

Abstract
Transgenic (Tg) mice expressing chimeras of mouse and human prion proteins (PrP) have shorter incubation periods for Creutzfeldt-Jakob disease (CJD) prions than mice expressing full-length human PrP. Increasing the sequence similarity of the chimeric PrP to mouse PrP, by reverting human residues to mouse, resulted in a Tg line, denoted Tg22372, which was susceptible to sporadic (s) CJD prions in ~110 days 1. Reversion of one additional residue (M111V) resulted in a new Tg line, termed Tg1014, susceptible to sCJD prions in ~75 days. Tg1014 mice also has shorter incubation periods for variant (v) CJD prions, providing a more tractable model for studying this prion strain. Transmission of vCJD prions to Tg1014 mice resulted in two different strains, determined by neuropathology and biochemical analysis, which correlated with the length of the incubation time. One strain had the biochemical, neuropathological, and transmission characteristics including longer incubation times of the inoculated vCJD strain; the second strain produced a phenotype resembling that of sCJD prions including relatively shorter incubation periods. Mice with intermediate incubation periods for vCJD prions had a mixture of the two strains. Both strains were serially transmitted in Tg1014 mice, which led to further reduction in incubation periods. Conversion of vCJD-like to sCJD-like strains was favored in Tg1014 mice more than in the Tg22372 line. The single amino acid difference therefore appears to offer selective pressure for propagation of the sCJD-like strain. These two Tg mouse lines provide relatively rapid models to study human prion diseases as well as the evolution of human prion strains.
INTRODUCTION
Human prion diseases, including Creutzfeldt-Jakob disease (CJD), are invariably fatal neurological illnesses, which are of sporadic, genetic or acquired origin. Analogous diseases are known in a number of agriculturally important animal species, including bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep, and chronic wasting disease in deer. In all cases, the host-encoded prion protein (PrP) is converted from its cellular isoform, PrPC, into the disease-causing PrPSc form by a conformational rearrangement. Subsequent propagation of PrPSc occurs in an autocatalytic, template-directed manner. Transmission of prions, both within and between species, requires the refolding of endogenous PrPC into the alternative isofom PrPSc 2.
Following the BSE epidemic in the United Kingdom, almost 200 variant (v) CJD cases have been identified in relatively young individuals, with a novel neuropathological profile 3. An etiologic relationship between BSE and vCJD was implicated by transmission studies in monkeys, cats and mice 4–6. Most compellingly, the incubation times, neuropathologic profiles, and patterns of PrPSc deposition in transgenic (Tg) mice expressing the bovine (Bo) PrP gene were indistinguishable whether the inocula originated from the brains of cattle with BSE or humans with vCJD 7–9.
Variant CJD has occurred almost exclusively in individuals homozygous for methionine at polymorphic PrP codon 129. Therefore, PrPSc found in the brains of these individuals has an identical sequence to that in the brains of individuals with the most common form of sporadic (s) CJD. However, the neuropathological profiles, biochemical behavior, and transmission characteristics clearly define vCJD and sCJD as different strains. The phenomenon of prion strains has been known for almost 50 years, when “scratchy” and “nervous” clinical phenotypes of scrapie were distinguished and serially passaged in goats 10. The concept of multiple strains was expanded by the work of Dickinson and colleagues, who transmitted scrapie prions from sheep and goats to rodents 11–14. Notably, further investigations revealed that many scrapie strains were actually re-isolations of the same strains 15, 16. The existence of different strains of scrapie prions argued for the existence of an independent genome 17, yet none has been found despite numerous efforts 18, 19. From studies of prion strains in mammals and fungi, considerable data argue that strain-specified properties are enciphered in the conformation of PrPSc 20–26.
One of the simplest ways to differentiate prion strains is by Western blotting following limited digestion with proteinase K (PK). For human prion strains, the sizes and intensities of the PK-resistant PrPSc glycoforms have led to different classification schemes 27, 28. However, recent reports of multiple strain types within a single brain make this classification more complicated 29–32. The most obvious difference between strains is the size of the unglycosylated, PK-resistant fragment. The most common form of sCJD has an unglycosylated PrPSc band of ~21-kDa, described as “type 1” in the classification system of Gambetti and colleagues 27. Conversely, all clinical vCJD cases to date are accompanied by unglycosylated PrPSc band of ~19 kDa, referred to as “type 2.”
Attempts to study the replication of sCJD and vCJD prions have been hampered by their poor transmission to wild-type mice. Therefore, we and others developed transgenic (Tg) mice expressing human (Hu) PrP to provide a more efficient replication environment with sequence similarity between PrPC in the host and PrPSc in the inoculum. Transmission of sCJD prions was only efficient in the absence of endogenous mouse (Mo) PrP expression 33, 34. Generation of additional Tg mouse lines expressing transgenes chimeric for MoPrP and HuPrP afforded reduced incubation periods for human prions 1, 34.
In some instances, prion isolates have been biologically “cloned” by limiting dilution and serial passage, to the point in which a single strain appears to be present. Subsequent passage to cells or serial passage in the same species has occasionally led to emergence of alternate strains 35, 36. Drug treatments can also lead to selection of drug-resistant substrains in both cells and mice 36, 37. Such changes can be explained using the language of evolution, where strains are converted or “mutated,” or alternatively selected from a pool based on external pressures. Mutation of prion strains does not imply a hypothetical nucleic acid component of the prion, and is consistent with the “protein-only” hypothesis of prion replication. Such mutations could arise from errors in fidelity of structural conversion between template PrPSc and substrate PrPC.
Previously, we showed that serial passage of vCJD prions in Tg mice expressing the chimeric MHu2M transgene resulted in two distinct strains 1. However, the extended incubation periods in these mice precluded more detailed study. Here we report serial passage of vCJD prions in two Tg lines expressing different chimeric Mo/Hu PrP transgenes, with substantially reduced incubation times. In some cases, two distinct prion strains appeared based on typing by Western immunoblotting and on neuropathogic analysis. Our data suggest that either vCJD isolates contain low levels of sCJD-like prions, which can be selected on serial passage, or that conversion occurs between these two strains. Moreover, the sequence of the transgene can influence the rate of selection or conversion, by as small a difference as a single conservative substitution.
MATERIALS AND METHODS
Construct nomenclature
The mature sequences of MoPrP and HuPrP differ at 17 residues, with a single residue insertion in HuPrP preceding residue 55 and a deletion of two residues following residue 226 (mouse numbering). In the chimeric MHu2M constructs, all numbering corresponds to the mouse sequence. The central region from residues 96 to 167 express the HuPrP sequence, corresponding to nine differences from MoPrP at residues 96, 108, 111, 137, 142, 144, 154, 165 and 167 34. This is the construct in the Tg(MHu2M)5378 line. Subsequent reversions of these HuPrP residues to the MoPrP sequence are denoted as mutations following “MHu2M,” i.e., MHu2M(M165V,E167Q) is the MHu2M transgene with codons 165 and 167 reverted to the respective MoPrP residues, and MHu2M(M111V,M165V,E167Q) has an additional reversion at residue 111 (Suppl. Fig. 1). Both Mo/Hu chimeric constructs described here encode M at position 129. All transgenic lines are on the PrP-ablated (Prnp0/0) background. For simplicity, transgenic lines are denoted by their line number: for example, Tg(MHu2M,M111V,M165V,E167Q)1014/Prnp0/0 is referred to as Tg1014.
CJD inocula
All human prion-infected tissues were obtained from neuropathologically confirmed cases, and in all instances, the full PrP open reading frame was sequenced including the polymorphism at residue 129. Strain typing was performed by Western blotting as described below. sCJD cases were all homozygous M129, type 1, denoted MM1. vCJD samples were obtained from the U.K. National CJD Surveillance Unit. Most samples have been described previously 1, 8.
Generation and inoculation of transgenic mouse lines
All animal procedures were performed under protocols approved by the Institutional Animal Care and Use Committee at the University of California San Francisco.
The Tg(MHu2M,M111V,M165V,E167Q)1014/Prnp0/0 mouse line was created as reported previously 38, 39. Briefly, the DNA construct was generated by site-directed mutagenesis using the QuickChange II kit (Stratagene, La Jolla, CA), then cloned into the Cos.tet vector and microinjected into PrP-ablated (Prnp0/0) zygotes. Potential founder mice were identified by binding of a radiolabeled probe, complementary to the vector sequence, to genomic DNA. Hemizygous Tg(MHu2M,M165V,E167Q)22372/Prnp0/0 mice, homozygous Tg(BoPrP+/+)4092/Prnp0/0, and Tg(MHu2M,M165V,E167Q+/+)22372/Prnp0/0 mice were reported previously 1, 9, 40.
Weanling mice were intracerebrally inoculated with 30 μl of 1% brain homogenate in phosphate-buffered saline (PBS) containing 5% bovine serum albumin, and monitored for onset of disease, as previously reported 41.
Survival analysis
Median incubation periods were calculated using the Kaplan-Meier function 42, and 95% confidence intervals (c.i.) were determined 43, which more accurately represent the survival data 40. Any mice with intercurrent illnesses were censored at the time they were euthanized. For comparison of the serial dilutions inoculated into Tg1014 and Tg22372+/+ mice, we used the semi-parametric Cox regression models from survival data. We compared the onset times of disease in Tg22372+/+ mice for each dilution to the serial dilution series for Tg1014 mice to relate the Tg22372+/+ results to an equivalent titer of brain homogenate in the Tg1014 mice. Specifically, to determine equivalent titers for dilutions into Tg22372+/+, we used terms for log dilution and transgenic line: λk(t) = λ(t) exp(β log dilution + δk treatment), for which k denotes the dilution for Tg22372+/+ mice. The equivalent Tg1014 dilution for the kth dilution in Tg22372+/+ is given by = δk/β. We determined 95% ci using Wald-based confidence intervals. All calculations were performed with Stata10 (Stata Corp., College Station, TX).
Sample preparation and Western blotting
Mice were euthanized and brains were frozen on dry ice, then stored at −80 °C. After thawing, 10% (wt/vol) homogenates were prepared in PBS using a Precellys 24 bead-beater (MO BIO, Carlsbad, CA). Samples were then treated with 100 μg/ml PK for 1 h at 37 °C, and digestion was terminated by the addition of 2 mM phenylmethylsulfonyl fluoride. Samples were resuspended in 2× lithium dodecyl sulfate sample buffer and boiled for 10 min before loading onto precast 10% NuPAGE Novex Bis-Tris gels using the Xcell4 SureLock Midi cell runner system (Invitrogen, Carlsbad, CA), with Novex sharp prestained protein standard markers (Invitrogen). Proteins were transferred to nitrocellulose using the iBlot Dry Blotting system (Invitrogen) for 7 min, then blocked with 10% nonfat milk in Tris-buffered saline with Tween-20, pH 7.5. PrP bands were detected with HuM-P Fab conjugated to horseradish peroxidase, and immunoreactivity was visualized by enhanced chemiluminescence (Amersham, Piscataway, NJ) using X-ray film (Eastman Kodak, Rochester, NY).
Neuropathology
Mice were euthanized and brains were immersion-fixed in 10% (vol/vol) buffered formalin. Fixed samples were paraffin embedded, sectioned and then stained with hematoxylin and eosin, or processed by immunohistochemistry for PrP with the R2 monoclonal antibody, as described previously 44. For each mouse, four sections were studied: caudate nucleus, hippocampus/thalamus, hippocampus/midbrain, and cerebellum/pons. Seven mice were analyzed for each round of serial transmission in Tg1014 mice.
To visualize the presence of amyloid plaques, brain tissue sections were stained with 0.05% Thioflavin S (Sigma, St Louis, MO) in 50% ethanol in the dark for 8 min. Residual Thioflavin S was removed by a rapid rinse in distilled water.
RESULTS
Transgenic mice susceptible to human prions
Tg22372 mice, expressing the MHu2M(M165V,E167Q) transgene, were susceptible to human sCJD prions in ~110 and vCJD prions in ~300 days 1. While this represented an important advance over wild-type mice and other Tg lines, repeated serial passage was still time consuming. In an attempt to abbreviate further the incubation times, we generated a range of Tg lines with either different leveIs of transgene expression or additional reversions in the MHu2M(M165V,E167Q) transgene (K.G. and S.B.P., in preparation). The MHu2M transgene encodes the mouse PrP sequence with the central region between residues 96–167 (mouse numbering) expressing the human PrP sequence. Reversions of the human sequence to mouse are indicated as mutations following “MHu2M.” One Tg line expressing the MHu2M(M111V,M165V,E167Q) transgene at ~4 × the level of wild-type PrP was particularly sensitive to inoculation with sCJD, while uninoculated mice did not develop any neurological signs of disease when monitored for more than 600 days.
Brain homogenates from multiple cases of sCJD were inoculated into the Tg(MHu2M,M111V,M165V,E167Q)1014/Prnp0/0 line, and mice were monitored for disease onset. Three isolates gave median incubation periods from 77 days (95% c.i. 74, 81) to 95 days (95% c.i. 75, 102) (Table 1). sCJD prions passaged in Tg1014 mice resulted in a PrPSc pattern on Western blot similar to the inoculum, with type 1 PrPSc retained and a small difference in the relative band intensities (Fig. 1A), as was previously reported for Tg22372 mice (Fig. 1B) 1.
Table 1.
Incubation periods for sCJD and vCJD prions in transgenic mice.*
| Tg22372† | Tg1014 | ||||
|---|---|---|---|---|---|
| MHu2M(M165V,E167Q) | MHu2M(M111V,M165V,E167Q) | ||||
| Inoculum | Case | Incubation period | n/n0 | Incubation period | n/n0 |
| sCJD | HU082 | 104 (99, 115) | 7/7 | 88 (85, 92) | 5/5 |
| HU108 | 112 (111, 122) | 7/7 | 95 (75, 102) | 7/7 | |
| HU178 | 107 (104, 111) | 27/27 | 77 (74, 81) | 16/16 | |
| vCJD | RU96/02 | 368 (368, 433) | 6/6 | 280 (228, 323) | 5/5 |
| RU96/07 | n.d. | 211 (146, 244) | 8/8 | ||
| RU96/45 | 279 (224, 363) | 13/13 | 176 (147, 242) | 13/13 | |
| RU96/80 | n.d. | 256 (222, 278) | 6/6 | ||
| RU96/110 | n.d. | 326 (264, 403) | 5/5 | ||
Incubation period reported as median time to disease onset in days, with 95% confidence intervals, calculated using Kaplan-Meier statistics;
n, number of mice with clinical signs of disease; n0, number of mice inoculated; mice with intercurrent illness excluded.
Includes previously reported data 1;
n.d., not determined.
Fig. 1.
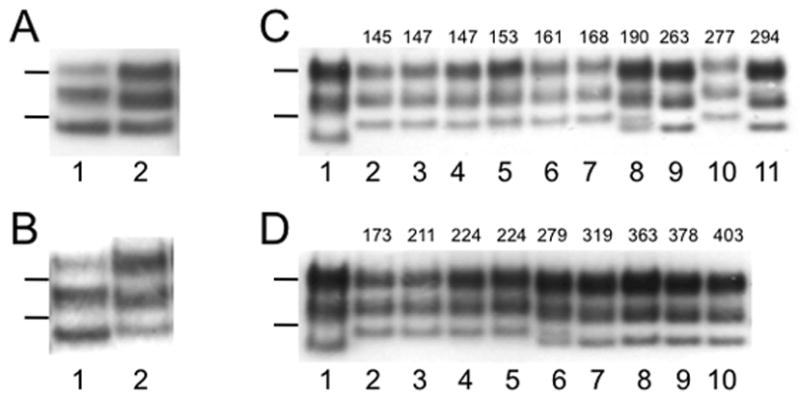
Western blotting of brain homogenates after limited digestion with PK shows strain typing of sCJD (A, B) and vCJD (C, D) prions. (A, B) sCJD prions in the brain of human sCJD case HU178 (lanes 1) retain type 1 PrPSc after passage in Tg1014 mice (A, lane 2) and Tg22372 mice (B, lane 2). (C, D) vCJD prions in the brain of human vCJD case RU96/45 before (lanes 1) and after passage in Tg1014 (C, lanes 2–11) and Tg22372 (D, lanes 2–10) mice. The incubation period, in days, for each mouse is listed above the respective lane. vCJD prions retained type 2 PrPSc in Tg mice with longer incubation periods, but changed to type 1 PrPSc in Tg mice with shorter incubation periods. Tg mice with intermediate incubation periods (190 d in C, 279 d in D) showed a mixture of type 1 and type 2 PrPSc. Molecular weight markers represent protein standards of 20 and 30 kilodaltons.
Because incubation times for each CJD case in Tg1014 mice were consistently ~25% shorter than in Tg22372 mice, we asked whether Tg1014 mice were more sensitive to sCJD by endpoint titration. Two different investigators each prepared dilution series consisting of three independent sets of 10 ten-fold dilutions of sCJD prions; each dilution was inoculated into a group of four Tg1014 mice. The survivor functions for each independent dilution series inoculated into Tg1014 mice were compared to confirm consistency of the dilutions. There were no significant differences either within the replicates of each dilution series (p=0.85, p=0.72) or between series (p=0.57), as determined by the log-rank test; therefore, the data were combined for further analysis. From the 10−1 dilution to the 10−6 dilution, increasing dilutions led to increased incubation periods. Dilutions greater than 10−7 had <50% of animals succumbing to disease, yet some mice succumbed to disease at these high dilutions (Fig. 2A). This yielded a titer of 3.5 × 107 ID50/ml from 10% sCJD brain homogenate for Tg1014 mice, as determined by the Spearman-Kärber method. This titer is slightly lower than the 7.0 × 107 ID50/ml obtained for the same sCJD case in Tg22372+/+ mice 40, 45
Fig. 2.

(A) Kaplan-Meier survival curves for serially diluted sCJD(MM1) prions (10 tenfold dilutions of 10% brain homogenate) inoculated into Tg1014 mice. (B) Graph comparing the sensitivity of Tg1014 and Tg22372+/+ mice to log dilutions of sCJD prions. Survival data for serial dilutions of sCJD in Tg22372+/+ mice were previously reported 40. To compare the survival curves for each dilution in the two Tg lines, Cox regression models were used to calculate equivalent log titers. In Tg22372 mice, the 10−1 dilution resulted in a survival curve similar to that of the ~10−4 dilution inoculated into Tg1014 mice; this difference is plotted on the ordinate. An equivalent calculation was performed for each dilution in Tg22372+/+ mice. The solid line represents the interpolation of each dilution, and dashed lines represent upper and lower 95% confidence intervals.
Titers calculated by the Spearman-Kärber method utilize only the highest dilution data, which is usually more variable than lower dilutions. To understand the apparent discrepancy between shorter incubation periods in Tg1014 mice, yet lower titer compared to Tg22372+/+ mice, we evaluated the complete dilution series for the two Tg lines. For each dilution of CJD brain homogenate inoculated into Tg22372+/+ mice, we compared survival data (time to onset of clinical disease for each mouse inoculated with a given dilution of sCJD prions), against the serial dilution survival data for the Tg1014 line. Cox proportional hazards models were used to calculate equivalent dilutions, enabling us to generate a measure of error in the form of 95% c.i. For example, survival data for the 10−1 (1 log) dilution of sCJD in Tg22372+/+ mice was equivalent to a 3.9 log dilution (95% c.i. 4.6, 2.9) in Tg1014 mice, a difference of 2.9 logs, or approximately 800-fold; the 10−2 dilution in Tg22372+/+ mice was equivalent to a 4.3 log dilution (95% c.i. 5.0, 3.5) in Tg1014 mice, a difference of 2.3 logs, or approximately 200-fold. This calculation was performed for each dilution of sCJD inoculated into Tg22372+/+ and plotted against the differences between the two lines (Fig 2B). At low dilutions of brain homogenate, the Tg1014 line was much more sensitive to sCJD prions, but at high dilutions, the sensitivity was similar for both lines.
Selection and separation of strains on passage of vCJD prions
vCJD isolates from five different patients were each inoculated into Tg1014 mice, resulting in median incubation periods ranging from 176 days (95% c.i. 147, 242) to 326 days (95% c.i. 264, 403) (Table 1). As demonstrated by the broad confidence intervals, each case had a large spread of incubation periods. Western blot analysis of the resulting brains showed distinctly different strain types depending on the incubation period. Mice succumbing to disease with shorter incubation periods had type 1 PrPSc, while those with longer incubation periods tended to have type 2 PrPSc (Fig. 1); a mouse with an intermediate incubation period contained a mixture of both type 1 and type 2 PrPSc (Fig. 1C, lane 8). Analogous results were observed for vCJD passaged in Tg22372 mice (Fig. 1D, lane 6). This phenomenon was not restricted to a single vCJD isolate but was found for other vCJD isolates (Suppl. Fig. 2).
Neuropathological phenotype correlates with biochemical strain type
Analysis of brain tissue from ill Tg1014 mice showed that neuropathology correlated with strain type. Tg1014 mice inoculated with sCJD had a neuropathology characterized by vacuolation that was similar in the cerebral cortex (Fig. 3A), the hippocampus and the caudate nucleus, and substantially less in the thalamus and brainstem (n=6 mice). Punctate deposition of PrPSc was found in association with the vacuoles (Fig. 3B, C). 0000Tg1014 mice inoculated with vCJD prions, resulting in type 1 PrPSc, exhibited neuropathological changes similar to those found after inoculation with sCJD prions: moderate vacuolation was observed in the cerebral cortex (Fig. 3E) and caudate nucleus but substantially less in the hippocampus, thalamus and brainstem (n=4 mice). Moderate numbers of kuru-type plaques were seen in the subcallosal region (Fig. 3F–G), which stained positively with Thioflavin-S, and thus were classified as amyloid deposits (Fig. 3H). In contrast, vCJD-inoculated Tg1014 mice with type 2 PrPSc had pathology characterized by little or no vacuolation in any brain region (Fig. 3I), but had moderate numbers of kuru-type, amyloid plaques, some of which were very large (n=3 mice) (Fig. 3J–L). In a Tg1014 mouse that had both type 1 and type 2 PrPSc, a third type of pathology was observed. The vacuolation was moderate in the caudate nucleus, cerebral cortex (Fig. 3M), hippocampus, thalamus and to a lesser extent in the brainstem. In addition, multiple amyloid plaques were found in the subcallosal region (Fig. 3N–P). A similar correlation between strain type by Western blot and neuropathology was observed in Tg22372 mice inoculated with vCJD (data not shown). Whereas amyloid plaques are found in only 10% of sCJD cases, formation of many PrP-immunopositive, amyloid plaques is a defining characteristic of vCJD in humans 3.
Fig. 3.

Neuropathological characterization of ill Tg1014 mice inoculated with sCJD prions (A–D) and vCJD prions (E–P). Brain sections were taken from the Tg1014 mouse inoculated with sCJD prions shown in Fig. 1A, lane 2, and Tg1014 mice inoculated with vCJD prions shown in Fig. 1C, lanes 3, 8 and 9. Tg1014 mice inoculated with sCJD prions showed moderate vacuolation (A) and punctate deposits of PrPSc (B, C), which were not amyloid (D). Tg1014 mice inoculated with vCJD, harboring type 1 PrPSc, showed moderate vacuolation similar to sCJD inoculation (E) and occasional kuru-type amyloid plaques (F–H). In mice with type 2 PrPSc, little vacuolation was observed (I) but the kuru-type amyloid plaques were large (J, K) and stained positively with Thioflavin S (L). Intermediate neuropathology was observed in Tg1014 mice with mixed type 1 and type 2 PrPSc: vacuolation was mild to moderate (M) and a few amyloid plaques were observed (N–P). Sections were stained with H&E to visualize vacuoles (left column), anti-PrP R2 antibody to visualize PrPSc deposition (middle column), and Thioflavin S to identify amyloid (right column). Bar in panel O represents 50 μm and applies panels in the three left columns; bar in panel P represents 50 μm and applies to the right column.
Transmission characteristics of mouse-passaged vCJD prions correlates with strain type
To determine whether vCJD prions passaged in Tg mice retained the transmission properties of the original vCJD strain, we prepared brain homogenates from ill Tg22372 mice that contained type 1 or type 2 prions. We injected these preparations into Tg(BoPrP+/+)4092/Prnp0/0 mice, which express bovine PrP. Tg4092+/+ mice were previously shown to be susceptible to human vCJD prions with incubation periods just slightly longer than those for BSE prions 9, but are resistant to inoculation with sCJD prions (Table 2). We observed median incubation periods of 277 days (95% c.i.: 239, 294) in Tg4092+/+ mice inoculated with type 2 PrPSc from the brains of Tg22372 mice after inoculation with vCJD prions. However, Tg4092+/+ mice inoculated with the type 1 PrPSc from Tg22372 mouse brains did not develop disease: in over 600 days of observation, only one mouse developed neurological dysfunction after 469 days (Table 2).
Table 2.
Incubation periods for vCJD prions passaged in Tg(BoPrP+/+)4092 mice.*
Strain selection on serial passage
Brain homogenates from ill, vCJD-inoculated Tg1014 mice containing type 1 PrPSc, type 2 PrPSc, or a mixture of type 1 and 2 PrPSc were prepared for serial passage. Brains from three mice with type 1 PrPSc all resulted in incubation times of ~75 dpi, despite incubation periods ranging from 147 to 277 days on first passage (Fig. 4, Suppl. Table 1). These mice had a vacuolation phenotype with few or no PrP amyloid plaques (Fig. 5A and B), similar to Tg1014 mice inoculated with sCJD prions. When the inoculum contained a mixture of type 1 and 2 PrPSc, the mice also developed neurological dysfunction in ~75 days and their brains harbored only type 1 PrPSc (Fig. 4). Second passage of the type 2 PrPSc was accompanied by a substantial reduction in the incubation period from 263 days to a median of 117 days (95% c.i.: 105, 118), but the type 2 PrPSc pattern persisted (Fig. 4, Suppl. Tables 1 and 2). Upon neuropathological examination, their brains showed minimal vacuolation characteristic of the type 2 strain, but an absence of amyloid plaques and sparse PrPSc deposits in the corpus callosum, which were not typical (Fig. 5E and F).
Fig 4.

Serial passage of vCJD prions in Tg1014 mice. Each box represents a single mouse, with its incubation period in days and PrPSc strain type indicated in color: type 1 (blue), type 2 (red), types 1 and 2 (purple), not determined (white). From center, inner ring shows the first passage with human vCJD brain homogenate, middle ring shows the second passage, and outer ring shows the third passage. For each serial passage, brain homogenate from the respective ill Tg1014 mice was prepared and inoculated into other Tg1014 mice.
Fig. 5.

Neuropathology resulting from serial passage of brain from vCJD-inoculated mice with either type 1 PrPSc (A–D) or type 2 PrPSc (E–H), in Tg1014 mice. For the type 1 strain, neuropathologic changes upon second (A, B) and third (C, D) passages were similar to first passage (Fig. 3E–G): moderate vacuolation (A, C) and few amyloid deposits (B, D) were observed. For type 2 PrPSc, second passage (E, F) showed little vacuolation (E) similar to first passage (Fig. 3I), but few amyloid plaques and sparse PrPSc deposits (F). Upon third passage of type 2 PrPSc (G, H), neuropathology resembled that of type 1 PrPSc with numerous vacuoles (G) and very few amyloid plaques (H). Sections stained with H & E (left column), and PrP immunohistochemistry (right column). Bar in panel H represents 50 μm and applies to all panels.
On third passage in Tg1014 mice, the type 1 PrPSc retained their characteristics (Fig 4, Fig. 5C and D) while the third passage of type 2 PrPSc was accompanied by a further reduction in the incubation time to 102 days (95% c.i.: 96, 109) as well as the appearance of the type 1 PrPSc pattern (Fig. 4). Brain sections showed numerous vacuoles usually associated with type 1 PrPSc; only one, kuru-type amyloid plaque was found in 1 of the 3 mice examined (Fig. 5G and H).
Brains of Tg22372 mice inoculated with vCJD were also serially passaged (Suppl. Tables 1 and 2). On second passage, two distinct strains were present, one with type 1 PrPSc and an incubation period of ~90 days, the other with type 2 PrPSc and an incubation period of ~300 days. Third passage was performed in the homozygous Tg22372+/+ line, which has slightly longer incubation periods for CJD prions than the hemizgous line 40. The brain homogenate from a mouse ill at 84 dpi, with type 1 PrPSc, caused disease with a median incubation period of ~115 days in Tg22372+/+ mice. Interestingly, two mice had type 1 PrPSc, and two had a mixture of both type 1 and type 2 PrPSc. We prepared a homogenate from one Tg22372+/+ mouse harboring type 1 and type 2 PrPSc and passaged it into additional Tg22372+/+ mice. All the resulting mice had a mixture of type 1 and type 2 PrPSc (Fig. 6). Third passage of type 2 PrPSc in the Tg22372+/+ line was accompanied by a large reduction in incubation period, but retention of the type 2 phenotype. Fourth passage of two brains resulted again in retention of type 2 PrPSc, with similar incubation periods (Fig. 6).
Fig. 6.

Serial passage of vCJD prions in Tg22372 mice. Each box represent a single mouse, with its incubation period in days and strain type indicated in color: type 1 (blue), type 2 (red), types 1 and 2 (purple), not determined (white). From center, inner ring shows the first passage with human vCJD brain homogenate, second ring shows second passage, third ring shows third passage, outer ring shows fourth passage. For first and second serial passage, brain homogenates from the respective ill Tg22372 mice were prepared and inoculated into other hemizygous Tg22372 mice. For third and fourth passage, inoculations were performed in homozygous Tg22372+/+ mice.
We also reviewed serial transmission of two separate isolates of vCJD in the Tg(MHu2M)5378/Prnp0/0 line. Although some samples were no longer available to analyze, a pattern similar to the Tg22372 line emerged, with both type 1 and type 2 strains being maintained upon serial passage (Suppl. Fig. 3).
DISCUSSION
A wealth of evidence argues that the sole component of mammalian prions is the conformational isoform PrPSc. To account for the different properties of prion strains, it has been suggested that variations in the conformation of PrPSc might be site where biological information for the prion is enciphered 46. There is now a large body of evidence to support this hypothesis for both mammalian 20, 21, 24 and yeast 23, 47, 48 prions. Varying the refolding conditions of recombinant PrP led to multiple novel strains of synthetic mammalian prions 22, 25.
The BSE epidemic was caused by feeding cattle contaminated meat and bone meal (MBM) prepared from sheep, cattle, pigs and chickens 49. As carcasses of infected but presymptomatic cattle were rendered into additional MBM, successive rounds of rendering may have played a role in the selection of a particularly resistant prion strain. To date, transmission of BSE prions to humans, resulting in vCJD, has to date only shown a single phenotype. However, our extensive serial passage data in transgenic mice suggest that prion strains can occasionally be modified upon serial passage.
Transmission of vCJD prions to Tg1014 or Tg22372 mice expressing a chimeric Mo/Hu PrP transgene resulted in two distinct strain types. Mice that succumbed to disease more rapidly had type 1 PrPSc, and neuropathology characterized by widespread vacuolation, with a few small amyloid plaques. This strain was similar to sCJD transmitted to the same Tg lines. Mice that had long incubation periods following inoculation with the same vCJD isolate tended to have type 2 PrPSc, little or no vacuolation, and large amyloid plaques. Mice with intermediate incubation periods showed both PrPSc strain types by biochemical analysis and had an intermediate pathology. The resulting type 2 strain not only more closely resembled the original inoculum but also transmitted to Tg(BoPrP) mice with very similar incubation periods to vCJD and BSE prions. Conversely, the type 1 strain resulting from passage of vCJD did not transmit to Tg(BoPrP) mice, analogous to sCJD.
On serial passage in Tg1014 mice, the type 1 strain ultimately predominated. In Tg22372 mice, two strains were clearly differentiated by second passage, and the type 2 strain was maintained after two additional passages. In contrast, the type 1 strain, in some mice, resulted in a mixture of type 1 and 2 PrPSc, which was maintained on further passage (Fig. 6). The only difference in the transgene between these two lines is the conservative substitution M111V, which appears to have a major impact on strain selection.
Changes in strain properties upon transmission to a different host have been known for many years 50. Indeed, the appearance of both vCJD-like and sCJD-like strains was observed upon passage of BSE into transgenic mice expressing HuPrP 51. We report here that changes in strains can also occur on serial passage in genetically identical hosts, and that the PrP sequence of the host can preferentially determine which strain is propagated.
Two possible mechanisms might account for the apparent changes in prion strain type. One model argues the strains are rarely clonal and that selected strains are replicated from mixtures. As another possibility, distinct prion strains may interconvert to alternative conformations 24. Our transmission data on the appearance of a mixed type 1 and 2 phenotype from passage of type 1 prions support the first hypothesis. Western blotting is usually performed on brain homogenates and is therefore an average; minor components may not be observed. Furthermore, neuropathologic analysis revealed occasional amyloid plaques in “type 1” mice inoculated with vCJD prions. Brain homogenates of these type 1 mice prepared for serial passage would have an uneven distribution of these occasional plaques, and therefore, some mice would receive more plaques than others. More dramatic was the strain change from type 2 PrPSc accompanied by limited vacuolation, to type 1 PrPSc with widespread vacuolation, which would appear to be more easily explained by a transformation to a more rapidly replicating strain. A similar strain transformation was observed with a synthetic prion strain, denoted MoSP1, upon serial passage independently in mice and in cell culture. In both cases, MoSP1, a type 2 strain, changed to a type 1 strain accompanied by shorter incubation periods 52.
The shift of prion strains to more rapidly replicating isolates has been observed previously. Serial passage of the 87A strain in mice resulted in the more rapidly replicating Me7 strain, on a number of independent occasions 17. Similar observations were made for prions passaged in hamsters: the DY strain with a longer incubation time converted to the HY strain 35, and the long-incubation-time 139H strain 53 has occasionally changed into the shorter-incubation-time Sc237 strain (D.G. and S.B.P., unpublished data). Conversely, interference of prion replication has been observed upon co-inoculation of multiple rodent-adapted strains 54, 55, which has been suggested to be due to competition for PrPC56.
The reasons are unclear why one prion strain changes whereas another remains stable upon passage. Assuming strains are distinct conformations, they should each occupy local minima on the energy folding landscape. Both absolute and relative energies between strains would likely be influenced by the amino acid sequence of the polypeptide chain. A mutant or chimeric PrP sequence may infer a different energy landscape compared to a wild-type species sequence. This might explain why we more readily observe strain changes in the Tg lines expressing chimeric mouse/human PrP described here, if the two strains have similar stabilities. Alternatively, if prion isolates are actually mixtures of strains, since prion growth is exponential, small changes in the replication of certain subtypes at or shortly after inoculation could have a large impact on the proportions of final strains observed in sick mice many months later.
Secondary transmission of vCJD has been reported from blood products derived from donors who later developed vCJD 57–59. The studies reported here suggest that second transmission of vCJD may not always produce the vCJD phenotype, and could be indistinguishable from sCJD. The large potential number of asymptomatic vCJD carriers 60 is a major concern to blood transfusion organizations. If second transmission does not present similar clinical signs and neuropathology as vCJD, it will be difficult to track the full extent of the epidemic.
In conclusion, the Tg mice described here that express chimeric human/mouse PrP genes provide a novel set of tools with which to study strains of human prions. These mice also provide a means of evaluating human prion infectivity relatively rapidly – such bioassays are likely to play a pivotal role in the development of therapeutics for treating patients dying of CJD.
Supplementary Material
Acknowledgments
The authors thank Dr. Pierre Lessard and the staff of the Hunter’s Point animal facility, and Ms. Hang Nguyen for expert editorial assistance. This work was supported by grants from the National Institutes of Health (AG02132, AG10770, and AG021601) as well as by gifts from the Sherman Fairchild and Lincy Foundations.
Abbreviations
- CJD
Creutzfeldt-Jakob disease
- sCJD
sporadic Creutzfeldt-Jakob disease
- vCJD
variant Creutzfeldt-Jakob disease
- PrP
prion protein
- Tg
transgenic
- Mo
mouse
- Hu
human
References
- 1.Korth C, Kaneko K, Groth D, et al. Abbreviated incubation times for human prions in mice expressing a chimeric mouse — human prion protein transgene. Proc Natl Acad Sci USA. 2003;100:4784–4789. doi: 10.1073/pnas.2627989100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Prusiner SB. Shattuck Lecture — Neurodegenerative diseases and prions. N Engl J Med. 2001;344:1516–1526. doi: 10.1056/NEJM200105173442006. [DOI] [PubMed] [Google Scholar]
- 3.Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347:921–925. doi: 10.1016/s0140-6736(96)91412-9. [DOI] [PubMed] [Google Scholar]
- 4.Lasmézas CI, Deslys J-P, Demaimay R, et al. BSE transmission to macaques. Nature. 1996;381:743–744. doi: 10.1038/381743a0. [DOI] [PubMed] [Google Scholar]
- 5.Collinge J, Sidle KCL, Meads J, et al. Molecular analysis of prion strain variation and the aetiology of "new variant" CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 6.Bruce ME, Will RG, Ironside JW, et al. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature. 1997;389:498–501. doi: 10.1038/39057. [DOI] [PubMed] [Google Scholar]
- 7.Scott MR, Safar J, Telling G, et al. Identification of a prion protein epitope modulating transmission of bovine spongiform encephalopathy prions to transgenic mice. Proc Natl Acad Sci US A. 1997;94:14279–14284. doi: 10.1073/pnas.94.26.14279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Scott MR, Will R, Ironside J, et al. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proc Natl Acad Sci USA. 1999;96:15137–15142. doi: 10.1073/pnas.96.26.15137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott MR, Peretz D, Nguyen H-OB, et al. Transmission barriers for bovine, ovine, and human prions in transgenic mice. J Virol. 2005;79:5259–5271. doi: 10.1128/JVI.79.9.5259-5271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pattison IH, Millson GC. Scrapie produced experimentally in goats with special reference to the clinical syndrome. J Comp Pathol. 1961;71:101–108. doi: 10.1016/s0368-1742(61)80013-1. [DOI] [PubMed] [Google Scholar]
- 11.Dickinson AG, Mackay JMK, Zlotnik I. Transmission by contact of scrapie in mice. J Comp Pathol. 1964;74:250–254. doi: 10.1016/s0368-1742(64)80030-8. [DOI] [PubMed] [Google Scholar]
- 12.Dickinson AG, Fraser H. An assessment of the genetics of scrapie in sheep and mice. In: Prusiner SB, Hadlow WJ, editors. Slow Transmissible Diseases of the Nervous System. Vol. 1. New York: Academic Press; 1979. pp. 367–386. [Google Scholar]
- 13.Bruce ME, Dickinson AG, Fraser H. Cerebral amyloidosis in scrapie in the mouse: effect of agent strain and mouse genotype. Neuropathol Appl Neurobiol. 1976;2:471–478. [Google Scholar]
- 14.Bruce ME, Dickinson AG. Genetic control of amyloid plaque production and incubation period in scrapie-infected mice. J Neuropathol Exp Neurol. 1985;44:285–294. doi: 10.1097/00005072-198505000-00006. [DOI] [PubMed] [Google Scholar]
- 15.Bruce ME, Dickinson AG. Biological stability of different classes of scrapie agent. In: Prusiner SB, Hadlow WJ, editors. Slow Transmissible Diseases of the Nervous System. Vol. 2. New York: Academic Press; 1979. pp. 71–86. [Google Scholar]
- 16.Ridley RM, Baker HF. To what extent is strain variation evidence for an independent genome in the agent of the transmissible spongiform encephalopathies? Neurodegeneration. 1996;5:219–231. doi: 10.1006/neur.1996.0030. [DOI] [PubMed] [Google Scholar]
- 17.Bruce ME, Dickinson AG. Biological evidence that the scrapie agent has an independent genome. J Gen Virol. 1987;68:79–89. doi: 10.1099/0022-1317-68-1-79. [DOI] [PubMed] [Google Scholar]
- 18.Meyer N, Rosenbaum V, Schmidt B, et al. Search for a putative scrapie genome in purified prion fractions reveals a paucity of nucleic acids. J Gen Virol. 1991;72:37–49. doi: 10.1099/0022-1317-72-1-37. [DOI] [PubMed] [Google Scholar]
- 19.Safar JG, Kellings K, Serban A, et al. Search for a prion-specific nucleic acid. J Virol. 2005;79:10796–10806. doi: 10.1128/JVI.79.16.10796-10806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66:2096–2101. doi: 10.1128/jvi.66.4.2096-2101.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274:2079–2082. doi: 10.1126/science.274.5295.2079. [DOI] [PubMed] [Google Scholar]
- 22.Legname G, Nguyen H-OB, Peretz D, et al. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci USA. 2006;103:19105–19110. doi: 10.1073/pnas.0608970103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toyama BH, Kelly MJ, Gross JD, Weissman JS. The structural basis of yeast prion strain variants. Nature. 2007;449:233–237. doi: 10.1038/nature06108. [DOI] [PubMed] [Google Scholar]
- 24.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318:930–936. doi: 10.1126/science.1138718. [DOI] [PubMed] [Google Scholar]
- 25.Colby DW, Giles K, Legname G, et al. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci USA. 2009;106:20417–20422. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colby DW, Wain R, Baskakov IV, et al. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778. doi: 10.1002/ana.410390613. [DOI] [PubMed] [Google Scholar]
- 28.Hill AF, Collinge J. Subclinical prion infection in humans and animals. Br Med Bull. 2003;66:161–170. doi: 10.1093/bmb/66.1.161. [DOI] [PubMed] [Google Scholar]
- 29.Uro-Coste E, Cassard H, Simon S, et al. Beyond PrPres type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008;4:e1000029. doi: 10.1371/journal.ppat.1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Levavasseur E, Laffont-Proust I, Morain E, et al. Regulating factors of PrP glycosylation in Creutzfeldt-Jakob disease--implications for the dissemination and the diagnosis of human prion strains. PLoS ONE. 2008;3:e2786. doi: 10.1371/journal.pone.0002786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Parchi P, Strammiello R, Notari S, et al. Incidence and spectrum of sporadic Creutzfeldt-Jakob disease variants with mixed phenotype and co-occurrence of PrP(Sc) types: an updated classification. Acta Neuropathol. 2009;118:659–671. doi: 10.1007/s00401-009-0585-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cali I, Castellani R, Alshekhlee A, et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: its effect on the phenotype and prion-type characteristics. Brain. 2009;132:2643–2658. doi: 10.1093/brain/awp196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Telling GC, Scott M, Hsiao KK, et al. Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci USA. 1994;91:9936–9940. doi: 10.1073/pnas.91.21.9936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Telling GC, Scott M, Mastrianni J, et al. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell. 1995;83:79–90. doi: 10.1016/0092-8674(95)90236-8. [DOI] [PubMed] [Google Scholar]
- 35.Bartz JC, Bessen RA, McKenzie D, et al. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol. 2000;74:5542–5547. doi: 10.1128/jvi.74.12.5542-5547.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li J, Browning S, Mahal SP, et al. Darwinian evolution of prions in cell culture. Science. 2010;327:869–872. doi: 10.1126/science.1183218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5:e1000673. doi: 10.1371/journal.ppat.1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott M, Foster D, Mirenda C, et al. Transgenic mice expressing hamster prion protein produce species-specific scrapie infectivity and amyloid plaques. Cell. 1989;59:847–857. doi: 10.1016/0092-8674(89)90608-9. [DOI] [PubMed] [Google Scholar]
- 39.Scott MR, Köhler R, Foster D, Prusiner SB. Chimeric prion protein expression in cultured cells and transgenic mice. Protein Sci. 1992;1:986–997. doi: 10.1002/pro.5560010804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giles K, Glidden DV, Beckwith R, et al. Resistance of bovine spongiform encephalopathy (BSE) prions to inactivation. PLoS Pathog. 2008;4:e1000206. doi: 10.1371/journal.ppat.1000206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carlson GA, Kingsbury DT, Goodman PA, et al. Linkage of prion protein and scrapie incubation time genes. Cell. 1986;46:503–511. doi: 10.1016/0092-8674(86)90875-5. [DOI] [PubMed] [Google Scholar]
- 42.Kaplan EL, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 43.Brookmeyer R, Crowley J. A confidence interval for the median survival time. Biometrics. 1982;38:29–41. [Google Scholar]
- 44.Muramoto T, DeArmond SJ, Scott M, et al. Heritable disorder resembling neuronal storage disease in mice expressing prion protein with deletion of an α-helix. Nat Med. 1997;3:750–755. doi: 10.1038/nm0797-750. [DOI] [PubMed] [Google Scholar]
- 45.Peretz D, Supattapone S, Giles K, et al. Inactivation of prions by acidic sodium dodecyl sulfate. J Virol. 2006;80:322–331. doi: 10.1128/JVI.80.1.322-331.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 47.Tanaka M, Chien P, Naber N, et al. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 49.Wilesmith JW, Ryan JBM, Atkinson MJ. Bovine spongiform encephalopathy: epidemiologic studies on the origin. Vet Rec. 1991;128:199–203. doi: 10.1136/vr.128.9.199. [DOI] [PubMed] [Google Scholar]
- 50.Pattison IH. Experiments with scrapie with special reference to the nature of the agent and the pathology of the disease. In: Gajdusek DC, Gibbs CJ Jr, Alpers MP, editors. Slow, Latent and Temperate Virus Infections, NINDB Monograph 2. Washington, D.C: U.S. Government Printing; 1965. pp. 249–257. [Google Scholar]
- 51.Asante EA, Linehan JM, Desbruslais M, et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–6366. doi: 10.1093/emboj/cdf653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghaemmaghami S, Nguyen H-OB, Watts J, et al. Biological adaptation of synthetic prion strains. PLoS Biol. In preparation. [Google Scholar]
- 53.Kimberlin RH, Walker CA. Evidence that the transmission of one source of scrapie agent to hamsters involves separation of agent strains from a mixture. J Gen Virol. 1978;39:487–496. doi: 10.1099/0022-1317-39-3-487. [DOI] [PubMed] [Google Scholar]
- 54.Dickinson AG, Fraser H, Meikle VMH, Outram GW. Competition between different scrapie agents in mice. Nature New Biol. 1972;237:244–245. doi: 10.1038/newbio237244a0. [DOI] [PubMed] [Google Scholar]
- 55.Schutt CR, Bartz JC. Prion interference with multiple prion isolates. Prion. 2008;2:61–63. doi: 10.4161/pri.2.2.6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shikiya RA, Ayers JI, Schutt CR, et al. Co-infecting prion strains compete for a limiting cellular resource. J Virol. doi: 10.1128/JVI.00243-10. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Llewelyn CA, Hewitt PE, Knight RS, et al. Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet. 2004;363:417–421. doi: 10.1016/S0140-6736(04)15486-X. [DOI] [PubMed] [Google Scholar]
- 58.Peden AH, Head MW, Ritchie DL, et al. Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet. 2004;364:527–529. doi: 10.1016/S0140-6736(04)16811-6. [DOI] [PubMed] [Google Scholar]
- 59.Health Protection Agency. Variant CJD and plasma products. 2009 http://www.hpa.org.uk/webw/HPAweb&HPAwebStandard/HPAweb_C/1195733818681?p=1225960597236.
- 60.Hilton DA, Ghani AC, Conyers L, et al. Prevalence of lymphoreticular prion protein accumulation in UK tissue samples. J Pathol. 2004;203:733–739. doi: 10.1002/path.1580. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.