Solution Structures of Cytosolic RNA Sensor MDA5 and LGP2 C-terminal Domains. Journal of Biological Chemistry. 2009;284:17465–17474. doi: 10.1074/jbc.M109.007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kirshner JR, Karpova AY, Kops M, Howley PM. Identification of TRAIL as an Interferon Regulatory Factor 3 Transcriptional Target. J. Virol. 2005;79:9320–9324. doi: 10.1128/JVI.79.14.9320-9324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Leaman DW, et al. Identification of X-linked Inhibitor of Apoptosis-associated Factor-1 as an Interferon-stimulated Gene That Augments TRAIL Apo2L-induced Apoptosis. J. Biol. Chem. 2002;277:28504–28511. doi: 10.1074/jbc.M204851200. [DOI] [PubMed] [Google Scholar]
- 41.Toth AM, Li Z, Cattaneo R, Samuel CE. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. Journal of Biological Chemistry. 2009;284:29350–29356. doi: 10.1074/jbc.M109.045146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.XuFeng R, et al. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proceedings of the National Academy of Sciences. 2009;106:17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blow M, et al. RNA editing of human microRNAs. Genome Biology. 2006;7:R27. doi: 10.1186/gb-2006-7-4-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kawai T, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 45.Wormington M, Searfoss AM, Hurney CA. Overexpression of poly(A) binding protein prevents maturation-specific deadenylation and translational inactivation in Xenopus oocytes. EMBO Journal. 1996;15:900–909. [PMC free article] [PubMed] [Google Scholar]
- 46.Du P, Kibbe WA, Lin SM. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.